Co-reporter:Thomas H. Allen, Takuji Kawamoto, Sean Gardner, Steven J. Geib, and Dennis P. Curran
Organic Letters July 7, 2017 Volume 19(Issue 13) pp:
Publication Date(Web):June 22, 2017
DOI:10.1021/acs.orglett.7b01777
Boron–hydrogen bond insertion reactions of N-heterocyclic carbene (NHC) boranes and diazoesters can be catalyzed by NHC-boryl iodides and produce stable α-NHC-boryl esters. The conditions of the reaction resemble the previous rhodium-catalyzed transformations (only the catalyst is different); however, the mechanisms of the two reactions are probably very different. The new boryl iodide catalyzed method is adept at producing α-substituted-α-NHC-boryl esters, and this has led to a family of NHC-boryl esters with amino acid and amino-acid-like side chains.
Co-reporter:Daniel A. Bolt and Dennis P. Curran
The Journal of Organic Chemistry December 15, 2017 Volume 82(Issue 24) pp:13746-13746
Publication Date(Web):November 8, 2017
DOI:10.1021/acs.joc.7b02730
1-Butyl-3-methylimidazol-2-ylidene borane has been synthesized directly from two inexpensive commercial reagents: 1-butyl-3-methylimidazolium bromide and sodium borohydride. This NHC-borane reagent is a stable, free-flowing liquid that shows promise for use in radical, ionic, and metal-catalyzed reactions.
Co-reporter:Takashi Watanabe, Steven J. Geib, Dennis P. Curran, and Tsuyoshi Taniguchi
The Journal of Organic Chemistry December 15, 2017 Volume 82(Issue 24) pp:13034-13034
Publication Date(Web):November 9, 2017
DOI:10.1021/acs.joc.7b01981
Thermal reactions of benzo[3,4]cyclodec-3-ene-1,5-diyne with N-heterocyclic carbene boranes (NHC-boranes) provided mixtures of 9-borylated 1,2,3,4-tetrahydroanthracenes along with 1,2,3,4-tetrahydroanthracene. These products indicate that NHC-boranes serve as hydrogen donors to a p-benzyne intermediate formed by the Masamune–Bergman reaction. Experimental results support a radical mechanism in nonpolar solvents, but suggest that ionic mechanisms compete in the production of 1,2,3,4-tetrahydroanthracene when the reaction is performed in a polar solvent.
Co-reporter:Timothy R. McFadden, Cheng Fang, Steven J. Geib, Everett Merling, Peng LiuDennis P. Curran
Journal of the American Chemical Society 2017 Volume 139(Issue 5) pp:1726-1729
Publication Date(Web):January 20, 2017
DOI:10.1021/jacs.6b09873
Reaction of bis-(2,6-diisopropylphenyl)-imidazol-2-ylidene borane with dimethyl acetylenedicarboxylate gives 80% yield of a stable borirane (boracyclopropane) formed by formal double hydroboration along with 5% of the (E)-alkenylborane. DFT calculations suggest a mechanism where divergence to the two products occurs after a common initial stage of hydride transfer from the NHC-borane to the acetylenedicarboxylate.
Co-reporter:Dennis P. Curran;Timothy R. McFadden
Journal of the American Chemical Society 2016 Volume 138(Issue 24) pp:7741-7752
Publication Date(Web):May 31, 2016
DOI:10.1021/jacs.6b04014
A unified kinetic theory for both initiation and autoxidation reactions of Et3B and O2 is put forth, and then divided into low-oxygen and high-oxygen experimental regimes for application of Et3B/O2 as an initiating system. In the low-oxygen regime, only long, efficient chains can be initiated. In the high-oxygen regime, less efficient chains can be initiated but they must compete with autoxidation. We apply the analysis along with new experimental results to show why AIBN and Et3B/O2 give different stereochemical results in hydrostannation reactions of propargyl silyl ethers. Counterintuitively, AIBN is the better initiator, initiating both the rapid chain hydrostannation and the subsequent slow E/Z isomerization. AIBN gives the thermodynamic results. Et3B/O2 is the poorer initiator, initiating only the hydrostannation and not the isomerization. Et3B gives the kinetic result. We further apply the analysis to understand recent results in Et3B/water reductions.
Co-reporter:Andrew J. Clark, Dennis P. Curran, David J. Fox, Franco Ghelfi, Collette S. Guy, Benjamin Hay, Natalie James, Jessica M. Phillips, Fabrizio Roncaglia, Philip B. Sellars, Paul Wilson, and Hanmo Zhang
The Journal of Organic Chemistry 2016 Volume 81(Issue 13) pp:5547-5565
Publication Date(Web):June 6, 2016
DOI:10.1021/acs.joc.6b00889
The barrier to rotation around the N-alkenyl bond of 38 N-alkenyl-N-alkylacetamide derivatives was measured (ΔG⧧ rotation varied between <8.0 and 31.0 kcal mol–1). The most important factor in controlling the rate of rotation was the level of alkene substitution, followed by the size of the nitrogen substituent and, finally, the size of the acyl substituent. Tertiary enamides with four alkenyl substituents exhibited half-lives for rotation between 5.5 days and 99 years at 298 K, sufficient to isolate enantiomerically enriched atropisomers. The radical cyclizations of a subset of N-alkenyl-N-benzyl-α-haloacetamides exhibiting relatively high barriers to rotation round the N-alkenyl bond (ΔG⧧ rotation >20 kcal mol–1) were studied to determine the regiochemistry of cyclization. Those with high barriers (>27 kcal mol–1) did not lead to cyclization, but those with lower values produced highly functionalized γ-lactams via a 5-endo-trig radical–polar crossover process that was terminated by reduction, an unusual cyclopropanation sequence, or trapping with H2O, depending upon the reaction conditions. Because elevated temperatures were necessary for cyclization, this precluded study of the asymmetric transfer in the reaction of individual atropisomers. However, enantiomerically enriched atropsiomeric enamides should be regarded as potential asymmetric building blocks for reactions that can be accomplished at room temperature.
Co-reporter:Thomas H. Allen and Dennis P. Curran
The Journal of Organic Chemistry 2016 Volume 81(Issue 5) pp:2094-2098
Publication Date(Web):February 12, 2016
DOI:10.1021/acs.joc.6b00091
Relative reactivities of a series of neutral ligated boranes L-BH3 (where L is NHC, amine, pyridine, or phosphine) and the cyanoborohydride anion have been assessed in Rh(II)-catalyzed B–H insertion reactions with methyl 2-phenyl-2-diazoacetate. Stable α-boryl ester products were isolated by flash chromatography in all cases except for the salt product from cyanoborohydride. All of the substrates were either comparable to or more reactive than 1,4-cyclohexadiene, which is one of the most reactive substrates in C–H insertion reactions. The range of reactivity between the most reactive pyridine-borane and the least reactive phosphine-borane is a factor of approximately 40.
Co-reporter:Thomas H. Allen, Steven J. Geib, and Dennis P. Curran
Organometallics 2016 Volume 35(Issue 17) pp:2975-2979
Publication Date(Web):August 25, 2016
DOI:10.1021/acs.organomet.6b00497
Reactions of NHC-boranes with several diazo compounds and one diazonium salt give new classes of N-substituted NHC-boranes. Reactions with diazo compounds and azo initiators such as AIBN provide N-(cyanoalkyl)-N-borylhydrazones. However, the reactant diazo 2,2-dimethyl-1,3-dioxane-4,6-dione adds directly to the NHC-borane to give an N-boryl hydrazone product arising formally from 1,1-hydroboration. A similar thermal reaction of an NHC-borane with a diazonium salt gives an intermediate that upon neutralization undergoes air oxidation to provide the first NHC-boryl azoxy compound.
Co-reporter:Takuji Kawamoto; Steven J. Geib
Journal of the American Chemical Society 2015 Volume 137(Issue 26) pp:8617-8622
Publication Date(Web):June 8, 2015
DOI:10.1021/jacs.5b04677
The observation that NHC-boryl radicals abstract cyano groups from various organic nitriles has been parlayed into two complementary transformations. In the main group chemistry aspect, reactions of various NHC-boranes with simple organic dinitriles selectively provide stable NHC-boryl mono- or dinitriles, depending on the nitrile source. In the organic synthesis aspect, reaction of malononitriles and related derivatives with readily available 1,3-dimethylimidazol-2-ylidene borane provides reductively decyanated products in good yields.
Co-reporter:Takashi Watanabe, Dennis P. Curran, and Tsuyoshi Taniguchi
Organic Letters 2015 Volume 17(Issue 14) pp:3450-3453
Publication Date(Web):June 26, 2015
DOI:10.1021/acs.orglett.5b01480
N-Heterocyclic carbene boranes (NHC-boranes) hydroborate arynes formed in situ by the hexadehydro-Diels–Alder (HDDA) reactions of triyne precursors. The reaction directly provides functionalized arylborane compounds. The unique feature of the NHC-boranes compared to other boranes is that they hydroborate only the aryne product and not the triyne precursor.
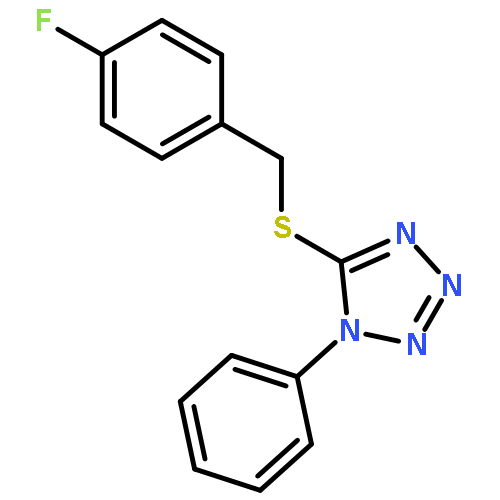
Co-reporter:Swapnil Nerkar and Dennis P. Curran
Organic Letters 2015 Volume 17(Issue 14) pp:3394-3397
Publication Date(Web):July 6, 2015
DOI:10.1021/acs.orglett.5b01101
Readily available NHC-arylboranes (NHC-BH2Ar) are converted in high yield to stable NHC-difluoro(aryl)boranes (NHC-BF2Ar) by treatment with 2 equiv of 1-chloromethyl-4-fluoro-1,4-diazonia-bicyclo[2.2.2]octane bis(tetrafluoroborate) (Selectfluor). In turn, the NHC-difluoro(aryl)boranes participate directly in Suzuki reactions under conditions previously used for anionic trifluoroborate salts. Accordingly, NHC-difluoroboranes are a new class of stable precursors for Suzuki reactions.
Co-reporter:Krishnan Damodaran, Xiben Li, Xiangcheng Pan, and Dennis P. Curran
The Journal of Organic Chemistry 2015 Volume 80(Issue 9) pp:4465-4469
Publication Date(Web):April 3, 2015
DOI:10.1021/acs.joc.5b00324
Dynamic NMR spectroscopy has been used to measure rotation barriers in five B,B-disubstituted 1,3-dimethylimidazol-2-ylidene boranes. The barriers are attributed to the sp2–sp3 bond between C(1) of the N-heterocyclic carbene ring and the boron atom. Bonds to boron atoms bearing a thexyl (1,1,2-trimethylpropyl) group show especially high barriers, ranging from 75–86 kJ mol–1. 2-Isopropyl-1,3,5-trimethylbenzene is used as a comparable to help understand the nature and magnitude of the barriers.
Co-reporter:Sean Gardner, Takuji Kawamoto, and Dennis P. Curran
The Journal of Organic Chemistry 2015 Volume 80(Issue 19) pp:9794-9797
Publication Date(Web):September 3, 2015
DOI:10.1021/acs.joc.5b01682
1,3-Dialkylimidazol-2-ylidene boranes have been made in moderate yields (typically 23–53%) from the corresponding N-alkyl imidazole, an alkylating agent (usually MeI), and sodium borohydride (NaBH4). The two-step, one-pot reaction sequence takes about 1 day to perform. The reagents are inexpensive, and the reactions are easy to conduct. The synthesis of 1,3-dimethylimidazol-2-ylidene borane has been conducted on scales up to 100 mmol and is especially convenient because the pure product can be isolated by direct recrystallization from water.
Co-reporter:E. Ben Hay; Hanmo Zhang
Journal of the American Chemical Society 2014 Volume 137(Issue 1) pp:322-327
Publication Date(Web):December 5, 2014
DOI:10.1021/ja510608u
1,1-Divinyl-2-phenylcyclopropanes are entry points to a rich area of rearrangement chemistry. With N,N-diallyl amide substrates, tandem radical cyclizations can be initiated at room temperature. Warming provides products of pure thermal rearrangements with acids, ester, and amides. These isomerizations give vinylcyclopentenes resulting from divinylcyclopropane rearrangements and more deeply rearranged tricyclic spirolactams resulting from aromatic Cope rearrangements followed by ene reactions. Conversion of the carbonyl group to an alcohol or ether opens retro-ene pathways followed by either tautomerization or Claisen rearrangement.
Co-reporter:Hanmo Zhang, Kyu Ok Jeon, E. Ben Hay, Steven J. Geib, Dennis P. Curran, and Matthew G. LaPorte
Organic Letters 2014 Volume 16(Issue 1) pp:94-97
Publication Date(Web):December 6, 2013
DOI:10.1021/ol403078e
A radical [3 + 2]-divinylcyclopropane annulation cascade has been extended to encompass five D-ring variants of the meloscine/epimeloscine core structure. Representative ABCD tetracyclic intermediates were further elaborated with novel substituted E-rings through subsequent transformations of advanced intermediates that provided opportunities for late-stage variation of the B-ring (lactam) N-substituents which were also developed.
Co-reporter:Xiangcheng Pan and Dennis P. Curran
Organic Letters 2014 Volume 16(Issue 10) pp:2728-2731
Publication Date(Web):May 6, 2014
DOI:10.1021/ol5010164
Newly discovered boryl sulfides and N-borylthioamides are shown to serve as neutral sources of sulfur nucleophiles in substitutions reactions. For example, heating of diMe-Imd-BH(SPh)2 with benzyl bromides, primary bromides, or acid chlorides provides the corresponding thioethers or thioesters in high yields. Likewise, N-phenyltetrazole thioethers/esters are made from a readily available N-borylthionotetrazole. The formation of the boryl sulfide and its onward nucleophilic substitution can be telescoped down to a one-pot reaction whose components are an NHC–borane (NHC-BH3), a disulfide, and an electrophile.
Co-reporter:Dr. Tsuyoshi Taniguchi;Dr. Dennis P. Curran
Angewandte Chemie International Edition 2014 Volume 53( Issue 48) pp:13150-13154
Publication Date(Web):
DOI:10.1002/anie.201408345
Abstract
Arynes were generated in situ from ortho-silyl aryl triflates and fluoride ions in the presence of stable N-heterocyclic carbene boranes (NHCBH3). Spontaneous hydroboration ensued to provide stable B-aryl-substituted NHC-boranes (NHCBH2Ar). The reaction shows good scope in terms of both the NHC-borane and aryne components and provides direct access to mono- and disubstituted NHC-boranes. The formation of unusual ortho regioisomers in the hydroboration of arynes with an electron-withdrawing group supports a hydroboration process with hydride-transfer character.
Co-reporter:Norazah Basar, Krishnan Damodaran, Hao Liu, Gareth A. Morris, Hasnah M. Sirat, Eric J. Thomas, and Dennis P. Curran
The Journal of Organic Chemistry 2014 Volume 79(Issue 16) pp:7477-7490
Publication Date(Web):July 14, 2014
DOI:10.1021/jo5012027
A systematic process is introduced to compare 13C NMR spectra of two (or more) candidate samples of known structure to a natural product sample of unknown structure. The process is designed for the case where the spectra involved can reasonably be expected to be very similar, perhaps even identical. It is first validated by using published 13C NMR data sets for the natural product 4,6,8,10,16,18-hexamethyldocosane. Then the stereoselective total syntheses of two candidate isomers of the related 4,6,8,10,16-pentamethyldocosane natural product are described, and the process is applied to confidently assign the configuration of the natural product as (4S,6R,8R,10S,16S). This is accomplished even though the chemical shift differences between this isomer and its (16R)-epimer are only ±5–10 ppb (±0.005–0.01 ppm).
Co-reporter:Edmund A.-H. Yeh ; Eveline Kumli ; Krishnan Damodaran
Journal of the American Chemical Society 2013 Volume 135(Issue 4) pp:1577-1584
Publication Date(Web):January 8, 2013
DOI:10.1021/ja311606u
All four diastereomers of a typical saturated oligoisoprenoid, 4,8,12-trimethylnonadecanol, are made by an iterative three-step cycle with the aid of traceless thionocarbonate fluorous tags to encode configurations. The tags have a minimum number of total fluorine atoms, starting at zero and increasing in increments of one. With suitable acquisition and data processing, each diastereomer exhibits characteristic chemical shifts of methyl resonances in its 1H and 13C NMR spectra. Together, these shifts provide a basis to predict the appearance of the methyl region of the spectrum of every stereoisomer of higher saturated oligoisoprenoids.
Co-reporter:Xiangcheng Pan ; Anne-Laure Vallet ; Stéphane Schweizer ; Karim Dahbi ; Bernard Delpech ; Nicolas Blanchard ; Bernadette Graff ; Steven J. Geib ; Dennis P. Curran ; Jacques Lalevée ;Emmanuel Lacôte
Journal of the American Chemical Society 2013 Volume 135(Issue 28) pp:10484-10491
Publication Date(Web):May 29, 2013
DOI:10.1021/ja403627k
Reactions of 1,3-dimethylimidazol-2-ylidene–borane (diMe-Imd-BH3) and related NHC–boranes with diaryl and diheteroaryl disulfides provide diverse NHC–boryl monosulfides (diMe-Imd-BH2SAr) and NHC–boryl disulfides (diMe-Imd-BH(SAr)2). Heating in the dark with 1 equiv of disulfide favors monosulfide formation, while irradiation with 2 equiv disulfide favors disulfide formation. With heteroaryl disulfides, the NHC–borane in the primary NHC–boryl sulfide product migrates from sulfur to nitrogen to give new products with a thioamide substructure. Most substitution reactions are thought to proceed through radical chains in which homolytic substitution of a disulfide by an NHC–boryl radical is a key step. However, with electrophilic disulfides under dark conditions, a competing ionic path may also be possible.
Co-reporter:Xiangcheng Pan ; Anne Boussonnière
Journal of the American Chemical Society 2013 Volume 135(Issue 38) pp:14433-14437
Publication Date(Web):August 27, 2013
DOI:10.1021/ja407678e
The hydroboration of alkenes of diverse structural types by assorted N-heterocyclic carbene boranes can be accomplished by addition of 5–10% diiodine. For example, reaction of 1,3-dimethylimidazol-2-ylidene borane (diMe-Imd-BH3) with 10% I2 followed by addition of 2,3-dimethyl-2-butene provided the corresponding thexyl NHC-borane (diMe-Imd-BH2thexyl) in 75% yield. This and related monohydroboration products are stable to chromatography and storage. The scope of the new reaction is described, and the mechanism is probed by 11B NMR experiments.
Co-reporter:Xiben Li and Dennis P. Curran
Journal of the American Chemical Society 2013 Volume 135(Issue 32) pp:12076-12081
Publication Date(Web):July 18, 2013
DOI:10.1021/ja4056245
Readily available rhodium(II) salts catalyze reactions between NHC-boranes (NHC-BH3) and diazocarbonyl compounds (N2CRCOR′). Stable α-NHC-boryl carbonyl compounds (NHC-BH2–CHRCOR′) are isolated in good yields. The reaction is a reliable way to make boron–carbon bonds with good tolerance for variation in both the NHC-borane and diazocarbonyl components. It presumably occurs by insertion of a transient rhodium carbene into a boron–hydrogen bond of the NHC-borane. Competitive experiments show that a typical NHC-borane is highly reactive toward rhodium carbenes.
Co-reporter:Hanmo Zhang ; E. Ben Hay ; Steven J. Geib
Journal of the American Chemical Society 2013 Volume 135(Issue 44) pp:16610-16617
Publication Date(Web):October 10, 2013
DOI:10.1021/ja408387d
Radical cyclizations of cyclic ene sulfonamides provide stable bicyclic and tricyclic aldimines and ketimines in good yields. Depending on the structure of the precursor, the cyclizations occur to provide fused and spirocyclic imines with five-, six-, and seven-membered rings. The initial radical cyclization produces an α-sulfonamidoyl radical that undergoes elimination to form the imine and a phenylsulfonyl radical. In a related method, 3,4-dihydroquinolines can also be produced by radical translocation reactions of N-(2-iodophenylsulfonyl)tetrahydroiso-quinolines. In either case, very stable sulfonamides are cleaved to form imines (rather than amines) under mild reductive conditions.
Co-reporter:Aleksandrs Prokofjevs ; Jeff W. Kampf ; Andrey Solovyev ; Dennis P. Curran ;Edwin Vedejs
Journal of the American Chemical Society 2013 Volume 135(Issue 42) pp:15686-15689
Publication Date(Web):October 2, 2013
DOI:10.1021/ja407458k
Hydride abstraction from monocationic hydride bridged salts [H(H2B–L)2]+ [B(C6F5)4]¯ (L = Lewis base) generates an observable primary borenium cation for L = iPr2NEt, but with L = Me3N, Me2NPr, or several N-heterocyclic carbenes, highly reactive dicationic dimers are formed.
Co-reporter:Abhishek Dewanji, Sandip Murarka, Dennis P. Curran, and Armido Studer
Organic Letters 2013 Volume 15(Issue 23) pp:6102-6105
Publication Date(Web):November 19, 2013
DOI:10.1021/ol402995e
A simple and efficient direct radical arylation of unactivated arenes is described which uses cheap and commercially available phenyl hydrazine as an initiator. The reaction occurs through a base promoted homolytic aromatic substitution (BHAS) mechanism involving aryl radicals and aryl radical anions as intermediates and offers a practical approach for preparation of an array of substituted biaryls.
Co-reporter:Xiangcheng Pan;Jacques Lalevée;Emmanuel Lacôte
Advanced Synthesis & Catalysis 2013 Volume 355( Issue 18) pp:3522-3526
Publication Date(Web):
DOI:10.1002/adsc.201300752
Co-reporter:Andrey Solovyev, Emmanuel Lacôte and Dennis P. Curran
Dalton Transactions 2013 vol. 42(Issue 3) pp:695-700
Publication Date(Web):07 Sep 2012
DOI:10.1039/C2DT31773G
Reaction of the triflate group in dipp-Imd–BH2OTf with nucleophiles generally occurs by direct substitution to give products dipp-Imd–BH2Nu. In contrast, reaction of this boryl triflate with aryloxides (ArO−) in THF resulted in insertion of THF with ring opening in between the NHC–boryl electrophile and the aryloxide to give products dipp-Imd–BH2O(CH2)4OAr. The incorporation of THF was observed with other nucleophiles such as ethylthiolate and trimethylsilanolate whose conjugate acids have pKa values similar to that of phenol. The cleavage of ethyl acetate and oxetane is reported as well. A possible mechanism is proposed.
Co-reporter:Anne Boussonnière, Xiangcheng Pan, Steven J. Geib, and Dennis P. Curran
Organometallics 2013 Volume 32(Issue 24) pp:7445-7450
Publication Date(Web):November 8, 2013
DOI:10.1021/om400932g
Borenium-catalyzed hydroboration reactions of a stable, readily available N-heterocyclic carbene–borane with allyl-, alkenyl-, and alkynylsilane substrates provides either standard 1,2-hydroboration products or rearranged 1,1-hydroboration products, depending on the structure of the substrate. A competent catalyst can be generated in situ by addition of bis(trifluoromethane)sulfonimide or diiodine. In a typical 1,2-hydroboration, reaction of 1,3-dimethylimidazol-2-ylidine–borane (diMe-Imd-BH3) with 1,2-bis(trimethylsilyl)ethene provides 1,3-dimethylimidazol-2-ylidine–(1,2-bis(trimethylsilyl)ethyl)borane (diMe-Imd-BH2CH(TMS)CH2TMS)) as a stable product. In a typical 1,1-hydroboration, the reaction of diMe-Imd-BH3 with bis(trimethylsilyl)ethyne provides 1,3-dimethylimidazol-2-ylidine–bis(2,2-bis(trimethylsilyl)ethenyl)borane (diMe-Imd-BH(CH═C(TMS)2)2 again as a stable product.
Co-reporter:Jeffrey Buter, Edmund A.-H. Yeh, Owen W. Budavich, Krishnan Damodaran, Adriaan J. Minnaard, and Dennis P. Curran
The Journal of Organic Chemistry 2013 Volume 78(Issue 10) pp:4913-4918
Publication Date(Web):April 12, 2013
DOI:10.1021/jo4005298
(4S,8S,12S,16S,20S)-Pentamethylheptacosan-1-ol has been synthesized and analyzed by resolution-enhanced NMR spectroscopy with the aid of a recent set predicted spectra of all its stereoisomers. The configuration was confirmed, but isomer purity of the sample (∼70%) was lower than expected. A truncated analogue, (2S,6S,10S,14S)-2,6,10,14-tetramethylhenicosan-1-ol TBDPS ether, was prepared from a late stage synthetic intermediate. Analysis of its spectra confirmed the configuration and showed that the sample was isomerically pure. The results suggest that a late-stage epimerization, not a failure of an asymmetric synthesis step, caused the formation of minor stereoisomers in the sample of pentamethylheptacosan-1-ol. The study shows the value of the predicted set of oligoisoprenoid spectra and further extends the predictive model to a new subclass of compounds.
Co-reporter:Jérémie Mandel, Xiaohong Pan, E. Ben Hay, Steven J. Geib, Craig S. Wilcox, and Dennis P. Curran
The Journal of Organic Chemistry 2013 Volume 78(Issue 8) pp:4083-4089
Publication Date(Web):March 28, 2013
DOI:10.1021/jo400385t
The rotational preferences of N-(2-bromo-4,6-dimethylphenyl)-N-methyl 2-phenylpropanamide were studied as a model of precursors for Hartwig asymmetric oxindole cyclizations. The atropisomers of this compound were separated by flash chromatography, and then the enantiomers were resolved and the interconversions of the stereocenter and the N–Ar axis were studied. Under thermal conditions, the axis is very stable. Under the basic conditions of the Hartwig cyclization, both the stereocenter and the chiral axis equilibrate via enolate formation. The N–Ar rotation barrier of a 2-phenylacetamide analogue was reduced from 31 kcal mol–1 in the precursor to 17 kcal mol–1 in the enolate. Reasons for this dramatic barrier reduction and implications of both N–Ar and amide C–N rotations for Hartwig cyclizations are discussed.
Co-reporter:Xiangcheng Pan ; Emmanuel Lacôte ; Jacques Lalevée
Journal of the American Chemical Society 2012 Volume 134(Issue 12) pp:5669-5674
Publication Date(Web):March 6, 2012
DOI:10.1021/ja300416f
Otherwise sluggish or completely ineffective radical reductions of alkyl and aryl halides by N-heterocyclic carbene boranes (NHC-boranes) are catalyzed by thiols. Reductions and reductive cyclizations with readily available 1,3-dimethylimidazol-2-ylidene borane and a water-soluble triazole relative are catalyzed by thiophenol and tert-dodecanethiol [C9H19C(CH3)2SH]. Rate constants for reaction of the phenylthiyl (PhS•) radical with two NHC-boranes have been measured to be ∼108 M–1 s–1 by laser flash photolysis experiments. An analysis of the available evidence suggests the operation of polarity reversal catalysis.
Co-reporter:Jared D. Moretti ; Xiao Wang
Journal of the American Chemical Society 2012 Volume 134(Issue 18) pp:7963-7970
Publication Date(Web):April 20, 2012
DOI:10.1021/ja302260d
Four mixtures of four fluorous-tagged quasiisomers have been synthesized, demixed, and detagged to make all 16 stereoisomers of the macrocyclic lactone natural product Sch725674. A new bare-minimum tagging pattern needs only two tags—one fluorous and one nonfluorous—to encode four isomers. The structure of Sch725674 is assigned as (5R,6S,8R,14R,E)-5,6,8-trihydroxy-14-pentyloxacyclotetradec-3-en-2-one. Various comparisons of spectra of 32 lactones (16 with tags, 16 without) and 16 ester precursors (8 with tags, 8 without) provide insights into when and why related compounds have the same or different spectra.
Co-reporter:Aleksandrs Prokofjevs ; Anne Boussonnière ; Linfeng Li ; Hélène Bonin ; Emmanuel Lacôte ; Dennis P. Curran ;Edwin Vedejs
Journal of the American Chemical Society 2012 Volume 134(Issue 29) pp:12281-12288
Publication Date(Web):June 19, 2012
DOI:10.1021/ja305061c
Treatment of alkenes such as 3-hexene, 3-octene, and 1-cyclohexyl-1-butene with the N-heterocyclic carbene (NHC)-derived borane 2 and catalytic HNTf2 (Tf = trifluoromethanesulfonyl (CF3SO2)) effects hydroboration at room temperature. With 3-hexene, surprisingly facile migration of the boron atom from C(3) of the hexyl group to C(2) was observed over a time scale of minutes to hours. Oxidative workup gave a mixture of alcohols containing 2-hexanol as the major product. A similar preference for the C(2) alcohol was observed after oxidative workup of the 3-octene and 1-cyclohexyl-1-butene hydroborations. NHC-borenium cations (or functional equivalents) are postulated as the species that accomplish the hydroborations, and the C(2) selective migrations are attributed to the four-center interconversion of borenium cations with cationic NHC-borane-olefin π-complexes.
Co-reporter:Markus Horn, Herbert Mayr, Emmanuel Lacôte, Everett Merling, Jordan Deaner, Sarah Wells, Timothy McFadden, and Dennis P. Curran
Organic Letters 2012 Volume 14(Issue 1) pp:82-85
Publication Date(Web):December 13, 2011
DOI:10.1021/ol202836p
The nucleophilicity parameters (N) of 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene borane and 1,3-dimethylimidazol-2-ylidene borane are 9.55 and 11.88. This places N-heterocyclic carbene boranes (NHC-boranes) among the most nucleophilic classes of neutral hydride donors. Reductions of highly electron-poor C═N and C═C bonds provide hydrogenation products along with new, stable borylated products. The results suggest that NHC-boranes have considerable untapped potential as neutral organic reductants.
Co-reporter:Everett Merling, Vladimir Lamm, Steven J. Geib, Emmanuel Lacôte, and Dennis P. Curran
Organic Letters 2012 Volume 14(Issue 11) pp:2690-2693
Publication Date(Web):May 22, 2012
DOI:10.1021/ol300851m
Thermal 1,3-dipolar cycloaddition reactions of 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene dihydridoboron azide occur smoothly with alkynes, nitriles, and alkenes bearing electron-withdrawing groups. New, stable NHC-boryl-substituted triazoles, tetrazoles, and triazolidines are formed in good to excellent yields.
Co-reporter:Tsuyoshi Taniguchi and Dennis P. Curran
Organic Letters 2012 Volume 14(Issue 17) pp:4540-4543
Publication Date(Web):August 16, 2012
DOI:10.1021/ol302010f
N-Heterocyclic carbene boranes (NHC-boranes) such as 1,3-dimethylimidazol-2-ylidine trihydridoborane (diMe-Imd-BH3) serve as practical hydride donors for the reduction of aldehydes and ketones in the presence of silica gel. Primary and secondary alcohols are formed in good yields under ambient conditions. Aldehydes are selectively reduced in the presence of ketones. One, two, or even all three of the boron hydrides can be transferred. The process is attractive because all the components are stable and easy to handle and because both the reaction and isolation procedures are convenient.
Co-reporter: Dennis P. Curran;Dr. Anne Boussonnière;Dr. Steven J. Geib;Dr. Emmanuel Lacôte
Angewandte Chemie 2012 Volume 124( Issue 7) pp:1634-1637
Publication Date(Web):
DOI:10.1002/ange.201107238
Co-reporter: Dennis P. Curran;Dr. Anne Boussonnière;Dr. Steven J. Geib;Dr. Emmanuel Lacôte
Angewandte Chemie International Edition 2012 Volume 51( Issue 7) pp:1602-1605
Publication Date(Web):
DOI:10.1002/anie.201107238
Co-reporter:Andrey Solovyev, Steven J. Geib, Emmanuel Lacôte, and Dennis P. Curran
Organometallics 2012 Volume 31(Issue 1) pp:54-56
Publication Date(Web):December 28, 2011
DOI:10.1021/om201260b
Reactions of NHC-BH2X (where X = Cl, OTf and NHC = 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene) with 1 equiv of triflic acid provide the stable products of acid/base reactions NHC-BH(OTf)2 and NHC-BH(OTf)Cl. Further reactions of these compounds with additional triflic acid (or direct reactions of NHC-BH3 with excess triflic acid) produce the isolable dihydroxyborenium triflate [NHC-B(OH)2]+TfO–. This first-in-class ligated borenium ion has been characterized by NMR spectroscopy and X-ray crystallography.
Co-reporter:Reena Bajpai
Journal of the American Chemical Society 2011 Volume 133(Issue 50) pp:20435-20443
Publication Date(Web):November 3, 2011
DOI:10.1021/ja2082679
Fluorous mixture synthesis provided all eight diastereomers of the phytophthora hormone α1 with the R configuration at C11 as individual samples after demixing and detagging. The library of all possible bis-Mosher esters (16) was then made by esterification. Complete sets of 1H, 13C, and (for the Mosher esters) 19F NMR spectra were recorded, assigned, and compared with each other and with published spectra. Not all of the spectra are unique, and the 1H NMR spectra of the Mosher esters provided the most information. The previous assignment of the natural sample as an “all-R” stereoisomer mixed with its 3S-epimer was confirmed.
Co-reporter:Hanmo Zhang
Journal of the American Chemical Society 2011 Volume 133(Issue 27) pp:10376-10378
Publication Date(Web):June 13, 2011
DOI:10.1021/ja2042854
The first stereoselective synthesis of epimeloscine has been accomplished in 13 total steps with a longest linear sequence of 10 steps. The core of the synthesis takes only five steps, the key ones being acylation, stereoselective tandem radical cyclization of a divinylcyclopropane to make two rings, and group-selective ring-closing metathesis of the resulting divinylcyclopentane to make the last ring.
Co-reporter:Achim Bruch ; Roland Fröhlich ; Stefan Grimme ; Armido Studer
Journal of the American Chemical Society 2011 Volume 133(Issue 40) pp:16270-16276
Publication Date(Web):September 1, 2011
DOI:10.1021/ja2070347
The paper describes examples of net diastereotopic-group-selective radical processes having the unusual feature that a single product is formed even though the key reaction of the two diastereotopic radical precursors is nonselective. For example, reaction of (R)-N-(cyclohex-2-en-1-yl)-N-(2,6-diiodo-4-methylphenyl)acetamide with tributyltin hydride produces 1-((4aR,9aR)-6-methyl-2,3,4,4a-tetrahydro-1H-carbazol-9(9aH)-yl)ethanone with high product selectivity and in high yield. Analysis of the concentration profiles of the closed-shell intermediates at the halfway point of the reaction shows that nonselective abstraction of diastereotopic iodides by tin radicals occurs, leading to diastereomeric aryl radicals. These isomeric intermediates evolve via two nonintersecting reaction pathways, cyclization and bimolecular trapping or vice versa, into the same final product. Origins of the selectivity are suggested on the basis of conformational analysis of the products using both X-ray crystallography and density functional theory calculations.
Co-reporter:Ashutosh S. Jogalekar ; Krishnan Damodaran ; Frederik H. Kriel ; Won-Hyuk Jung ; Ana A. Alcaraz ; Shi Zhong ; Dennis P. Curran ;James P. Snyder
Journal of the American Chemical Society 2011 Volume 133(Issue 8) pp:2427-2436
Publication Date(Web):February 7, 2011
DOI:10.1021/ja1023817
Dictyostatin (DCT, 1) is a complex, flexible polyketide macrolide that demonstrates potent microtubule-polymerization activity. Both a solution structure (2a) and a possible binding mode for DCT (Conf-1) have been proposed by earlier NMR experiments. In the present study, the conformational landscape of DCT in DMSO-d6 and methanol-d4 was explored using extensive force-field-based conformational searches combined with geometric parameters derived from solution NMR data. The results portray a diversity of conformations for dictyostatin that illustrates the molecule’s flexibility and excludes the previously suggested dominant solution conformation 2a. One conformation present in DMSO-d6 with a 7% population (Conf-2, 0.6 kcal/mol above the global minimum at 298°) also satisfies the TR-NOESY NMR parameters of Canales et al. that characterize the taxane binding-site interaction between DCT and assembled microtubules in water. Application of several docking methods (Glide, Autodock, and RosettaLigand) has identified a low-energy binding model of the DCT/β-tubulin complex (Pose-2/Conf-2) that is gratifyingly compatible with the emerging DCT structure−activity data.
Co-reporter:John C. Walton ; Malika Makhlouf Brahmi ; Julien Monot ; Louis Fensterbank ; Max Malacria ; Dennis P. Curran ;Emmanuel Lacôte
Journal of the American Chemical Society 2011 Volume 133(Issue 26) pp:10312-10321
Publication Date(Web):May 28, 2011
DOI:10.1021/ja2038485
Fifteen second-generation NHC-ligated boranes with aryl and alkyl substituents on boron were prepared, and their radical chemistry was explored by electron paramagnetic resonance (EPR) spectroscopy and calculations. Hydrogen atom abstraction from NHC–BH2Ar groups produced boryl radicals akin to diphenylmethyl with spin extensively delocalized across the NHC, BH, and aryl units. All of the NHC–B·HAr radicals studied abstracted Br-atoms from alkyl bromides. Radicals with bulky N,N′-dipp substituents underwent dimerization about 2 orders of magnitude more slowly than first-generation NHC-ligated trihydroborates. The evidence favored head-to-head coupling yielding ligated diboranes. The first ligated diboranyl radical, with a structure intermediate between that of ligated diboranes and diborenes, was spectroscopically characterized during photolysis of di-t-butyl peroxide with N,N′-di-t-butyl-imidazol-2-ylidene phenylborane. The reactive site of B-alkyl-substituted NHC–boranes switched from the boron center to the alkyl substituent for both linear and branched alkyl groups. The β-borylalkyl radicals obtained from N,N′-dipp-substituted boranes underwent exothermic β-scissions with production of dipp-Imd–BH2· radicals and alkenes. The reverse additions of NHC–boryl radicals to alkenes are probably endothermic for alkyl-substituted alkenes, but exothermic for conjugated alkenes (addition of an NHC–boryl radical to 1,1-diphenylethene was observed). A cyclopropylboryl radical was observed, but, unlike other α-cyclopropyl-substituted radicals, this showed no propensity for ring-opening.
Co-reporter:Andrey Solovyev, Emmanuel Lacôte, and Dennis P. Curran
Organic Letters 2011 Volume 13(Issue 22) pp:6042-6045
Publication Date(Web):October 24, 2011
DOI:10.1021/ol202516c
N,N′-Dialkyl and N,N′-diaryl imidazol-2-ylidene-boranes and trifluoroboranes are rapidly lithiated at C4 of the imidazole ring, and the resulting intermediates have been quenched with an assortment of electrophiles to provide ring-functionalized imidazol-2-ylidene-boranes. Further deprotonation and functionalization of C5 have been demonstrated. Deboronation of the products by treatment with triflic acid or iodine and then methanol opens a route to C4/C5 functionalized imidazolium salts and imidazol-2-ylidenes.
Co-reporter:Shau-Hua Ueng, Louis Fensterbank, Emmanuel Lacôte, Max Malacria and Dennis P. Curran
Organic & Biomolecular Chemistry 2011 vol. 9(Issue 9) pp:3415-3420
Publication Date(Web):26 Jan 2011
DOI:10.1039/C0OB01075H
1,3-Dimethylimidazol-2-ylidene borane and 2,4-dimethyl-1,2,4-triazol-3-ylidene borane are found to be useful reagents for the reduction of alkyl iodides and bromides bearing nearby electron withdrawing substituents. Signatures of radical chain reactions are seen in many cases, but ionic reductions may also be occurring with some substrates. The reagents are attractive because of their low molecular weight, their availability from inexpensive precursors, and their stability. Separation of the borane products from the target products is readily accomplished either with or without prior regeneration of the borane for later reuse. 2,4-Dimethyl-1,2,4-triazol-3-ylidene borane is versatile because both starting borane and its derived products can be removed by extraction with water.
Co-reporter:Venugopal Gudipati, Dennis P. Curran
Tetrahedron Letters 2011 Volume 52(Issue 17) pp:2254-2257
Publication Date(Web):27 April 2011
DOI:10.1016/j.tetlet.2011.01.086
Efficient syntheses of suitably functionalized top and bottom fragments of tetrafibricin are described. The bottom fragment is prepared by two consecutive Kocienski–Julia couplings, while the top fragment synthesis features a dithiane alkylation and a Horner–Wadsworth–Emmons reaction.
Co-reporter:Andrew J. Clark, Dennis P. Curran, Joanna V. Geden, Natalie James, and Paul Wilson
The Journal of Organic Chemistry 2011 Volume 76(Issue 11) pp:4546-4551
Publication Date(Web):April 20, 2011
DOI:10.1021/jo200343z
Barriers to rotation of the N-alkenyl bond in a series of N-cycloalkenyl-N-benzyl acetamide derivatives have been measured in different solvents by variable-temperature NMR experiments. The barriers range from 9.7 to 14.2 kcal/mol, depending on substituents on the acetamide acyl group. Polar solvents such as chloroform and methanol increase the barrier to rotation compared to nonpolar solvents such as toluene. The barrier to rotation of “mimics” for acetamide-based radicals are estimated. The relative order of the values of krot for different acyl groups parallels their reported Taft Es paramaters. For successful chirality transfer in 5-endo trig radical cyclization, it is evident that rotations would need to be significantly slower than those reported here.
Co-reporter: Dennis P. Curran;Andrey Solovyev;Dr. Malika MakhloufBrahmi; Louis Fensterbank; Max Malacria;Dr. Emmanuel Lacôte
Angewandte Chemie 2011 Volume 123( Issue 44) pp:10476-10500
Publication Date(Web):
DOI:10.1002/ange.201102717
Abstract
Borane sind weit verbreitete Lewis-Säuren, und N-heterocyclische Carbene (NHCs) sind beliebte Lewis-Basen. Dennoch war bis in die jüngste Zeit erstaunlich wenig über Komplexe dieser beiden Verbindungsklassen miteinander bekannt. NHC-Borane sind leicht herzustellen, und teilweise sind sie so stabil, dass sie weniger als Komplex, sondern vielmehr als eigenständige organische Verbindung betrachtet werden können. Sie verhalten sich nicht wie klassische Borane, und sie können ein breites Spektrum an funktionellen Gruppen enthalten. Als vielseitige Reaktionspartner, Reagentien und Katalysatoren verfügen NHC-Borane über ein immenses Potenzial für die organische Synthese und die Polymerchemie. Viele Reaktionen von NHC-Boranen laufen über neue reaktive Intermediate wie Boreniumkationen, Borylradikale und sogar Borylanionen ab. Hier geben wir eine umfassende Übersicht über die Synthese, Charakterisierung und Reaktionen von NHC-Boranen.
Co-reporter: Armido Studer; Dennis P. Curran
Angewandte Chemie International Edition 2011 Volume 50( Issue 22) pp:5018-5022
Publication Date(Web):
DOI:10.1002/anie.201101597
Co-reporter: Dennis P. Curran;Andrey Solovyev;Dr. Malika MakhloufBrahmi; Louis Fensterbank; Max Malacria;Dr. Emmanuel Lacôte
Angewandte Chemie International Edition 2011 Volume 50( Issue 44) pp:10294-10317
Publication Date(Web):
DOI:10.1002/anie.201102717
Abstract
Boranes are widely used Lewis acids and N-heterocyclic carbenes (NHCs) are popular Lewis bases, so it is remarkable how little was known about their derived complexes until recently. NHC-boranes are typically readily accessible and many are so stable that they can be treated like organic compounds rather than complexes. They do not exhibit “borane chemistry”, but instead are proving to have a rich chemistry of their own as reactants, as reagents, as initiators, and as catalysts. They have significant potential for use in organic synthesis and in polymer chemistry. They can be used to easily make unusual complexes with a broad spectrum of functional groups not usually seen in organoboron chemistry. Many of their reactions occur through new classes of reactive intermediates including borenium cations, boryl radicals, and even boryl anions. This Review provides comprehensive coverage of the synthesis, characterization, and reactions of NHC-boranes.
Co-reporter: Dennis P. Curran;Andrey Solovyev;Dr. Malika MakhloufBrahmi; Louis Fensterbank; Max Malacria;Dr. Emmanuel Lacôte
Angewandte Chemie International Edition 2011 Volume 50( Issue 44) pp:
Publication Date(Web):
DOI:10.1002/anie.201105618
Co-reporter: Dennis P. Curran;Andrey Solovyev;Dr. Malika MakhloufBrahmi; Louis Fensterbank; Max Malacria;Dr. Emmanuel Lacôte
Angewandte Chemie 2011 Volume 123( Issue 44) pp:
Publication Date(Web):
DOI:10.1002/ange.201105618
Co-reporter:Achim Bruch ; Andrea Ambrosius ; Roland Fröhlich ; Armido Studer ; David B. Guthrie ; Hanmo Zhang
Journal of the American Chemical Society 2010 Volume 132(Issue 33) pp:11452-11454
Publication Date(Web):July 28, 2010
DOI:10.1021/ja105070k
The rate constant for phosphanylation of an aryl radical with trimethylstannyl diphenylphosphane (Me3SnPPh2) has been measured as kphos ≈ 9 × 108 M−1 s−1. Aryl radicals derived from several axially chiral o-haloanilides are trapped by Me3SnPPh2 with complete retention of axial chirality as shown by oxidation of the phosphanes to give stable, easily analyzed phosphane oxides or sulfides. Double phosphanylations of o,o′-dihaloanilides followed by treatment with H2O2 or S8 in either order give enantiomers of a mixed diphosphane oxide sulfide. Chemodivergent trapping of diastereomers of an N-(cyclohex-2-enyl)anilide anilide is observed. For one isomer, the cyclization precedes the Me3SnPPh2 trapping, while for the other isomer direct trapping with Me3SnPPh2 supersedes the cyclization. The products are chiral triaryl phosphanes, oxides, and sulfides that are potentially interesting ligands in asymmetric catalysis.
Co-reporter:Kyoko Nozaki ; Yoshitaka Aramaki ; Makoto Yamashita ; Shau-Hua Ueng ; Max Malacria ; Emmanuel Lacôte
Journal of the American Chemical Society 2010 Volume 132(Issue 33) pp:11449-11451
Publication Date(Web):August 2, 2010
DOI:10.1021/ja105277u
Reaction of lithium 1,3-bis(2,6-diisopropylphenyl)-2,3-dihydro-1H-1,3,2-diazaborol-2-ide with borane·THF provides the first boryl-substituted borohydride: lithium [1,3-bis(2,6-diisopropylphenyl)-2,3-dihydro-1H-1,3,2-diazaborol-2-yl]trihydroborate. The compound is fully characterized by 11B, 1H, and 7Li NMR spectra and other means, and these data are compared to neutral and anionic benchmark compounds. The compound crystallizes as a dimer complexed to four THF molecules. The dimer lacks the bridging B−H bonds seen in neutral boranes and is instead held together by ionic Li---HB interactions. A preliminary scan of reactions with several iodides shows that the compound participates in an ionic reduction (with a primary-alkyl iodide), an organometallic reduction (Pd-catalyzed with an aryl iodide), and a radical reduction (AIBN-initiated with a sugar-derived iodide). Accordingly the new borylborohydride class may share properties of both traditional borohydrides and isoelectronic N-heterocyclic carbene boranes.
Co-reporter:Andrey Solovyev ; Qianli Chu ; Steven J. Geib ; Louis Fensterbank ; Max Malacria ; Emmanuel Lacôte
Journal of the American Chemical Society 2010 Volume 132(Issue 42) pp:15072-15080
Publication Date(Web):October 1, 2010
DOI:10.1021/ja107025y
Boryl halide, carboxylate and sulfonate complexes of 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene (dipp-Imd-BH2X, X = halide or sulfonate) have been prepared from the parent borane dipp-Imd-BH3 by (1) substitution reactions with R−X (X = halide or sulfonate), (2) reactions with electrophiles (like I2 or NIS), or (3) acid/base reactions with HX (provided that HX has a pKa of about 2 or less). Dipp-Imd-BH2I is most conveniently prepared by reaction with diiodine while dipp-Imd-BH2OTf is best prepared by reaction with triflic acid. These and other less reactive complexes behave as electrophiles and can be substituted by a wide range of heteroatom nucleophiles including halides, thiolates and other sulfur-based nucleophiles, isocyanate, azide, nitrite, and cyanide. The resulting products are remarkably stable, and many have been characterized by X-ray crystallography. Several are members of very rare classes of functionalized boron compounds (boron azide, nitro compound, nitrous ester, etc.).
Co-reporter:David B. Guthrie ; Steven J. Geib
Journal of the American Chemical Society 2010 Volume 133(Issue 1) pp:115-122
Publication Date(Web):December 10, 2010
DOI:10.1021/ja108795x
The outcomes of radical cyclizations and Heck reactions of N-(cyclohex-2-enyl)-N-(2-iodophenyl)acetamides depend critically on the configurations of the chiral axis and the stereocenter. In substrates without an ortho-methyl group, the diastereomeric precursors interconvert slowly at ambient temperatures. Cyclization of enriched mixtures of diastereomers provided similar yields of acetyl tetrahydrocarbazoles or dihydrocarbazoles, suggesting that interconversion of the radical or organometallic intermediates also occurs. Diastereomers of N-(cyclohex-2-enyl)-N-(2-iodo-4,6-dimethylphenyl)acetamides with an additional ortho-methyl group did not interconvert at ambient temperatures and were readily resolved. In radical cyclizations, syn diastereomers were prone to cyclize, while anti isomers were not. Strikingly, Heck reactions gave the opposite result; anti isomers were prone to cyclization and syn isomers were not. Heck reactions of allylic acetates occur with β-hydride elimination when acetate is trans to palladium and with β-acetoxy elimination when acetate is cis. This is surprising because prior studies have suggested that a trans relationship of palladium and acetoxy is essential for acetate elimination. Analyses of the results provide insights into mechanisms for radical cyclization and for insertion and elimination in the Heck reaction.
Co-reporter:John C. Walton ; Malika Makhlouf Brahmi ; Louis Fensterbank ; Emmanuel Lacôte ; Max Malacria ; Qianli Chu ; Shau-Hua Ueng ; Andrey Solovyev
Journal of the American Chemical Society 2010 Volume 132(Issue 7) pp:2350-2358
Publication Date(Web):January 27, 2010
DOI:10.1021/ja909502q
N-Heterocyclic carbene boranes (NHC−boranes) are a new “clean” class of reagents suitable for reductive radical chain transformations. Their structures are well suited for their reactivity to be tuned by inclusion of different NHC ring units and by appropriate placement of diverse substituents. EPR spectra were obtained for the boron-centered radicals generated on removal of one of the BH3 hydrogen atoms. This spectroscopic data, coupled with DFT computations, demonstrated that the NHC−BH2• radicals are planar π-delocalized species. tert-Butoxyl radicals abstracted hydrogen atoms from NHC−boranes more than 3 orders of magnitude faster than did C-centered radicals, although the rate decreased markedly for sterically shielded NHC−BH3 centers. Combinations of two NHC−boryl radicals afforded 1,2-bis-NHC−diboranes at rates which also depended strongly on steric shielding. The termination rate increased to the diffusion-controlled limit for sterically unhindered NHC−boryls. Bromine atoms were rapidly transferred to imidazole-based NHC−boryl radicals from alkyl, allyl, and benzyl bromides. Chlorine-atom abstraction was, however, much less efficient and only observed for sterically unhindered NHC−boryls reacting with allylic and benzylic chlorides. For an NHC−borane containing a bulky thexyl substituent at boron, the tertiary H atom of the thexyl group was selectively removed. The resulting β-boron-containing alkyl radical rapidly underwent β scission of the B−C bond with production of an NHC−boryl radical and an alkene.
Co-reporter:Wei Zhu ; María Jiménez ; Won-Hyuk Jung ; Daniel P. Camarco ; Raghavan Balachandran ; Andreas Vogt ; Billy W. Day
Journal of the American Chemical Society 2010 Volume 132(Issue 26) pp:9175-9187
Publication Date(Web):June 14, 2010
DOI:10.1021/ja103537u
The dictyostatins are a promising class of potential anti-cancer drugs because they are powerful microtubule-stabilizing agents, but the complexity of their chemical structures is a severe impediment to their further development. On the basis of both synthetic and medicinal chemistry analyses, 16-desmethyl-25,26-dihydrodictyostatin and its C6 epimer were chosen as potentially potent yet accessible dictyostatin analogues, and three new syntheses were developed. A relatively classical synthesis involving vinyllithium addition and macrocyclization gave way to a newer and more practical approach based on esterification and ring-closing metathesis reaction. Finally, aspects of these two approaches were combined to provide a third new synthesis based on esterification and Nozaki−Hiyama−Kishi reaction. This was used to prepare the target dihydro analogues and the natural product. All of the syntheses are streamlined because of their high convergency. The work provided several new analogues of dictyostatin, including a truncated macrolactone and a C10 E-alkene, which were 400- and 50-fold less active than (−)-dictyostatin, respectively. In contrast, the targeted 16-desmethyl-25,26-dihydrodictyostatin analogues retained almost complete activity in preliminary biological assays.
Co-reporter:Shau-Hua Ueng, Louis Fensterbank, Emmanuel Lacôte, Max Malacria and Dennis P. Curran
Organic Letters 2010 Volume 12(Issue 13) pp:3002-3005
Publication Date(Web):June 10, 2010
DOI:10.1021/ol101015m
Minimalist N-heterocyclic carbene boranes 1,3-dimethylimidazol-2-ylideneborane and 2,4-dimethyl-1,2,4-triazol-3-ylideneborane are readily available and have low molecular weights. They exhibit superior performance to first-generation NHC−boranes, providing improved yields in reductions of xanthates and related functional groups.
Co-reporter:Xiben Li and Dennis P. Curran
Organic Letters 2010 Volume 12(Issue 3) pp:612-614
Publication Date(Web):December 28, 2009
DOI:10.1021/ol902808m
Diverging chemoselective reactions of a pair of amide rotamers have been observed by separating the rotamers and then reacting them individually. Reduction of (Z)-N-allyl-2-(phenylselanyl)-N-(2,4,6-tri-tert-butylphenyl)acetamide with tributyltin hydride at room temperature provides only the product of 5-exo cyclization, 4-methyl-1-(2,4,6-tri-tert-butylphenyl)pyrrolidin-2-one. In contrast, reduction of the corresponding (E) amide rotational isomer under identical conditions provides only the reduced product, (E)-N-allyl-N-(2,4,6-tri-tert-butylphenyl)acetamide. Such diverging reactions of rotamers may be common in transformations involving reactive intermediates (carbenes, radicals, organometallic intermediates) that have low barriers to onward reactions relative to amide rotation.
Co-reporter:Dr. Julien Monot;Andrey Solovyev;Dr. Hélène Bonin-Dubarle;Dr. Étienne Derat; Dennis P. Curran; Marc Robert; Louis Fensterbank; Max Malacria;Dr. Emmanuel Lacôte
Angewandte Chemie International Edition 2010 Volume 49( Issue 48) pp:9166-9169
Publication Date(Web):
DOI:10.1002/anie.201004215
Co-reporter:Dr. Julien Monot;Andrey Solovyev;Dr. Hélène Bonin-Dubarle;Dr. Étienne Derat; Dennis P. Curran; Marc Robert; Louis Fensterbank; Max Malacria;Dr. Emmanuel Lacôte
Angewandte Chemie International Edition 2010 Volume 49( Issue 48) pp:
Publication Date(Web):
DOI:10.1002/anie.201005438
Co-reporter:Dr. Julien Monot;Andrey Solovyev;Dr. Hélène Bonin-Dubarle;Dr. Étienne Derat; Dennis P. Curran; Marc Robert; Louis Fensterbank; Max Malacria;Dr. Emmanuel Lacôte
Angewandte Chemie 2010 Volume 122( Issue 48) pp:9352-9355
Publication Date(Web):
DOI:10.1002/ange.201004215
Co-reporter:Dr. Julien Monot;Andrey Solovyev;Dr. Hélène Bonin-Dubarle;Dr. Étienne Derat; Dennis P. Curran; Marc Robert; Louis Fensterbank; Max Malacria;Dr. Emmanuel Lacôte
Angewandte Chemie 2010 Volume 122( Issue 48) pp:
Publication Date(Web):
DOI:10.1002/ange.201005438
Co-reporter:David B. Guthrie ; Steven J. Geib
Journal of the American Chemical Society 2009 Volume 131(Issue 42) pp:15492-15500
Publication Date(Web):October 2, 2009
DOI:10.1021/ja9066282
Radical cyclizations (Bu3SnH, Et3B/air, rt) of racemic α-halo-ortho-alkenyl anilides provide 3,4-dihydroquinolin-2-ones in high yield. Cyclizations of enantioenriched precursors occur in similarly high yields and with transfer of axial chirality to the new stereocenter of the products with exceptionally high fidelity (often >95%). Single and tandem cyclizations of α-halo-ortho-alkenyl anilides bearing an additional substituent on the α-carbon occur with high chirality transfer and high diastereoselectivity. Straightforward models are proposed to interpret both the chirality transfer and diastereoselectivity aspects. These first examples of an approach for axial chiral transfer from a reactive species in the amide to an acceptor suggest broad potential for extension both within and beyond radical reactions.
Co-reporter:Shau-Hua Ueng ; Andrey Solovyev ; Xinting Yuan ; Steven J. Geib ; Louis Fensterbank ; Emmanuel Lacôte ; Max Malacria ; Martin Newcomb ; John C. Walton
Journal of the American Chemical Society 2009 Volume 131(Issue 31) pp:11256-11262
Publication Date(Web):July 16, 2009
DOI:10.1021/ja904103x
Reduction of xanthates by N-heterocyclic carbene boranes (NHC-boranes) has been suggested to occur by a radical chain mechanism involving heretofore unknown NHC-boryl radicals. In support of this suggestion, both the expected borane dithiocarbonate product and an unexpected borane xanthate product have now been isolated. These are the first NHC-boranes with boron−sulfur bonds, and their structures have been secured by spectroscopic and crystallographic means. The first rate constants for H-atom transfer from an NHC borane complex were determined by using the ring opening of a substituted cyclobutylcarbinyl radical as a clock reaction. The rate constant for reaction of the NHC-borane with a secondary alkyl radical at ambient temperature is 4 × 104 M−1 s−1, and the Arrhenius function displayed an entropic term (log A term) that was typical for a bimolecular reaction. The B−H bond dissociation energy of an NHC-borane complex has been estimated at 88 kcal/mol. The putative NHC-boryl radical in these transformations has been detected by EPR spectroscopy. Spectral analysis suggests that it is a π-radical, analogous to the benzyl radical.
Co-reporter:Julien Monot, Malika Makhlouf Brahmi, Shau-Hua Ueng, Carine Robert, Marine Desage-El Murr, Dennis P. Curran, Max Malacria, Louis Fensterbank and Emmanuel Lacôte
Organic Letters 2009 Volume 11(Issue 21) pp:4914-4917
Publication Date(Web):October 2, 2009
DOI:10.1021/ol902012c
Complexes of triaryl- and trialkylboranes with N-heterocyclic carbenes (NHCs) participate in Suzuki−Miyaura cross-coupling reactions and provide coupled products in good yields under base-free conditions. The reaction can be applied to Csp2−Csp2 and Csp2−Csp3 carbon−carbon bond formation with triflates, iodides, bromides, and chlorides. These results enrich the utility of NHC−borane complexes, which can be added to the toolkit of Suzuki−Miyaura cross-couplings, along with boronic acids and organotrifluoroborates.
Co-reporter:Amador Garcia Sancho;Xiao Wang;Bin Sui ;DennisP. Curran
Advanced Synthesis & Catalysis 2009 Volume 351( Issue 7-8) pp:1035-1040
Publication Date(Web):
DOI:10.1002/adsc.200900061
Co-reporter:Qianli Chu Dr.;Malika MakhloufBrahmi;Andrey Solovyev;Shau-Hua Ueng Dr.;DennisP. Curran ;Max Malacria ;Louis Fensterbank ;Emmanuel Lacôte Dr.
Chemistry - A European Journal 2009 Volume 15( Issue 47) pp:12937-12940
Publication Date(Web):
DOI:10.1002/chem.200902450
Co-reporter:David B. Guthrie, Krishnan Damodaran, Dennis P. Curran, Paul Wilson and Andrew J. Clark
The Journal of Organic Chemistry 2009 Volume 74(Issue 11) pp:4262-4266
Publication Date(Web):May 4, 2009
DOI:10.1021/jo900491w
Barriers to rotation of the N-alkenyl bond in a series of N-cycloalkenyl-N-benzyl α-haloacetamide derivatives have been measured by variable-temperature NMR experiments. The barriers range from 10 to 18 kcal/mol, depending on ring size and on substituents on the cycloalkene and the amide. The observed trends aid in the design of substituent combinations that provide resolvable enantiomers or diastereomers at ambient temperature. The compounds undergo 4-exo and 5-endo radical cyclizations at rates that may be faster or slower than the estimated rate of N-alkenyl bond rotation in the derived radicals, depending on the substituents.
Co-reporter:Julie L. Eiseman ; Lihua Bai ; Won-Hyuk Jung ; Gustavo Moura-Letts ; Billy W. Day
Journal of Medicinal Chemistry 2008 Volume 51(Issue 21) pp:6650-6653
Publication Date(Web):October 8, 2008
DOI:10.1021/jm800979v
Structure−activity studies centered on the naturally occurring antitumor agent dictyostatin have recently identified several highly active epimers and analogues. From these compounds, 6-epi-dictyostatin was selected for scaleup preparation and evaluation in animals. Here we describe a new total synthesis that produced more than 30 mg of 6-epi-dictyostatin. The compound was found to have potent antitumor activity in SCID mice bearing MDA-MB231 human breast cancer xenografts.
Co-reporter:Won-Hyuk Jung;Sabrina Guyenne;Concepción Riesco-Fagundo;John Mancuso;Shuichi Nakamura ;DennisP. Curran Dr.
Angewandte Chemie 2008 Volume 120( Issue 6) pp:1146-1149
Publication Date(Web):
DOI:10.1002/ange.200704893
Co-reporter:Won-Hyuk Jung;Sabrina Guyenne;Concepción Riesco-Fagundo;John Mancuso;Shuichi Nakamura ;DennisP. Curran Dr.
Angewandte Chemie International Edition 2008 Volume 47( Issue 6) pp:1130-1133
Publication Date(Web):
DOI:10.1002/anie.200704893
Co-reporter:Dennis P. Curran;Reena Bajpai;Elizabeth Sanger
Advanced Synthesis & Catalysis 2006 Volume 348(Issue 12-13) pp:
Publication Date(Web):11 AUG 2006
DOI:10.1002/adsc.200606187
Solvent tuning and partition coefficient measurements have identified suitable reagent and solvent combinations for the purification of fluorous Mitsunobu reaction products. The crude reaction mixtures are partitioned between 2/1 HFE-7100/FC-72 and DMF/10 % water to provide reagent (fluorous) and product (organic) fractions.
Co-reporter:Dennis P. Curran, Gustavo Moura-Letts,Matthias Pohlman
Angewandte Chemie International Edition 2006 45(15) pp:2423-2426
Publication Date(Web):
DOI:10.1002/anie.200600041
Co-reporter:Dennis P. Curran Dr.;Gustavo Moura-Letts;Matthias Pohlman Dr.
Angewandte Chemie 2006 Volume 118(Issue 15) pp:
Publication Date(Web):9 MAR 2006
DOI:10.1002/ange.200600041
Doppelter Spaß: Eine neue Methode mit doppelter Markierung wurde verwendet, um acht individuelle Stereoisomere von Passifloricin über ein intermediär auftretendes Paar von quasi-isomeren Vierkomponentenmischungen zu synthetisieren.
Co-reporter:Raghuram S. Tangirala, Rachel Dixon, Danzhou Yang, Attila Ambrus, Smitha Antony, Keli Agama, Yves Pommier, Dennis P. Curran
Bioorganic & Medicinal Chemistry Letters 2005 Volume 15(Issue 21) pp:4736-4740
Publication Date(Web):1 November 2005
DOI:10.1016/j.bmcl.2005.07.074
Both enantiomers of 20-fluorocamptothecin and the racemate have been prepared by total synthesis. The (R)-enantiomer is essentially inactive in a topoisomerase-I/DNA assay, while the (S)-enantiomer is much less active than (20S)-camptothecin. The lactone ring of 20-fluorocamptothecin hydrolyzes more rapidly than that of camptothecin in PBS. The results provide insight into the role of the 20-hydroxy group in the binding of camptothecin to topoisomerase-I and DNA.Both enantiomers of 20-fluorocamptothecin and the racemate have been prepared by total synthesis. The (R)-enantiomer is essentially inactive in a topoisomerase-I/DNA assay, while the (S)-enantiomer is much less active than 20(S)-camptothecin. The lactone ring of 20-fluorocamptothecin hydrolyzes more rapidly than that of camptothecin in PBS.
Co-reporter:Craig S. Wilcox Dr.;Venugopal Gudipati;Hejun Lu;Serhan Turkyilmaz Dr.
Angewandte Chemie 2005 Volume 117(Issue 42) pp:
Publication Date(Web):5 OCT 2005
DOI:10.1002/ange.200501989
Doppelt markiert hält besser: Sechzehn Stereoisomere von Murisolin (siehe Schema) wurden gemeinsam synthetisiert und rein isoliert; dabei half ein Verfahren mit doppelter Markierung und Trennung. Eine Strategie für die Synthese von Stereoisomeren in Lösung durch doppeltes Markieren und Entmischen setzt auf Fluoralkyl- und Oligo(ethylenglycol)-Marker ((OCH2CH2)nOCH3, OEG).
Co-reporter:Craig S. Wilcox, Venugopal Gudipati, Hejun Lu, Serhan Turkyilmaz,Dennis P. Curran
Angewandte Chemie International Edition 2005 44(42) pp:6938-6940
Publication Date(Web):
DOI:10.1002/anie.200501989
Co-reporter:Qisheng Zhang
Chemistry - A European Journal 2005 Volume 11(Issue 17) pp:
Publication Date(Web):25 MAY 2005
DOI:10.1002/chem.200500076
The old adage “never mix pure organic compounds” holds in spades for enantiomers. After going to all the trouble to make enantiopure molecules, who in their right mind would ever mix them to make a racemate? Quasienantiomers are almost enantiomers, but not quite. Yet unlike enantiomers, the interest is not so much in separating them but in mixing them to make quasiracemates. This backwards thinking opens new possibilities for identification, analysis, separation and synthesis of enantiomers. A short history is provided, the terms are defined and illustrated, and recent applications of quasienantiomers, quasiracemates and related species are reviewed.
Co-reporter:Sivaraman Dapani Dr. and
Chemistry - A European Journal 2004 Volume 10(Issue 13) pp:
Publication Date(Web):26 MAY 2004
DOI:10.1002/chem.200400363
The Mitsunobu reaction is famous for its scope and power, but infamous for its separation headaches. Typically, the target product is enticed away from the reagent-derived byproducts by careful chromatography. The use of polymer-bound Mitsunobu reagents solves only half of the problem, because polymer-bound diethyl azodicarboxylate (DEAD) and phosphine reagents cannot be employed simultaneously. This article classifies, compares, and contrasts various emerging strategies for product isolation in Mitsunobu reactions. Because so many different strategies have been used, the Mitsunobu reaction is a microcosm for the new field of strategy level separations.
Co-reporter:Youseung Shin;Jean-Hugues Fournier Dr.;Yoshikazu Fukui Dr.;Arndt M. Brückner Dr. Dr.
Angewandte Chemie 2004 Volume 116(Issue 35) pp:
Publication Date(Web):1 JUL 2004
DOI:10.1002/ange.200460593
Das echte Dictyostatin bitte melden! Zum Schluss blieben fünf Kandidaten-Stereostrukturen für das wirksame Tumortherapeutikum Dictyostatin – sogar zehn, wenn man die Enantiomere einschließt. Die Totalsynthese von (−)-Dictyostatin (1) hat eine zehn Jahre andauernde Maskerade beendet: Der Gewinner ist die kürzlich von Paterson und Wright vorgeschlagene Struktur.
Co-reporter:Ian Paterson Dr.;Robert Britton Dr.;Oscar Delgado Dr.;Arndt Meyer;Karine G. Poullennec Dr.;Youseung Shin;Jean-Hugues Fournier Dr.;Yoshikazu Fukui Dr.;Arndt M. Brückner Dr. Dr.
Angewandte Chemie International Edition 2004 Volume 43(Issue 35) pp:
Publication Date(Web):1 SEP 2004
DOI:10.1002/anie.200490122
Co-reporter:Youseung Shin;Jean-Hugues Fournier Dr.;Yoshikazu Fukui Dr.;Arndt M. Brückner Dr. Dr.
Angewandte Chemie International Edition 2004 Volume 43(Issue 35) pp:
Publication Date(Web):1 JUL 2004
DOI:10.1002/anie.200460593
Will the real dictyostatin please stand up? There were five finalists as stereostructures for the potent anticancer agent dictyostatin; ten, if one were to include enantiomers. A total synthesis of (−)-dictyostatin (1) has ended the decade-old masquerade and identified the winner as a structure recently proposed by Paterson and Wright.
Co-reporter:Ian Paterson Dr.;Robert Britton Dr.;Oscar Delgado Dr.;Arndt Meyer;Karine G. Poullennec Dr.;Youseung Shin;Jean-Hugues Fournier Dr.;Yoshikazu Fukui Dr.;Arndt M. Brückner Dr. Dr.
Angewandte Chemie 2004 Volume 116(Issue 35) pp:
Publication Date(Web):1 SEP 2004
DOI:10.1002/ange.200490121
Co-reporter:Dennis P. Curran;Sivaraman Dandapani;Mario Jeske
PNAS 2004 Volume 101 (Issue 33 ) pp:12008-12012
Publication Date(Web):2004-08-17
DOI:10.1073/pnas.0401677101
All 16 stereoisomers of the sex pheromone of pine sawfly (3,7,11-trimethylundecanol propanoate ester) have been synthesized
on a 10- to 20-mg scale by a split-parallel fluorous mixture-synthesis approach. Spectral data obtained for all 32 compounds
(16 alcohols and the corresponding propionates) matched well with published data, thereby validating the fluorous-tag encoding
of diastereoisomers. This fluorous-tag encoding method is recommended for the efficient synthesis of multiple stereoisomers
for spectroscopic studies, biological tests, or other structure–function relationships.
Co-reporter:Dennis P. Curran;Qisheng Zhang
Advanced Synthesis & Catalysis 2003 Volume 345(Issue 3) pp:
Publication Date(Web):7 MAR 2003
DOI:10.1002/adsc.200390033
Brief microwave irradiation of mono-alkylated malonates and β-ketoesters at 160–200 °C in wet DMF induces smooth and selective decarboalkoxylation. Observations suggest that this reaction occurs by nucleophilic attack of water at the ester carbonyl carbon (hydrolysis) followed by decarboxylation of the resulting acid. The process occurs despite the absence of traditional acid, base or nucleophile catalysts or reagents.
Co-reporter:José M. Mı́nguez, Sun-Young Kim, Kenneth A. Giuliano, Raghavan Balachandran, Charitha Madiraju, Billy W. Day, Dennis P. Curran
Bioorganic & Medicinal Chemistry 2003 Volume 11(Issue 15) pp:3335-3357
Publication Date(Web):31 July 2003
DOI:10.1016/S0968-0896(03)00186-X
An efficient, convergent and stereocontrolled synthesis of simplified analogues of the potent antimitotic agent (+)-discodermolide has been achieved and several small libraries have been prepared. In all the libraries, the discodermolide methyl groups at C14 and C16 and the C7 hydroxy group were removed and the lactone was replaced by simple esters. Other modifications introduced in each series of analogues were related to C11, C17 and C19 of the natural product. Key elements of the synthetic strategy included (a) elaboration of the main subunits from a common intermediate and (b) fragment couplings using Wittig reactions to install the (Z)-olefins. Library components were analyzed for microtubule-stabilizing actions in vitro, for displacement of [3H]paclitaxel from its binding site on tubulin, for antiproliferative activity against human carcinoma cells, and for cell signaling and mitotic spindle alterations by a multiparameter fluorescence cell-based screening technique. The results show that even significant structural simplification can lead to analogues with actions related to microtubule targeting.Graphic
Co-reporter:Jun Terauchi, Dennis P. Curran
Tetrahedron: Asymmetry 2003 Volume 14(Issue 5) pp:587-592
Publication Date(Web):7 March 2003
DOI:10.1016/S0957-4166(03)00041-7
The first catalytic asymmetric synthesis of axially chiral anilides has been carried out under palladium-catalyzed allylation reaction conditions with chiral phosphine ligands. Allylation of anilides bearing an orthotert-butyl group with a palladium catalyst and (S)-BINAP as a chiral ligand gave axially chiral anilides in about 50% ee.GraphicN-Allyl-N-(2-tert-butylphenyl)acetamideC17H27NOEe=53%[α]D23=+70.6 (c 0.565, CHCl3)Source of chirality: asymmetric N-allylationAbsolute configuration: PN-Allyl-N-[(2-methoxymethyl-1,1-dimethylethyl)phenyl]phenylacetamideC22H27NO2[α]D23=+51.7 (c 0.41, CHCl3)Source of chirality: asymmetric N-allylationAbsolute configuration: PN-Allyl-N-(2-tert-butylphenyl)phenylacetamideC22H27NO2Ee=53%[α]D23=+73.4 (c 0.415, CHCl3)Source of chirality: asymmetric N-allylationAbsolute configuration: PN-Allyl-N-(2-tert-butylphenyl)acrylamideC16H21NOEe=43%[α]D23=+54.5 (c 0.415, CHCl3)Source of chirality: asymmetric N-allylationAbsolute configuration: PN-Allyl-N-(2-tert-butylphenyl)benzamideC20H23NOEe=36%[α]D23=+12.4 (c 0.41, CHCl3)Source of chirality: asymmetric N-allylationAbsolute configuration: PN-Allyl-N-(2-tert-butylphenyl)pivamideC17H27NOEe=12%[α]D23=+14.6 (c 0.705, CHCl3)Source of chirality: asymmetric N-allylationAbsolute configuration: P
Co-reporter:Wu Du, Dennis P Curran, Robert L Bevins, Stephen G Zimmer, Junhong Zhang, Thomas G Burke
Bioorganic & Medicinal Chemistry 2002 Volume 10(Issue 1) pp:103-110
Publication Date(Web):January 2002
DOI:10.1016/S0968-0896(01)00252-8
The synthesis of a novel E-ring modified keto ether analogue of camptothecin and homocamptothecin by the cascade radical annulation route is reported. The analogue, Du1441, is an isomer of homocamptothecin, but includes the α-hydroxy carbonyl functionality that camptothecin possesses and homocamptothecin lacks. Despite these similarities, the new keto ether analogue is inactive in cell assays, and implications for the structure/activity relationship are discussed.Graphic
Co-reporter:Shau-Hua Ueng, Louis Fensterbank, Emmanuel Lacôte, Max Malacria and Dennis P. Curran
Organic & Biomolecular Chemistry 2011 - vol. 9(Issue 9) pp:NaN3420-3420
Publication Date(Web):2011/01/26
DOI:10.1039/C0OB01075H
1,3-Dimethylimidazol-2-ylidene borane and 2,4-dimethyl-1,2,4-triazol-3-ylidene borane are found to be useful reagents for the reduction of alkyl iodides and bromides bearing nearby electron withdrawing substituents. Signatures of radical chain reactions are seen in many cases, but ionic reductions may also be occurring with some substrates. The reagents are attractive because of their low molecular weight, their availability from inexpensive precursors, and their stability. Separation of the borane products from the target products is readily accomplished either with or without prior regeneration of the borane for later reuse. 2,4-Dimethyl-1,2,4-triazol-3-ylidene borane is versatile because both starting borane and its derived products can be removed by extraction with water.
Co-reporter:Andrey Solovyev, Emmanuel Lacôte and Dennis P. Curran
Dalton Transactions 2013 - vol. 42(Issue 3) pp:NaN700-700
Publication Date(Web):2012/09/07
DOI:10.1039/C2DT31773G
Reaction of the triflate group in dipp-Imd–BH2OTf with nucleophiles generally occurs by direct substitution to give products dipp-Imd–BH2Nu. In contrast, reaction of this boryl triflate with aryloxides (ArO−) in THF resulted in insertion of THF with ring opening in between the NHC–boryl electrophile and the aryloxide to give products dipp-Imd–BH2O(CH2)4OAr. The incorporation of THF was observed with other nucleophiles such as ethylthiolate and trimethylsilanolate whose conjugate acids have pKa values similar to that of phenol. The cleavage of ethyl acetate and oxetane is reported as well. A possible mechanism is proposed.
.jpg)










![[4-(4,4,5,5,6,6,7,7,8,8,9,9,9-tridecafluorononoxy)phenyl]methanol](http://img.cochemist.com/ccimg/957300/957206-65-4.png)
![[4-(4,4,5,5,6,6,7,7,8,8,9,9,9-tridecafluorononoxy)phenyl]methanol](http://img.cochemist.com/ccimg/957300/957206-65-4_b.png)




![[4-(4,4,5,5,6,6,7,7,7-nonafluoroheptoxy)phenyl]methanol](http://img.cochemist.com/ccimg/892200/892154-51-7.png)
![[4-(4,4,5,5,6,6,7,7,7-nonafluoroheptoxy)phenyl]methanol](http://img.cochemist.com/ccimg/892200/892154-51-7_b.png)
![1H-Tetrazole, 1-phenyl-5-[[3-(trimethylsilyl)-2-propynyl]thio]-](http://img.cochemist.com/ccimg/880900/880885-69-8.png)
![1H-Tetrazole, 1-phenyl-5-[[3-(trimethylsilyl)-2-propynyl]thio]-](http://img.cochemist.com/ccimg/880900/880885-69-8_b.png)


![1H-Tetrazole, 1-phenyl-5-[[[4-(trifluoromethyl)phenyl]methyl]thio]-](http://img.cochemist.com/ccimg/875700/875654-53-8.png)
![1H-Tetrazole, 1-phenyl-5-[[[4-(trifluoromethyl)phenyl]methyl]thio]-](http://img.cochemist.com/ccimg/875700/875654-53-8_b.png)




![Rhodium, bis[m-[a,a,a',a'-tetramethyl-1,3-benzenedipropanoato(2-)-kO1,kO'3:kO3,kO'1]]di-, (Rh-Rh)](http://img.cochemist.com/ccimg/819100/819050-89-0.png)
![Rhodium, bis[m-[a,a,a',a'-tetramethyl-1,3-benzenedipropanoato(2-)-kO1,kO'3:kO3,kO'1]]di-, (Rh-Rh)](http://img.cochemist.com/ccimg/819100/819050-89-0_b.png)


![5-Hexen-3-ol, 1-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-4-methyl-, (3S,4S)-](/data/chemimg/1965700/756834-72-7.png)
![5-Hexen-3-ol, 1-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-4-methyl-, (3S,4S)-](/data/chemimg/1965700/756834-72-7_b.png)


![Heptanal, 7-[(4-methoxyphenyl)methoxy]-](http://img.cochemist.com/ccimg/667500/667419-29-6.png)
![Heptanal, 7-[(4-methoxyphenyl)methoxy]-](http://img.cochemist.com/ccimg/667500/667419-29-6_b.png)


![1-HEPTANOL, 7-[(4-METHOXYPHENYL)METHOXY]-](http://img.cochemist.com/ccimg/627900/627871-77-6.png)
![1-HEPTANOL, 7-[(4-METHOXYPHENYL)METHOXY]-](http://img.cochemist.com/ccimg/627900/627871-77-6_b.png)


![Pentanal, 5-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2-methyl-, (2S)-](http://img.cochemist.com/ccimg/529500/529494-83-5.png)
![Pentanal, 5-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2-methyl-, (2S)-](http://img.cochemist.com/ccimg/529500/529494-83-5_b.png)
![1H-Imidazolium, 1,3-bis[2,6-bis(1-methylethyl)phenyl]-, iodide](http://img.cochemist.com/ccimg/524800/524742-02-7.png)
![1H-Imidazolium, 1,3-bis[2,6-bis(1-methylethyl)phenyl]-, iodide](http://img.cochemist.com/ccimg/524800/524742-02-7_b.png)


![BENZENE, 1-(BROMOMETHYL)-4-[(4,4,5,5,6,6,7,7,7-NONAFLUOROHEPTYL)OXY]-](http://img.cochemist.com/ccimg/444700/444682-61-5.png)
![BENZENE, 1-(BROMOMETHYL)-4-[(4,4,5,5,6,6,7,7,7-NONAFLUOROHEPTYL)OXY]-](http://img.cochemist.com/ccimg/444700/444682-61-5_b.png)













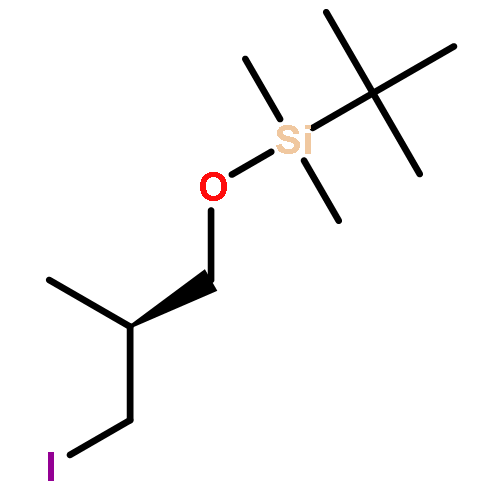


![Propanamide,N-[(1S,2S)-2-hydroxy-1-methyl-2-phenylethyl]-N-methyl-](http://img.cochemist.com/ccimg/159300/159213-03-3.png)
![Propanamide,N-[(1S,2S)-2-hydroxy-1-methyl-2-phenylethyl]-N-methyl-](http://img.cochemist.com/ccimg/159300/159213-03-3_b.png)








![1-Pentanol, 5-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2,4-dimethyl-,(2S,4R)-](http://img.cochemist.com/ccimg/147800/147782-80-7.png)
![1-Pentanol, 5-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2,4-dimethyl-,(2S,4R)-](http://img.cochemist.com/ccimg/147800/147782-80-7_b.png)












![Oxirane, 2-[[(4-methoxyphenyl)methoxy]methyl]-, (2R)-](/data/chemimg/3749100/134733-19-0.png)
![Oxirane, 2-[[(4-methoxyphenyl)methoxy]methyl]-, (2R)-](/data/chemimg/3749100/134733-19-0_b.png)


![2H-Pyran-2-one,6-[(2S,3Z,5S,6S,7S,8Z,11S,12R,13S,14S,15S,16Z)-14-[(aminocarbonyl)oxy]-2,6,12-trihydroxy-5,7,9,11,13,15-hexamethyl-3,8,16,18-nonadecatetraen-1-yl]tetrahydro-4-hydroxy-3,5-dimethyl-,(3R,4S,5R,6S)-](http://img.cochemist.com/ccimg/128000/127943-53-7.png)
![2H-Pyran-2-one,6-[(2S,3Z,5S,6S,7S,8Z,11S,12R,13S,14S,15S,16Z)-14-[(aminocarbonyl)oxy]-2,6,12-trihydroxy-5,7,9,11,13,15-hexamethyl-3,8,16,18-nonadecatetraen-1-yl]tetrahydro-4-hydroxy-3,5-dimethyl-,(3R,4S,5R,6S)-](http://img.cochemist.com/ccimg/128000/127943-53-7_b.png)










![5-Hexen-3-ol, 1-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2,4-dimethyl-,(2S,3R,4S)-](http://img.cochemist.com/ccimg/106400/106357-22-6.png)
![5-Hexen-3-ol, 1-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2,4-dimethyl-,(2S,3R,4S)-](http://img.cochemist.com/ccimg/106400/106357-22-6_b.png)


![6-[(4-METHOXYPHENYL)METHOXY]HEXAN-1-OL](/data/chemimg/334700/105192-33-4.png)
![6-[(4-METHOXYPHENYL)METHOXY]HEXAN-1-OL](/data/chemimg/334700/105192-33-4_b.png)

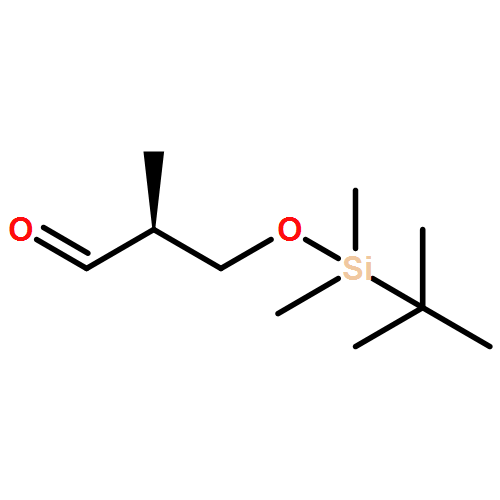


![Benzenemethanamine, N-[3-(trimethylsilyl)-2-propynyl]-](http://img.cochemist.com/ccimg/102000/101911-12-0.png)
![Benzenemethanamine, N-[3-(trimethylsilyl)-2-propynyl]-](http://img.cochemist.com/ccimg/102000/101911-12-0_b.png)


![Silane, (1,1-dimethylethyl)[(1,1-dimethyl-2-propynyl)oxy]dimethyl-](http://img.cochemist.com/ccimg/98800/98733-45-0.png)
![Silane, (1,1-dimethylethyl)[(1,1-dimethyl-2-propynyl)oxy]dimethyl-](http://img.cochemist.com/ccimg/98800/98733-45-0_b.png)
![Propanal, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2-methyl-, (R)-](http://img.cochemist.com/ccimg/97900/97826-89-6.png)
![Propanal, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2-methyl-, (R)-](http://img.cochemist.com/ccimg/97900/97826-89-6_b.png)
![2-Pentynoic acid, 5-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, ethyl ester](http://img.cochemist.com/ccimg/90800/90708-75-1.png)
![2-Pentynoic acid, 5-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, ethyl ester](http://img.cochemist.com/ccimg/90800/90708-75-1_b.png)




![Benzoic acid,4-[(phenylthio)methyl]-, methyl ester](http://img.cochemist.com/ccimg/88400/88393-07-1.png)
![Benzoic acid,4-[(phenylthio)methyl]-, methyl ester](http://img.cochemist.com/ccimg/88400/88393-07-1_b.png)


![2-PENTEN-1-OL, 5-[[(1,1-DIMETHYLETHYL)DIMETHYLSILYL]OXY]-3-METHYL-, (Z)-](http://img.cochemist.com/ccimg/86500/86462-83-1.png)
![2-PENTEN-1-OL, 5-[[(1,1-DIMETHYLETHYL)DIMETHYLSILYL]OXY]-3-METHYL-, (Z)-](http://img.cochemist.com/ccimg/86500/86462-83-1_b.png)







![SILANE, TRIMETHYL[3-(PHENYLTHIO)-1-PROPYNYL]-](http://img.cochemist.com/ccimg/82800/82707-19-5.png)
![SILANE, TRIMETHYL[3-(PHENYLTHIO)-1-PROPYNYL]-](http://img.cochemist.com/ccimg/82800/82707-19-5_b.png)

![BENZENE, 1-IODO-2-[(PHENYLTHIO)METHYL]-](http://img.cochemist.com/ccimg/73100/73038-47-8.png)
![BENZENE, 1-IODO-2-[(PHENYLTHIO)METHYL]-](http://img.cochemist.com/ccimg/73100/73038-47-8_b.png)


![2,4-Hexadien-1-ol, 6-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (E,E)-](http://img.cochemist.com/ccimg/72300/72297-15-5.png)
![2,4-Hexadien-1-ol, 6-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (E,E)-](http://img.cochemist.com/ccimg/72300/72297-15-5_b.png)





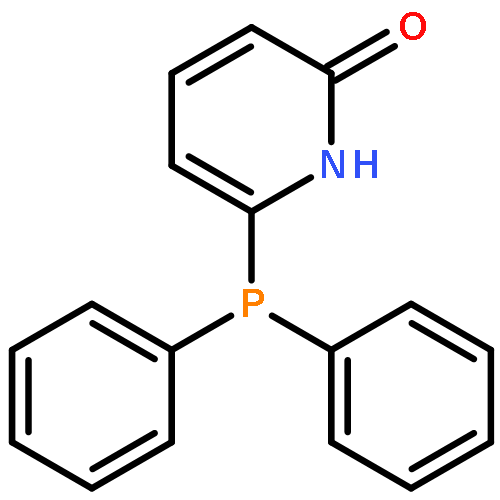






![Benzene, [(hexyloxy)methyl]-](http://img.cochemist.com/ccimg/61200/61103-84-2.png)
![Benzene, [(hexyloxy)methyl]-](http://img.cochemist.com/ccimg/61200/61103-84-2_b.png)

![Benzene, [(5-hexenyloxy)methyl]-](http://img.cochemist.com/ccimg/59200/59137-50-7.png)
![Benzene, [(5-hexenyloxy)methyl]-](http://img.cochemist.com/ccimg/59200/59137-50-7_b.png)













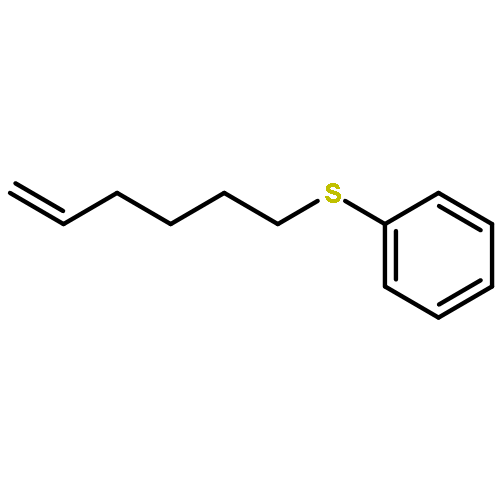


![Benzene, 1-[(phenylthio)methyl]-4-(trifluoromethyl)-](http://img.cochemist.com/ccimg/31000/30932-37-7.png)
![Benzene, 1-[(phenylthio)methyl]-4-(trifluoromethyl)-](http://img.cochemist.com/ccimg/31000/30932-37-7_b.png)
![Benzene,[(3-phenylpropyl)thio]-](http://img.cochemist.com/ccimg/30200/30134-12-4.png)
![Benzene,[(3-phenylpropyl)thio]-](http://img.cochemist.com/ccimg/30200/30134-12-4_b.png)







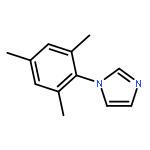
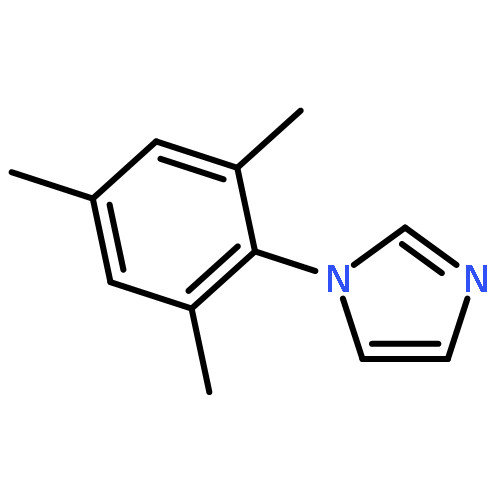






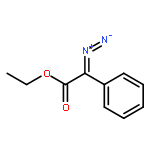
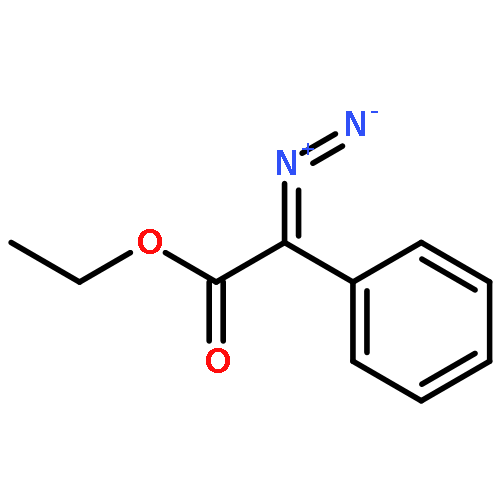
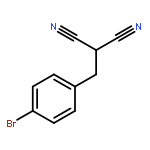
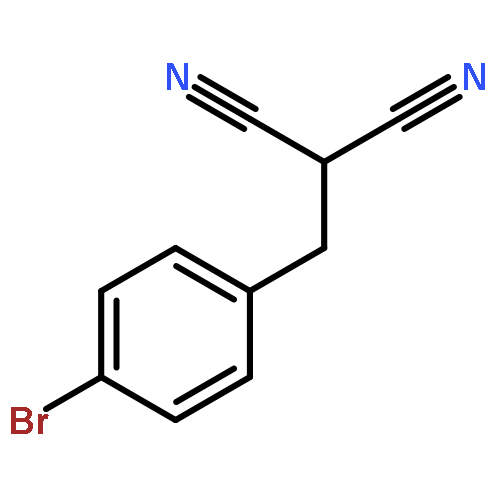
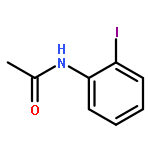
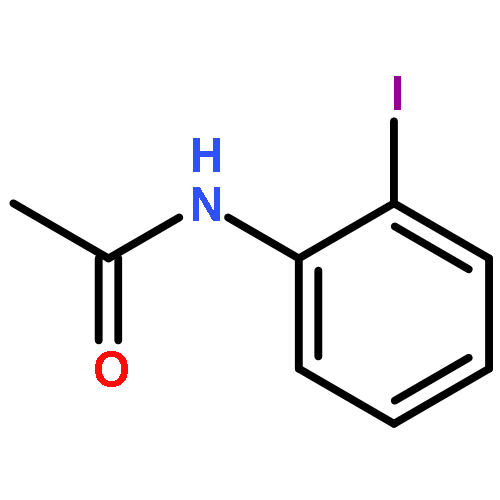
![trimethyl[(Z)-2-phenylethenyl]silane](http://img.cochemist.com/ccimg/19400/19319-11-0.png)
![trimethyl[(Z)-2-phenylethenyl]silane](http://img.cochemist.com/ccimg/19400/19319-11-0_b.png)










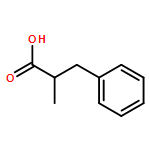
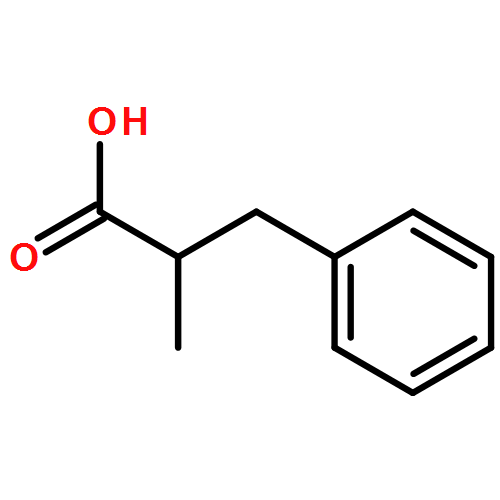
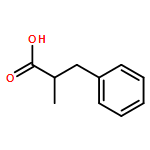
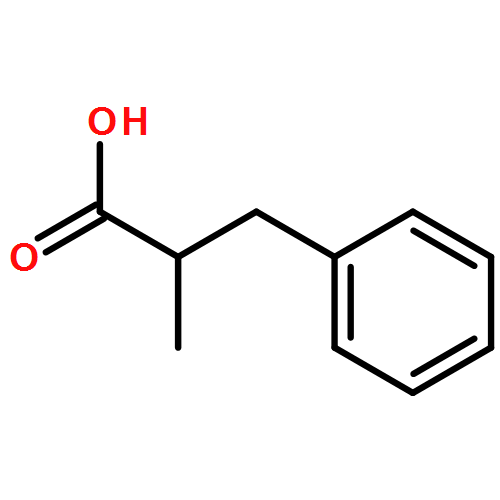


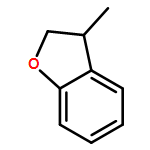

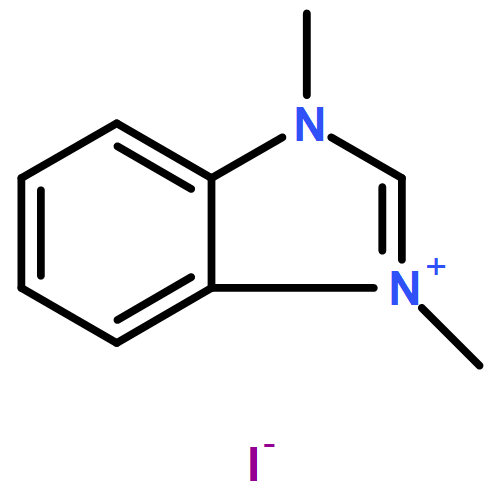


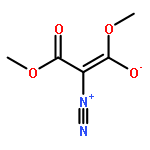
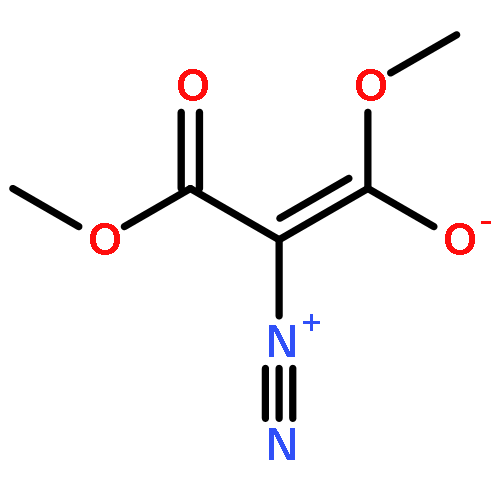
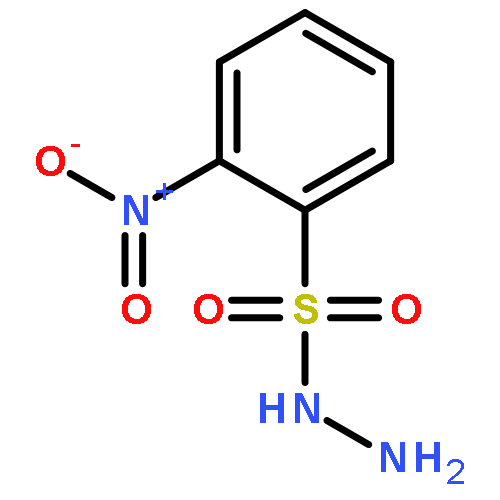


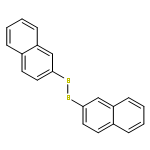
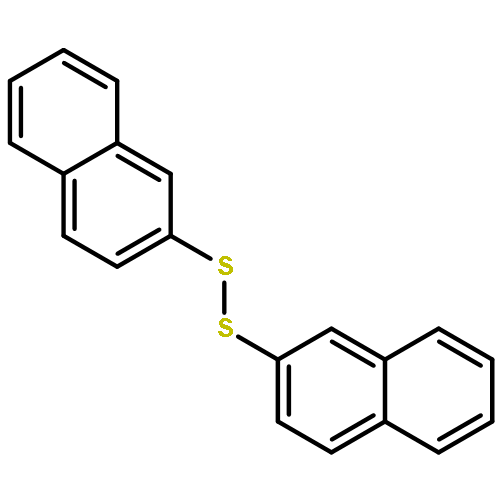
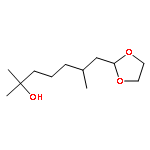







![1H-Tetrazole, 1-phenyl-5-[(phenylmethyl)thio]-](http://img.cochemist.com/ccimg/3300/3206-48-2.png)
![1H-Tetrazole, 1-phenyl-5-[(phenylmethyl)thio]-](http://img.cochemist.com/ccimg/3300/3206-48-2_b.png)
![Propanedinitrile,2-[(4-chlorophenyl)methyl]-](http://img.cochemist.com/ccimg/3000/2964-33-2.png)
![Propanedinitrile,2-[(4-chlorophenyl)methyl]-](http://img.cochemist.com/ccimg/3000/2964-33-2_b.png)
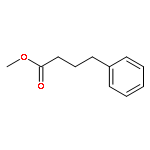

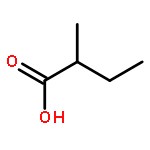

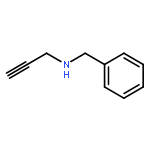
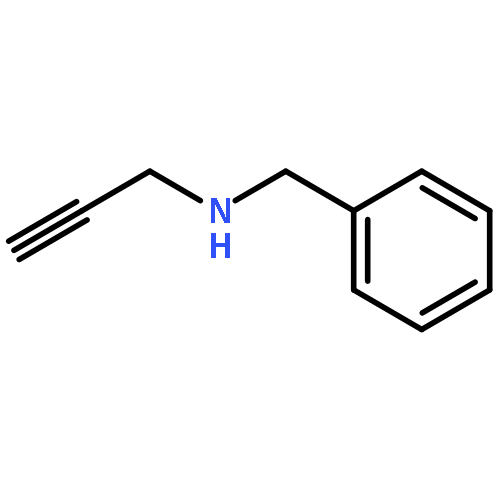






![Benzene,1-fluoro-4-[(phenylthio)methyl]-](http://img.cochemist.com/ccimg/400/351-66-6.png)
![Benzene,1-fluoro-4-[(phenylthio)methyl]-](http://img.cochemist.com/ccimg/400/351-66-6_b.png)
![2-Azaspiro[4.4]nonane](http://img.cochemist.com/ccimg/200/175-94-0.png)
![2-Azaspiro[4.4]nonane](http://img.cochemist.com/ccimg/200/175-94-0_b.png)




![2-Hexenoic acid, 5-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-4-[(2-methoxyethoxy)methoxy]-, ethyl ester, (2E,4R,5S)-](/data/chemimg/1851300/566203-43-8.png)
![2-Hexenoic acid, 5-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-4-[(2-methoxyethoxy)methoxy]-, ethyl ester, (2E,4R,5S)-](/data/chemimg/1851300/566203-43-8_b.png)


![N-[4-(1H,1H,2H,2H-PERFLUORODECYL)BENZYLOXYCARBONYLOXY]SUCCINIMIDE](http://img.cochemist.com/ccimg/356100/356056-15-0.png)
![N-[4-(1H,1H,2H,2H-PERFLUORODECYL)BENZYLOXYCARBONYLOXY]SUCCINIMIDE](http://img.cochemist.com/ccimg/356100/356056-15-0_b.png)






![1,3-Dioxolane, 4-[2-[(4-methoxyphenyl)methoxy]ethyl]-2,2-dimethyl-,(4R)-](http://img.cochemist.com/ccimg/214000/213978-60-0.png)
![1,3-Dioxolane, 4-[2-[(4-methoxyphenyl)methoxy]ethyl]-2,2-dimethyl-,(4R)-](http://img.cochemist.com/ccimg/214000/213978-60-0_b.png)




![2-Hexenoic acid, 5-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-4-[(2-methoxyethoxy)methoxy]-, (2E,4R,5S)-](/data/chemimg/1779700/196926-52-0.png)
![2-Hexenoic acid, 5-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-4-[(2-methoxyethoxy)methoxy]-, (2E,4R,5S)-](/data/chemimg/1779700/196926-52-0_b.png)


![Propanamide, N-[(1R,2R)-2-hydroxy-1-methyl-2-phenylethyl]-N-methyl-](http://img.cochemist.com/ccimg/192100/192060-67-6.png)
![Propanamide, N-[(1R,2R)-2-hydroxy-1-methyl-2-phenylethyl]-N-methyl-](http://img.cochemist.com/ccimg/192100/192060-67-6_b.png)
![Oxacyclodocosa-3,5,11-trien-2-one, 8,10,14,20-tetrahydroxy-7,13,15,17,21-pentamethyl-22-[(1S,2Z)-1-methyl-2,4-pentadien-1-yl]-, (3Z,5E,7R,8S,10S,11Z,13S,14R,15S,17S,20R,21S,22S)-](/data/chemimg/3766500/156312-07-1.png)
![Oxacyclodocosa-3,5,11-trien-2-one, 8,10,14,20-tetrahydroxy-7,13,15,17,21-pentamethyl-22-[(1S,2Z)-1-methyl-2,4-pentadien-1-yl]-, (3Z,5E,7R,8S,10S,11Z,13S,14R,15S,17S,20R,21S,22S)-](/data/chemimg/3766500/156312-07-1_b.png)






![Butanoic acid, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (3S)-](http://img.cochemist.com/ccimg/105800/105729-41-7.png)
![Butanoic acid, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (3S)-](http://img.cochemist.com/ccimg/105800/105729-41-7_b.png)
![Butanoic acid, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, methyl ester, (3R)-](/data/chemimg/1042900/104524-19-8.png)
![Butanoic acid, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, methyl ester, (3R)-](/data/chemimg/1042900/104524-19-8_b.png)
![Benzene, 1-bromo-2-[(octyloxy)methyl]-](http://img.cochemist.com/ccimg/103100/103009-97-8.png)
![Benzene, 1-bromo-2-[(octyloxy)methyl]-](http://img.cochemist.com/ccimg/103100/103009-97-8_b.png)
![Butanoic acid, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (3R)-](http://img.cochemist.com/ccimg/101600/101515-46-2.png)
![Butanoic acid, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (3R)-](http://img.cochemist.com/ccimg/101600/101515-46-2_b.png)
![Pyrrolidine, 3-methyl-1-[(4-methylphenyl)sulfonyl]-](/data/chemimg/964400/86553-37-9.png)
![Pyrrolidine, 3-methyl-1-[(4-methylphenyl)sulfonyl]-](/data/chemimg/964400/86553-37-9_b.png)














![Benzene,[(octyloxy)methyl]-](http://img.cochemist.com/ccimg/54900/54852-64-1.png)
![Benzene,[(octyloxy)methyl]-](http://img.cochemist.com/ccimg/54900/54852-64-1_b.png)







