Co-reporter:Trevor M. Penning
Chemical Research in Toxicology January 17, 2017 Volume 30(Issue 1) pp:
Publication Date(Web):November 3, 2016
DOI:10.1021/acs.chemrestox.6b00319
Human aldo-keto reductases (AKRs) are NAD(P)H-dependent oxidoreductases that convert aldehydes and ketones to primary and secondary alcohols for subsequent conjugation reactions and can be referred to as “phase 1” enzymes. Among all the human genes regulated by the Keap1/Nrf2 pathway, they are consistently the most overexpressed in response to Nrf2 activators. Although these enzymes play clear cytoprotective roles and deal effectively with carbonyl stress, their upregulation by the Keap1/Nrf2 pathway also has a potential dark-side, which can lead to chemotherapeutic drug resistance and the metabolic activation of lung carcinogens (e.g., polycyclic aromatic hydrocarbons). They also play determinant roles in 4-(methylnitrosoamino)-1-(3-pyridyl)-1-butanone metabolism to R- and S-4-(methylnitrosoamino)-1-(3-pyridyl)-1-butanol. The overexpression of AKR genes as components of the “smoking gene” battery raises the issue as to whether this is part of a smoking stress response or acquired susceptibility to lung cancer. Human AKR genes also regulate retinoid, prostaglandin, and steroid hormone metabolism and can regulate the local concentrations of ligands available for nuclear receptors (NRs). The prospect exists that signaling through the Keap1/Nrf2 system can also effect NR signaling, but this has remained largely unexplored. We present the case that chemoprevention through the Keap1/Nrf2 system may be context dependent and that the Nrf2 “dose-response curve” for electrophilic and redox balance may not be monotonic.
Co-reporter:Adegoke Adeniji, Md. Jashim Uddin, Tianzhu Zang, Daniel Tamae, Phumvadee Wangtrakuldee, Lawrence J. Marnett, and Trevor M. Penning
Journal of Medicinal Chemistry 2016 Volume 59(Issue 16) pp:7431-7444
Publication Date(Web):August 3, 2016
DOI:10.1021/acs.jmedchem.6b00160
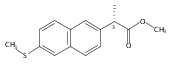
Type 5 17β-hydroxysteroid dehydrogenase, aldo-keto reductase 1C3 (AKR1C3) converts Δ4-androstene-3,17-dione and 5α-androstane-3,17-dione to testosterone (T) and 5α-dihydrotestosterone, respectively, in castration resistant prostate cancer (CRPC). In CRPC, AKR1C3 is implicated in drug resistance, and enzalutamide drug resistance can be surmounted by indomethacin a potent inhibitor of AKR1C3. We examined a series of naproxen analogues and find that (R)-2-(6-methoxynaphthalen-2-yl)butanoic acid (in which the methyl group of R-naproxen was replaced by an ethyl group) acts as a potent AKR1C3 inhibitor that displays selectivity for AKR1C3 over other AKR1C enzymes. This compound was devoid of inhibitory activity on COX isozymes and blocked AKR1C3 mediated production of T and induction of PSA in LNCaP-AKR1C3 cells as a model of a CRPC cell line. R-Profens are substrate selective COX-2 inhibitors and block the oxygenation of endocannabinoids and in the context of advanced prostate cancer R-profens could inhibit intratumoral androgen synthesis and act as analgesics for metastatic disease.
Co-reporter:Meng Huang, Clementina Mesaros, Suhong Zhang, Ian A. Blair, and Trevor M. Penning
Chemical Research in Toxicology 2016 Volume 29(Issue 6) pp:991
Publication Date(Web):April 7, 2016
DOI:10.1021/acs.chemrestox.6b00036
Exposure to polycyclic aromatic hydrocarbons (PAHs) is the major human health hazard associated with the Deepwater Horizon oil spill. C2-Chrysenes are representative PAHs present in crude oil and could contaminate the food chain. We describe the metabolism of a C2-chrysene regioisomer, 6-ethylchrysene (6-EC), in human HepG2 cells. The structures of the metabolites were identified by HPLC-UV-fluorescence detection and LC-MS/MS. 6-EC-tetraol isomers were identified as signature metabolites of the diol-epoxide pathway. O-Monomethyl-O-monosulfonated-6-EC-catechol, its monohydroxy products, and N-acetyl-l-cysteine(NAC)-6-EC-ortho-quinone were discovered as signature metabolites of the ortho-quinone pathway. Potential dual metabolic activation of 6-EC involving the formation of bis-electrophiles, i.e., a mono-diol-epoxide and a mono-ortho-quinone within the same structure, bis-diol-epoxides, and bis-ortho-quinones was observed as well. The identification of 6-EC-tetraol, O-monomethyl-O-monosulfonated-6-EC-catechol, its monohydroxy products, and NAC-6-EC-ortho-quinone supports potential metabolic activation of 6-EC by P450 and AKR enzymes followed by metabolic detoxification of the ortho-quinone through interception of its redox cycling capability by catechol-O-methyltransferase and sulfotransferase enzymes. The tetraols and catechol conjugates could be used as biomarkers of human exposure to 6-EC resulting from oil spills.
Co-reporter:Meng Huang, Li Zhang, Clementina Mesaros, Linda C. Hackfeld, Richard P. Hodge, Ian A. Blair, and Trevor M. Penning
Chemical Research in Toxicology 2015 Volume 28(Issue 10) pp:2045
Publication Date(Web):September 22, 2015
DOI:10.1021/acs.chemrestox.5b00256
Exposure to polycyclic aromatic hydrocarbons (PAHs) in the food chain is the major human health hazard associated with the Deepwater Horizon oil spill. C1-chrysenes are representative PAHs present in the crude oil and have been detected in contaminated sea food in amounts that exceed their permissible safety thresholds. We describe the metabolism of the most carcinogenic C1-chrysene regioisomer, 5-methylchrysene (5-MC), in human HepG2 cells. The structures of the metabolites were identified by HPLC-UV-fluorescence detection and LC-MS/MS. 5-MC-tetraol, a signature metabolite of the diol-epoxide pathway, was identified as reported previously. Novel O-monosulfonated-5-MC-catechol isomers and O-monomethyl-O-monosulfonated-5-MC-catechol were discovered, and evidence for their precursor ortho-quinones was obtained. The identities of O-monosulfonated-5-MC-1,2-catechol, O-monomethyl-O-monosulfonated-5-MC-1,2-catechol, and 5-MC-1,2-dione were validated by comparison to authentic synthesized standards. Dual metabolic activation of 5-MC involving the formation of bis-electrophiles, i.e., a mono-diol-epoxide and a mono-ortho-quinone within the same structure, bis-diol-epoxides, and bis-ortho-quinones is reported for the first time. Evidence was also obtained for minor metabolic conversion of 5-MC to form monohydroxylated-quinones and bis-phenols. The identification of 5-MC-tetraol, O-monosulfonated-5-MC-1,2-catechol, O-monomethyl-O-monosulfonated-5-MC-1,2-catechol, and 5-MC-1,2-dione supports metabolic activation of 5-MC by P450 and AKR isozymes followed by metabolic detoxification of the ortho-quinone through interception of redox cycling by COMT and SULT isozymes. The major metabolites, O-monosulfonated-catechols and tetraols, could be used as biomarkers of human exposure to 5-MC resulting from oil spills.
Co-reporter:Mo Chen, Yi Jin, and Trevor M. Penning
Biochemistry 2015 Volume 54(Issue 41) pp:6343-6351
Publication Date(Web):September 29, 2015
DOI:10.1021/acs.biochem.5b00816
Human steroid-5β-reductase (aldo-keto reductase 1D1, AKR1D1) stereospecifically reduces Δ4-3-ketosteroids to 5β-dihydrosteroids and is essential for steroid hormone metabolism and bile acid biosynthesis. Genetic defects in AKR1D1 cause bile acid deficiency that leads to life threatening neonatal hepatitis and cholestasis. The disease-associated P133R mutation caused significant decreases in catalytic efficiency with both the representative steroid (cortisone) and the bile acid precursor (7α-hydroxycholest-4-en-3-one) substrates. Pro133 is a second shell residue to the steroid binding channel and is distal to both the cofactor binding site and the catalytic center. Strikingly, the P133R mutation caused over a 40-fold increase in Kd values for the NADP(H) cofactors and increased the rate of release of NADP+ from the enzyme by 2 orders of magnitude when compared to the wild type enzyme. By contrast the effect of the mutation on Kd values for steroids were 10-fold or less. The reduced affinity for the cofactor suggests that the mutant exists largely in the less stable cofactor-free form in the cell. Using stopped-flow spectroscopy, a significant reduction in the rate of the chemical step was observed in multiple turnover reactions catalyzed by the P133R mutant, possibly due to the altered position of NADPH. Thus, impaired NADPH binding and hydride transfer is the molecular basis for bile acid deficiency in patients with the P133R mutation. Results revealed that optimal cofactor binding is vulnerable to distant structural perturbation, which may apply to other disease-associated mutations in AKR1D1, all of which occur at conserved residues and are unstable.
Co-reporter:Meng Huang, Li Zhang, Clementina Mesaros, Suhong Zhang, Michael A. Blaha, Ian A. Blair, and Trevor M. Penning
Chemical Research in Toxicology 2014 Volume 27(Issue 5) pp:852
Publication Date(Web):March 19, 2014
DOI:10.1021/tx500031p
Exposure to polycyclic aromatic hydrocarbons (PAHs) in the food chain is the major human health hazard associated with the Deepwater Horizon oil spill. Phenanthrene is a representative PAH present in crude oil, and it undergoes biological transformation, photooxidation, and chemical oxidation to produce its signature oxygenated derivative, phenanthrene-9,10-quinone. We report the downstream metabolic fate of phenanthrene-9,10-quinone in HepG2 cells. The structures of the metabolites were identified by HPLC–UV–fluorescence detection and LC–MS/MS. O-mono-Glucuronosyl-phenanthrene-9,10-catechol was identified, as reported previously. A novel bis-conjugate, O-mono-methyl-O-mono-sulfonated-phenanthrene-9,10-catechol, was discovered for the first time, and evidence for both of its precursor mono conjugates was obtained. The identities of these four metabolites were unequivocally validated by comparison to authentic enzymatically synthesized standards. Evidence was also obtained for a minor metabolic pathway of phenanthrene-9,10-quinone involving bis-hydroxylation followed by O-mono-sulfonation. The identification of 9,10-catechol conjugates supports metabolic detoxification of phenanthrene-9,10-quinone through interception of redox cycling by UGT, COMT, and SULT isozymes and indicates the possible use of phenanthrene-9,10-catechol conjugates as biomarkers of human exposure to oxygenated PAH.
Co-reporter:Trevor M. Penning
Chemical Research in Toxicology 2014 Volume 27(Issue 11) pp:1901
Publication Date(Web):October 3, 2014
DOI:10.1021/tx500298n
Aldo-keto reductases (AKRs) are promiscuous NAD(P)(H) dependent oxidoreductases implicated in the metabolic activation of polycyclic aromatic hydrocarbons (PAH). These enzymes catalyze the oxidation of non-K-region trans-dihydrodiols to the corresponding o-quinones with the concomitant production of reactive oxygen species (ROS). The PAH o-quinones are Michael acceptors and can form adducts but are also redox-active and enter into futile redox cycles to amplify ROS formation. Evidence exists to support this metabolic pathway in humans. The human recombinant AKR1A1 and AKR1C1–AKR1C4 enzymes all catalyze the oxidation of PAH trans-dihydrodiols to PAH o-quinones. Many human AKRs also catalyze the NADPH-dependent reduction of the o-quinone products to air-sensitive catechols, exacerbating ROS formation. Moreover, this pathway of PAH activation occurs in a panel of human lung cell lines, resulting in the production of ROS and oxidative DNA damage in the form of 8-oxo-2′-deoxyguanosine. Using stable-isotope dilution liquid chromatography tandem mass spectrometry, this pathway of benzo[a]pyrene (B[a]P) metabolism was found to contribute equally with the diol-epoxide pathway to the activation of this human carcinogen in human lung cells. Evaluation of the mutagenicity of anti-B[a]P-diol epoxide with B[a]P-7,8-dione on p53 showed that the o-quinone produced by AKRs was the more potent mutagen, provided that it was permitted to redox cycle, and that the mutations observed were G to T transversions, reminiscent of those observed in human lung cancer. It is concluded that there is sufficient evidence to support the role of human AKRs in the metabolic activation of PAH in human lung cell lines and that they may contribute to the causation of human lung cancer.
Co-reporter:Andy J. Liedtke ; Adegoke O. Adeniji ; Mo Chen ; Michael C. Byrns ; Yi Jin ; David W. Christianson ; Lawrence J. Marnett ;Trevor M. Penning
Journal of Medicinal Chemistry 2013 Volume 56(Issue 6) pp:2429-2446
Publication Date(Web):February 22, 2013
DOI:10.1021/jm3017656
Castrate-resistant prostate cancer (CRPC) is a fatal, metastatic form of prostate cancer. CRPC is characterized by reactivation of the androgen axis due to changes in androgen receptor signaling and/or adaptive intratumoral androgen biosynthesis. AKR1C3 is upregulated in CRPC where it catalyzes the formation of potent androgens. This makes AKR1C3 a target for the treatment of CRPC. AKR1C3 inhibitors should not inhibit AKR1C1/AKR1C2, which inactivate 5α-dihydrotestosterone. Indomethacin, used to inhibit cyclooxygenase, also inhibits AKR1C3 and displays selectivity over AKR1C1/AKR1C2. Parallel synthetic strategies were used to generate libraries of indomethacin analogues, which exhibit reduced cyclooxygenase inhibitory activity but retain AKR1C3 inhibitory potency and selectivity. The lead compounds inhibited AKR1C3 with nanomolar potency, displayed >100-fold selectivity over AKR1C1/AKR1C2, and blocked testosterone formation in LNCaP-AKR1C3 cells. The AKR1C3·NADP+·2′-des-methyl-indomethacin crystal structure was determined, and it revealed a unique inhibitor binding mode. The compounds reported are promising agents for the development of therapeutics for CRPC.
Co-reporter:Meng Huang, Ian A. Blair, and Trevor M. Penning
Chemical Research in Toxicology 2013 Volume 26(Issue 5) pp:685
Publication Date(Web):April 15, 2013
DOI:10.1021/tx300476m
Metabolic activation of the proximate carcinogen benzo[a]pyrene-7,8-trans-dihydrodiol (B[a]P-7,8-trans-dihydrodiol) by aldo-keto reductases (AKRs) leads to B[a]P-7,8-dione that is both electrophilic and redox-active. B[a]P-7,8-dione generates reactive oxygen species resulting in oxidative DNA damage in human lung cells. However, information on the formation of stable B[a]P-7,8-dione-DNA adducts in these cells is lacking. We studied stable DNA adduct formation of B[a]P-7,8-dione in human lung adenocarcinoma A549 cells, human bronchoalveolar H358 cells, and immortalized human bronchial epithelial HBEC-KT cells. After treatment with 2 μM B[a]P-7,8-dione, the cellular DNA was extracted from the cell pellets subjected to enzyme hydrolysis and subsequent analysis by LC-MS/MS. Several stable DNA adducts of B[a]P-7,8-dione were only detected in A549 and HBEC-KT cells. In A549 cells, the structures of stable B[a]P-7,8-dione-DNA adducts were identified as hydrated-B[a]P-7,8-dione-N2-2′-deoxyguanosine and hydrated-B[a]P-7,8-dione-N1-2′-deoxyguanosine. In HBEC-KT cells, the structures of stable B[a]P-7,8-dione-DNA adducts were identified as hydrated-B[a]P-7,8-dione-2′-deoxyadenosine, hydrated-B[a]P-7,8-dione-N1- or N3-2′-deoxyadenosine, and B[a]P-7,8-dione-N1- or N3-2′-deoxyadenosine. In each case, adduct structures were characterized by MSn spectra. Adduct structures were also compared to those synthesized from reactions of B[a]P-7,8-dione with either deoxyribonucleosides or salmon testis DNA in vitro but were found to be different.
Co-reporter:Li Zhang, Meng Huang, Ian A. Blair, and Trevor M. Penning
Chemical Research in Toxicology 2013 Volume 26(Issue 10) pp:1570
Publication Date(Web):September 18, 2013
DOI:10.1021/tx400268q
Polycyclic aromatic hydrocarbons (PAHs) are environmental and tobacco carcinogens. Proximate carcinogenic PAH trans-dihydrodiols are activated by human aldo-keto reductases (AKRs) to yield electrophilic and redox-active o-quinones. Interconversion among benzo[a]pyrene (B[a]P)-7,8-dione, a representative PAH o-quinone, and its corresponding catechol generates a futile redox-cycle with the concomitant production of reactive oxygen species (ROS). We investigated whether glucuronidation of B[a]P-7,8-catechol by human UDP glucuronosyltransferases (UGTs) could intercept the catechol in three different human lung cells. RT-PCR showed that UGT1A1, 1A3, and 2B7 were only expressed in human lung adenocarcinoma A549 cells. The corresponding recombinant UGTs were examined for their kinetic constants and product profile using B[a]P-7,8-catechol as a substrate. B[a]P-7,8-dione was reduced to B[a]P-7,8-catechol by dithiothreitol under anaerobic conditions and then further glucuronidated by the UGTs in the presence of uridine-5′-diphosphoglucuronic acid as a glucuronic acid group donor. UGT1A1 catalyzed the glucuronidation of B[a]P-7,8-catechol and generated two isomeric O-monoglucuronsyl-B[a]P-7,8-catechol products that were identified by RP-HPLC and by LC-MS/MS. By contrast, UGT1A3 and 2B7 catalyzed the formation of only one monoglucuronide, which was identical to that formed in A549 cells. The kinetic profiles of three UGTs followed Michaelis–Menten kinetics. On the basis of the expression levels of UGT1A3 and UGT2B7 and the observation that a single monoglucuronide was produced in A549 cells, we suggest that the major UGT isoforms in A549 cells that can intercept B[a]P-7,8-catechol are UGT1A3 and 2B7.
Co-reporter:Adegoke O. Adeniji ; Barry M. Twenter ; Michael C. Byrns ; Yi Jin ; Mo Chen ; Jeffrey D. Winkler ;Trevor M. Penning
Journal of Medicinal Chemistry 2012 Volume 55(Issue 5) pp:2311-2323
Publication Date(Web):January 20, 2012
DOI:10.1021/jm201547v
Aldo–keto reductase 1C3 (AKR1C3; type 5 17β-hydroxysteroid dehydrogenase) is overexpressed in castration resistant prostate cancer (CRPC) and is implicated in the intratumoral biosynthesis of testosterone and 5α-dihydrotestosterone. Selective AKR1C3 inhibitors are required because compounds should not inhibit the highly related AKR1C1 and AKR1C2 isoforms which are involved in the inactivation of 5α-dihydrotestosterone. NSAIDs, N-phenylanthranilates in particular, are potent but nonselective AKR1C3 inhibitors. Using flufenamic acid, 2-{[3-(trifluoromethyl)phenyl]amino}benzoic acid, as lead compound, five classes of structural analogues were synthesized and evaluated for AKR1C3 inhibitory potency and selectivity. Structure–activity relationship (SAR) studies revealed that a meta-carboxylic acid group relative to the amine conferred pronounced AKR1C3 selectivity without loss of potency, while electron withdrawing groups on the phenylamino B-ring were optimal for AKR1C3 inhibition. Lead compounds did not inhibit COX-1 or COX-2 but blocked the AKR1C3 mediated production of testosterone in LNCaP-AKR1C3 cells. These compounds offer promising leads toward new therapeutics for CRPC.
Co-reporter:Meng Huang, Xiaojing Liu, Sankha S. Basu, Li Zhang, Mary E. Kushman, Ronald G. Harvey, Ian A. Blair, and Trevor M. Penning
Chemical Research in Toxicology 2012 Volume 25(Issue 5) pp:993
Publication Date(Web):April 5, 2012
DOI:10.1021/tx200463s
Benzo[a]pyrene-7,8-dione (B[a]P-7,8-dione) is produced in human lung cells by the oxidation of (±)-B[a]P-7,8-trans-dihydrodiol, which is catalyzed by aldo-keto reductases (AKRs). However, information relevant to the cell-based metabolism of B[a]P-7,8-dione is lacking. We studied the metabolic fate of 2 μM 1,3-[3H2]-B[a]P-7,8-dione in human lung adenocarcinoma A549 cells, human bronchoalveolar H358 cells, and immortalized human bronchial epithelial HBEC-KT cells. In these three cell lines, 1,3-[3H2]-B[a]P-7,8-dione was rapidly consumed, and radioactivity was distributed between the organic and aqueous phase of ethyl acetate-extracted media, as well as in the cell lysate pellets. After acidification of the media, several metabolites of 1,3-[3H2]-B[a]P-7,8-dione were detected in the organic phase of the media by high performance liquid chromatography–ultraviolet–radioactivity monitoring (HPLC-UV-RAM). The structures of B[a]P-7,8-dione metabolites varied in the cell lines and were identified as B[a]P-7,8-dione conjugates with glutathione (GSH) and N-acetyl-l-cysteine (NAC), 8-O-monomethylated-catechol, catechol monosulfate, and monoglucuronide, and monohydroxylated-B[a]P-7,8-dione by liquid chromatography–tandem mass spectrometry (LC-MS/MS). We also obtained evidence for the first time for the formation of an adenine adduct of B[a]P-7,8-dione. Among these metabolites, the identity of the GSH-B[a]P-7,8-dione and the NAC-B[a]P-7,8-dione was further validated by comparison to authentic synthesized standards. The pathways of B[a]P-7,8-dione metabolism in the three human lung cell lines are formation of GSH and NAC conjugates, reduction to the catechol followed by phase II conjugation reactions leading to its detoxification, monohydroxylation, as well as formation of the adenine adduct.
Co-reporter:Mo Chen, Adegoke O. Adeniji, Barry M. Twenter, Jeffrey D. Winkler, David W. Christianson, Trevor M. Penning
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 10) pp:3492-3497
Publication Date(Web):15 May 2012
DOI:10.1016/j.bmcl.2012.03.085
Castrate resistant prostate cancer (CRPC) is associated with increased androgen receptor (AR) signaling often brought about by elevated intratumoral androgen biosynthesis and AR amplification. Inhibition of androgen biosynthesis and/or AR antagonism should be efficacious in the treatment of CRPC. AKR1C3 catalyzes the formation of potent AR ligands from inactive precursors and is one of the most upregulated genes in CRPC. AKR1C3 inhibitors should not inhibit the related isoforms, AKR1C1 and AKR1C2 that are involved in 5α-dihydrotestosterone inactivation in the prostate. We have previously developed a series of flufenamic acid analogs as potent and selective AKR1C3 inhibitors [Adeniji, A. O. et al., J. Med. Chem.2012, 55, 2311]. Here we report the X-ray crystal structure of one lead compound 3-((4-(trifluoromethyl)phenyl) amino)benzoic acid (1) in complex with AKR1C3. Compound 1 adopts a similar binding orientation as flufenamic acid, however, its phenylamino ring projects deeper into a subpocket and confers selectivity over the other AKR1C isoforms. We exploited the observation that some flufenamic acid analogs also act as AR antagonists and synthesized a second generation inhibitor, 3-((4-nitronaphthalen-1-yl)amino)benzoic acid (2). Compound 2 retained nanomolar potency and selective inhibition of AKR1C3 but also acted as an AR antagonist. It inhibited 5α-dihydrotestosterone stimulated AR reporter gene activity with an IC50 = 4.7 μM and produced a concentration dependent reduction in androgen receptor levels in prostate cancer cells. The in vitro and cell-based effects of compound 2 make it a promising lead for development of dual acting agent for CRPC. To illuminate the structural basis of AKR1C3 inhibition, we also report the crystal structure of the AKR1C3·NADP+·2 complex, which shows that compound 2 forms a unique double-decker structure with AKR1C3.Superimposed structures of AKR1C3 in complex with two N-aryl(amino)benzoic acid analogs, compound 1 (magenta) and compound 2 (blue).
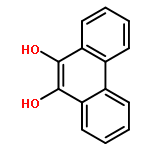
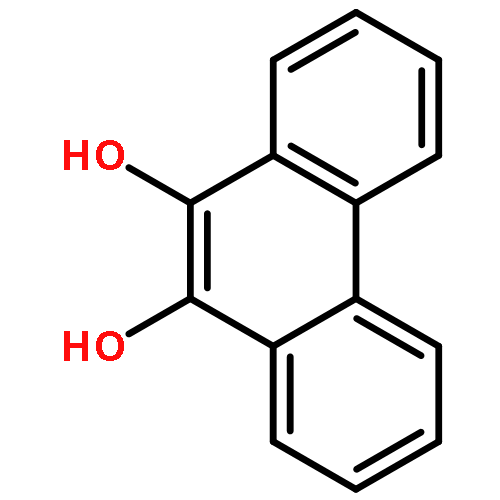
Co-reporter:Carol A. Shultz, Amy M. Quinn, Jong-Heum Park, Ronald G. Harvey, Judy L. Bolton, Edmund Maser, and Trevor M. Penning
Chemical Research in Toxicology 2011 Volume 24(Issue 12) pp:2153
Publication Date(Web):September 12, 2011
DOI:10.1021/tx200294c
Polycyclic aromatic hydrocarbons (PAHs) are suspect human lung carcinogens and can be metabolically activated to remote quinones, for example, benzo[a]pyrene-1,6-dione (B[a]P-1,6-dione) and B[a]P-3,6-dione by the action of either P450 monooxygenase or peroxidases, and to non-K region o-quinones, for example B[a]P-7,8-dione, by the action of aldo keto reductases (AKRs). B[a]P-7,8-dione also structurally resembles 4-hydroxyequilenin o-quinone. These three classes of quinones can redox cycle, generate reactive oxygen species (ROS), and produce the mutagenic lesion 8-oxo-dGuo and may contribute to PAH- and estrogen-induced carcinogenesis. We compared the ability of a complete panel of human recombinant AKRs to catalyze the reduction of PAH o-quinones in the phenanthrene, chrysene, pyrene, and anthracene series. The specific activities for NADPH-dependent quinone reduction were often 100–1000 times greater than the ability of the same AKR isoform to oxidize the cognate PAH-trans-dihydrodiol. However, the AKR with the highest quinone reductase activity for a particular PAH o-quinone was not always identical to the AKR isoform with the highest dihydrodiol dehydrogenase activity for the respective PAH-trans-dihydrodiol. Discrete AKRs also catalyzed the reduction of B[a]P-1,6-dione, B[a]P-3,6-dione, and 4-hydroxyequilenin o-quinone. Concurrent measurements of oxygen consumption, superoxide anion, and hydrogen peroxide formation established that ROS were produced as a result of the redox cycling. When compared with human recombinant NAD(P)H:quinone oxidoreductase (NQO1) and carbonyl reductases (CBR1 and CBR3), NQO1 was a superior catalyst of these reactions followed by AKRs and last CBR1 and CBR3. In A549 cells, two-electron reduction of PAH o-quinones causes intracellular ROS formation. ROS formation was unaffected by the addition of dicumarol, suggesting that NQO1 is not responsible for the two-electron reduction observed and does not offer protection against ROS formation from PAH o-quinones.
Co-reporter:Ding Lu, Ronald G. Harvey, Ian A. Blair, and Trevor M. Penning
Chemical Research in Toxicology 2011 Volume 24(Issue 11) pp:1905
Publication Date(Web):September 30, 2011
DOI:10.1021/tx2002614
Polycyclic aromatic hydrocarbons (PAHs) are ubiquitous environmental pollutants and are carcinogenic in multiple organs and species. Benzo[a]pyrene (B[a]P) is a representative PAH and has been studied extensively for its carcinogenicity and toxicity. B[a]P itself is chemically inert and requires metabolic activation to exhibit its toxicity and carcinogenicity. Three major metabolic pathways have been well documented. The signature metabolites generated from the radical cation (peroxidase or monooxygenase mediated) pathway are B[a]P-1,6-dione and B[a]P-3,6-dione, the signature metabolite generated from the diol-epoxide (P450 mediated) pathway is B[a]P-r-7,t-8,t-9,c-10-tetrahydrotetrol (B[a]P-tetrol-1), and the signature metabolite generated from the o-quinone (aldo-keto reductase mediated) pathway is B[a]P-7,8-dione. The contributions of these different metabolic pathways to cancer initiation and the exploitation of this information for cancer prevention are still under debate. With the availability of a library of [13C4]-labeled B[a]P metabolite internal standards, we developed a sensitive stable isotope dilution atmospheric pressure chemical ionization tandem mass spectrometry method to address this issue by quantitating B[a]P metabolites from each metabolic pathway in human lung cells. This analytical method represents a 500-fold increased sensitivity compared with that of a method using HPLC-radiometric detection. The limit of quantitation (LOQ) was determined to be 6 fmol on column for 3-hydroxybenzo[a]pyrene (3-OH-B[a]P), the generally accepted biomarker for B[a]P exposure. This high level of sensitivity and robustness of the method was demonstrated in a study of B[a]P metabolic profiles in human bronchoalveolar H358 cells induced or uninduced with the AhR ligand, 2,3,7,8-tetrachlorodibenzodioxin (TCDD). All the signature metabolites were detected and successfully quantitated. Our results suggest that all three metabolic pathways contribute equally in the overall metabolism of B[a]P in H358 cells with or without TCDD induction. The sensitivity of the method should permit the identification of cell-type differences in B[a]P activation and detoxication and could also be used for biomonitoring human exposure to PAH.
Co-reporter:Adegoke O. Adeniji, Barry M. Twenter, Michael C. Byrns, Yi Jin, Jeffrey D. Winkler, Trevor M. Penning
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 5) pp:1464-1468
Publication Date(Web):1 March 2011
DOI:10.1016/j.bmcl.2011.01.010
Aldo-keto reductase 1C3 (AKR1C3) also known as type 5 17β-hydroxysteroid dehydrogenase has been implicated as one of the key enzymes driving the elevated intratumoral androgen levels observed in castrate resistant prostate cancer (CRPC). AKR1C3 inhibition therefore presents a rational approach to managing CRPC. Inhibitors should be selective for AKR1C3 over other AKR1C enzymes involved in androgen metabolism. We have synthesized 2-, 3-, and 4-(phenylamino)benzoic acids and identified 3-(phenylamino)benzoic acids that have nanomolar affinity and exhibit over 200-fold selectivity for AKR1C3 versus other AKR1C isoforms. The AKR1C3 inhibitory potency of the 4′-substituted 3-(phenylamino)benzoic acids shows a linear correlation with both electronic effects of substituents and the pKa of the carboxylic acid and secondary amine groups, which are interdependent. These compounds may be useful in treatment and/or prevention of CRPC as well as understanding the role of AKR1C3 in endocrinology.
Co-reporter:Adegoke O. Adeniji, Mo Chen, Trevor M. Penning
The Journal of Steroid Biochemistry and Molecular Biology (September 2013) Volume 137() pp:136-149
Publication Date(Web):1 September 2013
DOI:10.1016/j.jsbmb.2013.05.012
•Superior therapeutic agents to Abiraterone and Enzalutamide are required to treat CRPC.•AKR1C3 synthesizes potent ligands for the androgen receptor locally.•AKR1C3 is upregulated by androgen deprivation in CRPC.•Structurally diverse, potent and selective AKR1C3 inhibitors have been developed.•AKR1C3 inhibitors need to be tested in xenograft models and taken into preclinical development.Aberrant androgen receptor (AR) activation is the major driver of castrate resistant prostate cancer (CRPC). CRPC is ultimately fatal and more therapeutic agents are needed to treat this disease. Compounds that target the androgen axis by inhibiting androgen biosynthesis and or AR signaling are potential candidates for use in CRPC treatment and are currently being pursued aggressively. Aldo-keto reductase 1C3 (AKR1C3) plays a pivotal role in androgen biosynthesis within the prostate. It catalyzes the 17-ketoreduction of weak androgen precursors to give testosterone and 5α-dihydrotestosterone. AKR1C3 expression and activity has been implicated in the development of CRPC, making it a rational target. Selective inhibition of AKR1C3 will be important, however, due to the presence of closely related isoforms, AKR1C1 and AKR1C2 that are also involved in androgen inactivation. We examine the evidence that supports the vital role of AKR1C3 in CRPC and recent developments in the discovery of potent and selective AKR1C3 inhibitors.This article is part of a Special Issue entitled ‘CSR 2013’.Download full-size image
Co-reporter:Daniel Tamae, Michael Byrns, Brett Marck, Elahe A. Mostaghel, Peter S. Nelson, Paul Lange, Daniel Lin, Mary-Ellen Taplin, Steven Balk, William Ellis, Larry True, Robert Vessella, Bruce Montgomery, Ian A. Blair, Trevor M. Penning
The Journal of Steroid Biochemistry and Molecular Biology (November 2013) Volume 138() pp:281-289
Publication Date(Web):1 November 2013
DOI:10.1016/j.jsbmb.2013.06.014
•Androgen deprivation therapy (ADT) is an effective treatment for prostate cancer.•We present a method for quantification of free and conjugated keto-androgens using SID-LC/ESI/SRM/MS.•This method was validated using standards set by the United States Food and Drug Administration (US FDA).•This was applied to patients enrolled in the Targeted Androgen Pathway Suppression (TAPS) clinical trial.•The method can be used to interrogate personalized response and resistance to ADT.Prostate cancer is the most frequently diagnosed form of cancer in males in the United States. The disease is androgen driven and the use of orchiectomy or chemical castration, known as androgen deprivation therapy (ADT) has been employed for the treatment of advanced prostate cancer for over 70 years. Agents such as GnRH agonists and non-steroidal androgen receptor antagonists are routinely used in the clinic, but eventually relapse occurs due to the emergence of castration-resistant prostate cancer. With the appreciation that androgen signaling still persists in these patients and the development of new therapies such as abiraterone and enzalutamide that further suppresses androgen synthesis or signaling, there is a renewed need for sensitive and specific methods to quantify androgen precursor and metabolite levels to assess drug efficacy. We describe the development, validation and application of a stable isotope dilution liquid chromatography electrospray ionization selected reaction monitoring mass spectrometry (SID-LC/ESI/SRM/MS) method for quantification of serum keto-androgens and their sulfate and glucuronide conjugates using Girard-T oxime derivatives. The method is robust down to 0.2–4 pg on column, depending on the androgen metabolite quantified, and can also quantify dehydroepiandrosterone sulfate (DHEA-S) in only 1 μL of serum. The clinical utility of this method was demonstrated by analyzing serum androgens from patients enrolled in a clinical trial assessing combinations of pharmacological agents to maximally suppress gonadal and adrenal androgens (Targeted Androgen Pathway Suppression, TAPS clinical trial). The method was validated by correlating the results obtained with a hydroxylamine derivatization procedure coupled with tandem mass spectrometry using selected reaction monitoring that was conducted in an independent laboratory.Download full-size image
Co-reporter:Mo Chen, Trevor M. Penning
Steroids (May 2014) Volume 83() pp:17-26
Publication Date(Web):1 May 2014
DOI:10.1016/j.steroids.2014.01.013
•5β-Reduced steroids are metabolites of Δ4-3-ketosteroids.•5β-Reduced androstanes, pregnanes, and cholanes perform biological functions.•All mammalian bile-acids are 5β-cholanes.•Aldo–keto reductase (AKR) 1D1 is the only human 5β-reductase.•Structure–function studies on AKR1D1 provide a basis for bile-acid deficiency.5β-Reduced steroids are non-planar steroids that have a 90° bend in their structure to create an A/B cis-ring junction. This novel property is required for bile-acids to act as emulsifiers, but in addition 5β-reduced steroids have remarkable physiology and may act as potent tocolytic agents, endogenous cardiac glycosides, neurosteroids, and can act as ligands for orphan and membrane bound receptors. In humans there is only a single 5β-reductase gene AKR1D1, which encodes Δ4-3-ketosteroid-5β-reductase (AKR1D1). This enzyme is a member of the aldo–keto reductase superfamily, but possesses an altered catalytic tetrad, in which Glu120 replaces the conserved His residue. This predominant liver enzyme generates all 5β-dihydrosteroids in the C19–C27 steroid series. Mutations exist in the AKR1D1 gene, which result in loss of protein stability and are causative in bile-acid deficiency.Download full-size image
Co-reporter:Michael C. Byrns, Rebekka Mindnich, Ling Duan, Trevor M. Penning
The Journal of Steroid Biochemistry and Molecular Biology (May 2012) Volume 130(Issues 1–2) pp:7-15
Publication Date(Web):1 May 2012
DOI:10.1016/j.jsbmb.2011.12.012
Type 5 17β-hydroxysteroid dehydrogenase (AKR1C3) is the major enzyme in the prostate that reduces 4-androstene-3,17-dione (Δ4-Adione) to the androgen receptor (AR) ligand testosterone. AKR1C3 is upregulated in prostate cancer (PCa) and castrate resistant prostate cancer (CRPC) that develops after androgen deprivation therapy. PCa and CRPC often depend on intratumoral androgen biosynthesis and upregulation of AKR1C3 could contribute to intracellular synthesis of AR ligands and stimulation of proliferation through AR signaling. To test this hypothesis, we developed an LNCaP prostate cancer cell line overexpressing AKR1C3 (LNCaP-AKR1C3) and compared its metabolic and proliferative responses to Δ4-Adione treatment with that of the parental, AKR1C3 negative LNCaP cells. In LNCaP and LNCaP-AKR1C3 cells, metabolism proceeded via 5α-reduction to form 5α-androstane-3,17-dione and then (epi)androsterone-3-glucuronide. LNCaP-AKR1C3 cells made significantly higher amounts of testosterone-17β-glucuronide. When 5α-reductase was inhibited by finasteride, the production of testosterone-17β-glucuronide was further elevated in LNCaP-AKR1C3 cells. When AKR1C3 activity was inhibited with indomethacin the production of testosterone-17β-glucuronide was significantly decreased. Δ4-Adione treatment stimulated cell proliferation in both cell lines. Finasteride inhibited LNCaP cell proliferation, consistent with 5α-androstane-3,17-dione acting as the major metabolite that stimulates growth by binding to the mutated AR. However, LNCaP-AKR1C3 cells were resistant to the growth inhibitory properties of finasteride, consistent with the diversion of Δ4-Adione metabolism from 5α-reduced androgens to increased formation of testosterone. Indomethacin did not result in differences in Δ4-Adione induced proliferation since this treatment led to the same metabolic profile in LNCaP and LNCaP-AKR1C3 cells. We conclude that AKR1C3 overexpression diverts androgen metabolism to testosterone that results in proliferation in androgen sensitive prostate cancer. This effect is seen despite high levels of uridine glucuronosyl transferases suggesting that AKR1C3 activity can surmount the effects of this elimination pathway. Treatment options in prostate cancer that target 5α-reductase where AKR1C3 co-exists may be less effective due to the diversion of Δ4-Adione to testosterone.Graphical abstractDownload full-size imageHighlights► AKR1C3 is one of the most overexpressed genes in castrate resistant prostate cancer. ► AKR1C3 was stably transfected into LNCaP cells which are AKR1C3 null and its effect on Δ4-Adione metabolism and Δ4-Adione mediated cell growth was studied. ► LNCaP-AKR1C3 cells showed significant conversion of Δ4-Adione to testosterone-17β-glucuronide that was inhibited by indomethacin. ► Proliferation of LNCaP-AKR1C3 cells stimulated by Δ4-Adione was resistant to finasteride inhibition consistent with a re-direction of metabolism to testosterone. ► When AKR1C3 and 5α-reductase co-exist in prostate cancer the use of 5α-reductase inhibitors may be deleterious due to the unintended re-diversion of Δ4-Adione to testosterone.
Co-reporter:Michael C. Byrns, Yi Jin, Trevor M. Penning
The Journal of Steroid Biochemistry and Molecular Biology (May 2011) Volume 125(Issues 1–2) pp:95-104
Publication Date(Web):1 May 2011
DOI:10.1016/j.jsbmb.2010.11.004
There is considerable interest in the development of an inhibitor of aldo–keto reductase (AKR) 1C3 (type 5 17β-hydroxysteroid dehydrogenase and prostaglandin F synthase) as a potential therapeutic for both hormone-dependent and hormone-independent cancers. AKR1C3 catalyzes the reduction of 4-androstene-3,17-dione to testosterone and estrone to 17β-estradiol in target tissues, which will promote the proliferation of hormone dependent prostate and breast cancers, respectively. AKR1C3 also catalyzes the reduction of prostaglandin (PG) H2 to PGF2α and PGD2 to 9α,11β-PGF2, which will limit the formation of anti-proliferative prostaglandins, including 15-deoxy-Δ12,14-PGJ2, and contribute to proliferative signaling. AKR1C3 is overexpressed in a wide variety of cancers, including breast and prostate cancer. An inhibitor of AKR1C3 should not inhibit the closely related isoforms AKR1C1 and AKR1C2, as they are involved in other key steroid hormone biotransformations in target tissues. Several structural leads have been explored as inhibitors of AKR1C3, including non-steroidal anti-inflammatory drugs, steroid hormone analogues, flavonoids, cyclopentanes, and benzodiazepines. Inspection of the available crystal structures of AKR1C3 with multiple ligands bound, along with the crystal structures of the other AKR1C isoforms, provides a structural basis for the rational design of isoform specific inhibitors of AKR1C3. We find that there are subpockets involved in ligand binding that are considerably different in AKR1C3 relative to the closely related AKR1C1 or AKR1C2 isoforms. These pockets can be used to further improve the binding affinity and selectivity of the currently available AKR1C3 inhibitors.Article from the special issue on Targeted Inhibitors.Research highlights► AKR1C3 produces steroids and prostaglandins for receptor mediated growth. ► AKR1C3 inhibition is desirable for hormone dependent and independent cancers. ► NSAIDs, steroids, flavonoids, cyclopentanes, and benzodiazepines inhibit AKR1C3. ► Crystal structures identify subpockets for rational design of AKR1C3 inhibitors. ► Differences in AKR1C subpockets can be exploited for selective inhibition.
Co-reporter:Trevor M. Penning
The Journal of Steroid Biochemistry and Molecular Biology (May 2011) Volume 125(Issues 1–2) pp:46-56
Publication Date(Web):1 May 2011
DOI:10.1016/j.jsbmb.2011.01.009
Hydroxysteroid dehydrogenases (HSDs) represent a major class of NAD(P)(H) dependent steroid hormone oxidoreductases involved in the pre-receptor regulation of hormone action. This is achieved by HSDs working in pairs so that they can interconvert ketosteroids with hydroxysteroids resulting in a change in ligand potency for nuclear receptors. HSDs belong to two protein superfamilies the aldo–keto reductases and the short-chain dehydrogenase/reductases. In humans, many of the important enzymes have been thoroughly characterized including the elucidation of their three-dimensional structures. Because these enzymes play fundamental roles in steroid hormone action they can be considered to be drug targets for a variety of steroid driven diseases, e.g. metabolic syndrome and obesity, inflammation, and hormone dependent malignancies of the endometrium, prostate and breast. This article will review how fundamental knowledge of these enzymes can be exploited in the development of isoform specific HSD inhibitors from both protein superfamilies.Article from the Special issue on Targeted Inhibitors.Research highlights► Hydroxysteroid dehydrogenases (HSDs) belong to two protein superfamilies (AKRs and SDRs). ► Pairs of HSDs regulate ligand access to nuclear receptors. ► Knowledge of HSD structure, function, kinetic and catalytic mechanism can aid inhibitor design. ► Type 1 11β-HSD inhibitors can target metabolic syndrome and diabetes. ► 17β-HSD inhibitors can target hormonal dependent malignancies.
Co-reporter:Trevor M. Penning
The Journal of Steroid Biochemistry and Molecular Biology (July 2016) Volume 161() pp:5-12
Publication Date(Web):1 July 2016
DOI:10.1016/j.jsbmb.2015.10.016
•Stopped-flow techniques dissect reactions catalyzed by steroid transforming enzymes.•Ligand binding reactions can identify isomerization steps and new enzyme forms.•Single turnover experiments isolate the chemistry step.•Multiple turnover experiments identify burst or linear phase kinetics.•The technique determines the effects of mutations with precision.Structure-function studies on steroid transforming enzymes often use site-directed mutagenesis to inform mechanisms of catalysis and effects on steroid binding, and data are reported in terms of changes in steady state kinetic parameters kcat, Km and kcat/Km. However, this dissection of function is limited since kcat is governed by the rate-determining step and Km is a complex macroscopic kinetic constant. Often site-directed mutagenesis can lead to a change in the rate-determining step which cannot be revealed by just reporting a decrease in kcat alone. These issues are made more complex when it is considered that many steroid transforming enzymes have more than one substrate and product. We present the case for using transient-kinetics performed with stopped-flow spectrometry to assign rate constants to discrete steps in these multi-substrate reactions and their use to interpret enzyme mechanism and the effects of disease and engineered mutations. We demonstrate that fluorescence kinetic transients can be used to measure ligand binding that may be accompanied by isomerization steps, revealing the existence of new enzyme intermediates. We also demonstrate that single-turnover reactions can provide a klim for the chemical step and Ks for steroid-substrate binding and that when coupled with kinetic isotope effect measurements can provide information on transition state intermediates. We also demonstrate how multiple turnover experiments can provide evidence for either “burst-phase” kinetics, which can reveal a slow product release step, or linear-phase kinetics, in which the chemical step can be rate-determining. With these assignments it becomes more straightforward to analyze the effects of mutations. We use examples from the hydroxysteroid dehydrogenases (AKR1Cs) and human steroid 5β-reductase (AKR1D1) to illustrate the utility of the approach, which are members of the aldo-keto reductase (AKR) superfamily.Download full-size image
Co-reporter:Tianzhu Zang, Daniel Tamae, Clementina Mesaros, Qingqing Wang, Meng Huang, Ian A. Blair, Trevor M. Penning
The Journal of Steroid Biochemistry and Molecular Biology (January 2017) Volume 165(Part B) pp:342-355
Publication Date(Web):1 January 2017
DOI:10.1016/j.jsbmb.2016.08.001
•Stereoselective enzymatic synthesis of stable isotope labeled androstanediols.•Simultaneous quantitation of nine hydroxy-androgens in human serum by LC–MS/MS.•Quantitation of 5-androstenediol and its conjugates in human serum using LC–MS/MS.•Quantitation of 3α- and 3β-androstanediol and their conjugates in human serum using LC–MS/MS.Castration resistant prostate cancer (CRPC), the fatal form of prostate cancer, remains androgen dependent despite castrate levels of circulating testosterone (T) and 5α-dihydrotestosterone (DHT). To investigate mechanisms by which the tumor can synthesize its own androgens and develop resistance to abiraterone acetate and enzalutamide, methods to measure a complete androgen profile are imperative. Here, we report the development and validation of a stable isotope dilution liquid chromatography electrospray ionization tandem mass spectrometric (SID-LC-ESI–MS/MS) method to quantify nine human hydroxy-androgens as picolinates, simultaneously with requisite specificity and sensitivity. In the established method, the fragmentation patterns of all nine hydroxy-androgen picolinates were identified, and [13C3]-5α-androstane-3α, 17β-diol and [13C3]-5α-androstane-3β, 17β-diol used as internal standards were synthesized enzymatically. Intra-day and inter-day precision and accuracy corresponds to the U.S. Food and Drug Administration Criteria for Bioanalytical Method Validation. The lower limit of quantitation (LLOQ) of nine hydroxy-androgens is 1.0 pg to 2.5 pg on column. Diols which have been infrequently measured: 5-androstene-3β, 17β-diol and 5α-androstane-3α, 17β-diol can be determined in serum at values as low as 1.0 pg on column. The method also permits the quantitation of conjugated hydroxy-androgens following enzymatic digestion. While direct detection of steroid conjugates by electrospray-ionization tandem mass spectrometry has advantages the detection of unconjugated and conjugated steroids would require separate methods for each set of analytes. Our method was applied to pooled serum from male and female donors to provide reference values for both unconjugated and conjugated hydroxy-androgens. This method will allow us to interrogate the involvement of the conversion of 5-androstene-3β, 17β-diol to T, the backdoor pathway involving the conversion of 5α-androstane-3α, 17β-diol to DHT and the inactivation of DHT to 5α-androstane-3β, 17β-diol in advanced prostate cancer.Download high-res image (157KB)Download full-size image
Co-reporter:Tea Lanišnik Rižner, Trevor M. Penning
Steroids (January 2014) Volume 79() pp:49-63
Publication Date(Web):1 January 2014
DOI:10.1016/j.steroids.2013.10.012
•The aldo–keto reductase superfamily contains five human steroid-metabolizing enzymes.•AKR1C isoforms act as 3-, 17- and 20-ketosteroid reductases.•AKR1D1 is the sole steroid 5β-reductase in humans.•AKR enzymes control ligand access to nuclear and membrane bound receptors.•Expression profiles, inherited mutations and SNP support their roles in human disease.Human aldo–keto reductases AKR1C1–AKR1C4 and AKR1D1 play essential roles in the metabolism of all steroid hormones, the biosynthesis of neurosteroids and bile acids, the metabolism of conjugated steroids, and synthetic therapeutic steroids. These enzymes catalyze NADPH dependent reductions at the C3, C5, C17 and C20 positions on the steroid nucleus and side-chain. AKR1C1–AKR1C4 act as 3-keto, 17-keto and 20-ketosteroid reductases to varying extents, while AKR1D1 acts as the sole Δ4-3-ketosteroid-5β-reductase (steroid 5β-reductase) in humans. AKR1 enzymes control the concentrations of active ligands for nuclear receptors and control their ligand occupancy and trans-activation, they also regulate the amount of neurosteroids that can modulate the activity of GABAA and NMDA receptors. As such they are involved in the pre-receptor regulation of nuclear and membrane bound receptors. Altered expression of individual AKR1C genes is related to development of prostate, breast, and endometrial cancer. Mutations in AKR1C1 and AKR1C4 are responsible for sexual development dysgenesis and mutations in AKR1D1 are causative in bile-acid deficiency.Download full-size image
Co-reporter:Trevor M. Penning, Seon-Hwa Lee, Yi Jin, Alejandro Gutierrez, Ian A. Blair
The Journal of Steroid Biochemistry and Molecular Biology (August 2010) Volume 121(Issues 3–5) pp:546-555
Publication Date(Web):1 August 2010
DOI:10.1016/j.jsbmb.2010.01.005
Advances in liquid chromatography–mass spectrometry (LC–MS) can be used to measure steroid hormone metabolites in vitro and in vivo. We find that LC–electrospray ionization (ESI)-MS using a LCQ ion trap mass spectrometer in the negative ion mode can be used to monitor the product profile that results from 5α-dihydrotestosterone (DHT)-17β-glucuronide, DHT-17β-sulfate, and tibolone-17β-sulfate reduction catalyzed by human members of the aldo–keto reductase (AKR) 1C subfamily and assign kinetic constants to these reactions. We also developed a stable isotope dilution LC–electron capture atmospheric pressure chemical ionization (ECAPCI)-MS method for the quantitative analysis of estrone (E1) and its metabolites as pentafluorobenzyl (PFB) derivatives in human plasma in the attomole range. The limit of detection for E1-PFB was 740 attomole on column. Separations can be performed using normal-phase LC because ionization takes place in the gas phase rather than in solution. This permits efficient separation of the regioisomeric 2- and 4-methoxy-E1. The method was validated for the simultaneous analysis of plasma E2 and its metabolites: 2-methoxy-E2, 4-methoxy-E2, 16α-hydroxy-E2, estrone (E1), 2-methoxy-E1, 4-methoxy-EI, and 16α-hydroxy-E1 from 5 pg/mL to 2000 pg/mL. Our LC–MS methods have sufficient sensitivity to detect steroid hormone levels in prostate and breast tumors and should aid their molecular diagnosis and treatment.
Co-reporter:Trevor M. Penning, Seon-Hwa Lee, Yi Jin, Alejandro Gutierrez, Ian A. Blair
The Journal of Steroid Biochemistry and Molecular Biology (August 2010) Volume 121(Issues 3–5) pp:546-555
Publication Date(Web):1 August 2010
DOI:10.1016/j.jsbmb.2010.01.005
Advances in liquid chromatography–mass spectrometry (LC–MS) can be used to measure steroid hormone metabolites in vitro and in vivo. We find that LC–electrospray ionization (ESI)-MS using a LCQ ion trap mass spectrometer in the negative ion mode can be used to monitor the product profile that results from 5α-dihydrotestosterone (DHT)-17β-glucuronide, DHT-17β-sulfate, and tibolone-17β-sulfate reduction catalyzed by human members of the aldo–keto reductase (AKR) 1C subfamily and assign kinetic constants to these reactions. We also developed a stable isotope dilution LC–electron capture atmospheric pressure chemical ionization (ECAPCI)-MS method for the quantitative analysis of estrone (E1) and its metabolites as pentafluorobenzyl (PFB) derivatives in human plasma in the attomole range. The limit of detection for E1-PFB was 740 attomole on column. Separations can be performed using normal-phase LC because ionization takes place in the gas phase rather than in solution. This permits efficient separation of the regioisomeric 2- and 4-methoxy-E1. The method was validated for the simultaneous analysis of plasma E2 and its metabolites: 2-methoxy-E2, 4-methoxy-E2, 16α-hydroxy-E2, estrone (E1), 2-methoxy-E1, 4-methoxy-EI, and 16α-hydroxy-E1 from 5 pg/mL to 2000 pg/mL. Our LC–MS methods have sufficient sensitivity to detect steroid hormone levels in prostate and breast tumors and should aid their molecular diagnosis and treatment.
Co-reporter:Mo Chen, Jason E. Drury, Trevor M. Penning
Steroids (April 2011) Volume 76(Issue 5) pp:484-490
Publication Date(Web):1 April 2011
DOI:10.1016/j.steroids.2011.01.003
Human steroid 5β-reductase (aldo-keto reductase 1D1) catalyzes the stereospecific NADPH-dependent reduction of the C4–C5 double bond of Δ4-ketosteroids to yield an A/B cis-ring junction. This cis-configuration is crucial for bile acid biosynthesis and plays important roles in steroid metabolism. The biochemical properties of the enzyme have not been thoroughly studied and conflicting data have been reported, partially due to the lack of highly homogeneous protein. In the present study, we systematically determined the substrate specificity of homogeneous human recombinant AKR1D1 using C18, C19, C21, and C27 Δ4-ketosteroids and assessed the pH-rate dependence of the enzyme. Our results show that AKR1D1 proficiently reduced all the steroids tested at physiological pH, indicating AKR1D1 is the only enzyme necessary for all the 5β-steroid metabolites present in humans. Substrate inhibition was observed with C18 to C21 steroids provided that the C11 position was unsubstituted. This structure activity relationship can be explained by the existence of a small alternative substrate binding pocket revealed by the AKR1D1 crystal structure. Non-steroidal anti-inflammatory drugs which are potent inhibitors of the related AKR1C enzymes do not inhibit AKR1D1. By contrast chenodeoxycholate and ursodeoxycholate were found to be potent non-competitive inhibitors suggesting that bile-acids may regulate their own synthesis at the level of AKR1D1 inhibition.Research highlights► AKR1D1 proficiently catalyzes the 5β-reduction of a series of Δ4-3-ketosteroids. ► Bile-acids are potent non-competitive inhibitors of AKR1D1 and may regulate their own synthesis via this mechanism. ► AKR1D1 is the only enzyme required to account for the spectrum of 5β-reduced steroids in humans.
Co-reporter:Rebekka Mindnich, Jason E. Drury, Trevor M. Penning
Chemico-Biological Interactions (30 May 2011) Volume 191(Issues 1–3) pp:250-254
Publication Date(Web):30 May 2011
DOI:10.1016/j.cbi.2010.12.020
The stereospecific 5β-reduction of Δ4-3-ketosterols is very difficult to achieve chemically and introduces a 90° bend between ring A and B of the planar steroid. In mammals, the reaction is catalyzed by steroid 5β-reductase, a member of the aldo-keto reductase (AKR) family. The human enzyme, AKR1D1, plays an essential role in bile-acid biosynthesis since the 5β-configuration is required for the emulsifying properties of bile. Deficient 5β-reductase activity can lead to cholestasis and neo-natal liver failure and is often lethal if it remains untreated. In five patients with 5β-reductase deficiency, sequencing revealed individual, non-synonymous point mutations in the AKR1D1 gene: L106F, P133R, G223E, P198L and R261C. However, mapping these mutations to the AKR1D1 crystal structure failed to reveal any obvious involvement in substrate or cofactor binding or catalytic mechanism, and it remained unclear whether these mutations could be causal for the observed disease. We analyzed the positions of the reported mutations and found that they reside in highly conserved portions of AKR1D1 and hypothesized that they would likely lead to changes in protein folding, and hence enzyme activity. Attempts to purify the mutant enzymes for further characterization by over-expression in Escherichia coli yielded sufficient amounts of only one mutant (P133R). This enzyme exhibited reduced Km and kcat values with the bile acid intermediate Δ4-cholesten-7α-ol-3-one as substrate reminiscent of uncompetitive inhibition. In addition, P133R displayed no change in cofactor affinity but was more thermolabile as judged by CD-spectroscopy. When all AKR1D1 mutants were expressed in HEK 293 cells, protein expression levels and enzyme activity were dramatically reduced. Furthermore, cycloheximide treatment revealed decreased stability of several of the mutants compared to wild type. Our data show, that all five mutations identified in patients with functional bile acid deficiency strongly affected AKR1D1 enzyme functionality and therefore may be causal for this disease.
Co-reporter:Oleg A. Barski, Rebekka Mindnich, Trevor M. Penning
Chemico-Biological Interactions (25 February 2013) Volume 202(Issues 1–3) pp:153-158
Publication Date(Web):25 February 2013
DOI:10.1016/j.cbi.2012.12.012
The aldo–keto reductase superfamily contains 173 proteins which are present in all phyla. Examination of the human and mouse genomes has identified that in some instances a single AKR gene can give rise to alternatively spliced mRNA variants which in some cases can give rise to more than one protein isoform. This is currently well documented in the AKR6A subfamily which contains the β-subunits of the voltage-gated potassium ion channels. With the emergence of second generation sequencing it is likely that the occurrence of transcript variants and protein isoforms from a single AKR gene may become common place. To deal with this issue we recommend that the Ensembl data-base nomenclature be used to annotate the transcript variants from a single AKR gene. However, since multiple transcript variants could give rise to either the same or multiple protein isoforms from the same AKR gene we also propose to expand the nomenclature of the AKR protein superfamily, so that when a protein isoform is shown to be expressed and is functional it would be assigned the standard AKR name followed by a “period or full-stop” and a number for that unique isoform. Numbers will be assigned chronologically and linked to the respective transcripts annotated in Ensembl e.g. AKR6A5.1 (Kvβ2.1) (AKR6A5-001, -006 and -201), followed by AKR6A5.2 (Kvβ2.2) (AKR6A5-002,-202). This nomenclature is expandable and it enables multiple protein isoforms to be assigned to their respective transcripts when they arise from the same AKR gene or for a single protein isoform to be assigned to multiple transcripts when the transcripts encode the same AKR protein.
Co-reporter:Michael C. Byrns, Ling Duan, Seon Hwa Lee, Ian A. Blair, Trevor M. Penning
The Journal of Steroid Biochemistry and Molecular Biology (15 February 2010) Volume 118(Issue 3) pp:177-187
Publication Date(Web):15 February 2010
DOI:10.1016/j.jsbmb.2009.12.009
Aldo-keto reductase (AKR) 1C3 (type 5 17β-hydroxysteroid dehydrogenase and prostaglandin F synthase), may stimulate proliferation via steroid hormone and prostaglandin (PG) metabolism in the breast. Purified recombinant AKR1C3 reduces PGD2 to 9α,11β-PGF2, Δ4-androstenedione to testosterone, progesterone to 20α-hydroxyprogesterone, and to a lesser extent, estrone to 17β-estradiol. We established MCF-7 cells that stably express AKR1C3 (MCF-7-AKR1C3 cells) to model its over-expression in breast cancer. AKR1C3 expression increased steroid conversion by MCF-7 cells, leading to a pro-estrogenic state. Unexpectedly, estrone was reduced fastest by MCF-7-AKR1C3 cells when compared to other substrates at 0.1 μM. MCF-7-AKR1C3 cells proliferated three times faster than parental cells in response to estrone and 17β-estradiol. AKR1C3 therefore represents a potential target for attenuating estrogen receptor α induced proliferation. MCF-7-AKR1C3 cells also reduced PGD2, limiting its dehydration to form PGJ2 products. The AKR1C3 product was confirmed as 9α,11β-PGF2 and quantified with a stereospecific stable isotope dilution liquid chromatography–mass spectrometry method. This method will allow the examination of the role of AKR1C3 in endogenous prostaglandin formation in response to inflammatory stimuli. Expression of AKR1C3 reduced the anti-proliferative effects of PGD2 on MCF-7 cells, suggesting that AKR1C3 limits peroxisome proliferator activated receptor γ (PPARγ) signaling by reducing formation of 15-deoxy-Δ12,14-PGJ2 (15dPGJ2).

































![2-Propenoic acid, 3-[4-(acetyloxy)-3-(3-methyl-2-butenyl)phenyl]-, (E)-](http://img.cochemist.com/ccimg/107400/107390-20-5.png)
![2-Propenoic acid, 3-[4-(acetyloxy)-3-(3-methyl-2-butenyl)phenyl]-, (E)-](http://img.cochemist.com/ccimg/107400/107390-20-5_b.png)








![(2E)-3-[4-hydroxy-3-(3-methylbut-2-en-1-yl)phenyl]prop-2-enoic acid](http://img.cochemist.com/ccimg/53800/53755-58-1.png)
![(2E)-3-[4-hydroxy-3-(3-methylbut-2-en-1-yl)phenyl]prop-2-enoic acid](http://img.cochemist.com/ccimg/53800/53755-58-1_b.png)














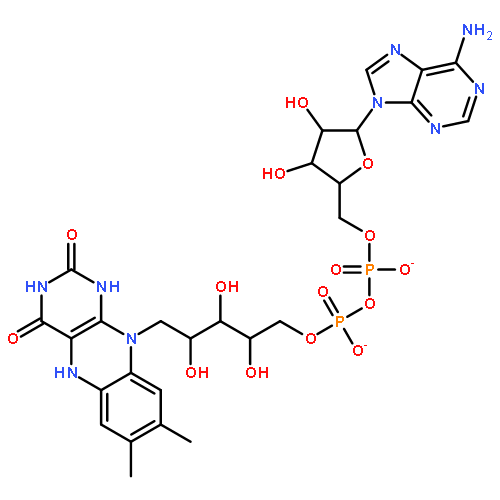
![5'-O-[hydroxy(sulfooxy)phosphoryl]adenosine 3'-(dihydrogen phosphate)](http://img.cochemist.com/ccimg/500/482-67-7.png)
![5'-O-[hydroxy(sulfooxy)phosphoryl]adenosine 3'-(dihydrogen phosphate)](http://img.cochemist.com/ccimg/500/482-67-7_b.png)