Co-reporter:Rie Ueda, Takayoshi Suzuki, Koshiki Mino, Hiroki Tsumoto, Hidehiko Nakagawa, Makoto Hasegawa, Ryuzo Sasaki, Tamio Mizukami and Naoki Miyata
Journal of the American Chemical Society December 9, 2009 Volume 131(Issue 48) pp:17536-17537
Publication Date(Web):November 16, 2009
DOI:10.1021/ja907055q
Lysine specific demethylase 1 (LSD1) plays a key role in the regulation of gene expression by removing the methyl groups from methylated Lys4 of histone H3 (H3K4). Here we report the identification of the first small-molecule LSD1-selective inhibitors. These inhibitors show in vivo H3K4-methylating activity and antiproliferative activity and should be useful as lead structures for anticancer drugs and as tools for studying the biological roles of LSD1.
Co-reporter:Prima R. Tatum, Hideyuki Sawada, Yosuke Ota, Yukihiro Itoh, Peng Zhan, Naoya Ieda, Hidehiko Nakagawa, Naoki Miyata, Takayoshi Suzuki
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 8) pp:1871-1874
Publication Date(Web):15 April 2014
DOI:10.1016/j.bmcl.2014.03.026
A series of 114 SIRT inhibitor candidates was assembled using ‘click chemistry’, by reacting two alkynes bearing 2-anilinobenzamide pharmacophore with 57 azide building blocks in the presence of Cu(I) catalyst. Screening identified two SIRT2-selective inhibitors, which were more SIRT2-selective than AGK2, a known SIRT2 inhibitor. These findings will be useful for further development of SIRT2-selective inhibitors.To find novel SIRT2-selective inhibitors, we designed and prepared a library of SIRT inhibitor candidates derived from CuAAC reaction between propargyl 2-anilinobenzamides A and a series of azides B. Screening identified SIRT2-selective inhibitors A1B11 and A2B57, which were more SIRT2-selective than AGK2 (1), a known SIRT2 inhibitor. These findings will be useful in the further development of SIRT2-selective inhibitors.
Co-reporter: Takayoshi Suzuki;Nobusuke Muto;Dr. Masashige Bo;Dr. Yukihiro Itoh;Ayako Masaki;Dr. Masaki Ri;Yosuke Ota; Hidehiko Nakagawa; Shinsuke Iida; Katsuhiko Shirahige; Naoki Miyata
ChemMedChem 2014 Volume 9( Issue 3) pp:657-664
Publication Date(Web):
DOI:10.1002/cmdc.201300414
Abstract
We recently discovered N-hydroxy-3-[1-(phenylthio)methyl-1H-1,2,3-triazol-4-yl]benzamide (NCC149) as a potent and selective histone deacetylase 8 (HDAC8) inhibitor from a 151-member triazole compound library using a click chemistry approach. In this work, we present a series of NCC149 derivatives bearing various aromatic linkers that were designed and synthesized as HDAC8-selective inhibitors. A series of in vitro assays were used to evaluate the newly synthesized compounds, four of which showed HDAC8 inhibitory activity similar to that of NCC149, and one of which displayed HDAC8 selectivity superior to that of NCC149. In addition, these top four compounds induced the increase of acetylated cohesin (an HDAC8 substrate) in HeLa cells in a dose-dependent manner, indicating inhibition of HDAC8 in the cells. While none of these compounds enhanced the acetylation of H3K9 (a substrate of HDAC1 and 2), only one compound refrained from increasing α-tubulin acetylation, a substrate of HDAC6, indicating that this compound is more selective for HDAC8 than the other derivatives. Furthermore, this HDAC8-selective inhibitor suppressed the growth of T-cell lymphoma cells more potently than did NCC149. These findings are useful for the further development of HDAC8-selective inhibitors.
Co-reporter:Takayoshi Suzuki ; Hiroki Ozasa ; Yukihiro Itoh ; Peng Zhan ; Hideyuki Sawada ; Koshiki Mino ; Louise Walport ; Rei Ohkubo ; Akane Kawamura ; Masato Yonezawa ; Yuichi Tsukada ▽; Anthony Tumber ; Hidehiko Nakagawa ; Makoto Hasegawa ; Ryuzo Sasaki ; Tamio Mizukami ; Christopher J. Schofield
Journal of Medicinal Chemistry 2013 Volume 56(Issue 18) pp:7222-7231
Publication Date(Web):August 21, 2013
DOI:10.1021/jm400624b
Histone Nε-methyl lysine demethylases KDM2/7 have been identified as potential targets for cancer therapies. On the basis of the crystal structure of KDM7B, we designed and prepared a series of hydroxamate analogues bearing an alkyl chain. Enzyme assays revealed that compound 9 potently inhibits KDM2A, KDM7A, and KDM7B, with IC50s of 6.8, 0.2, and 1.2 μM, respectively. While inhibitors of KDM4s did not show any effect on cancer cells tested, the KDM2/7-subfamily inhibitor 9 exerted antiproliferative activity, indicating the potential for KDM2/7 inhibitors as anticancer agents.
Co-reporter:Yukihiro Itoh, Takayoshi Suzuki and Naoki Miyata
Molecular BioSystems 2013 vol. 9(Issue 5) pp:873-896
Publication Date(Web):28 Feb 2013
DOI:10.1039/C3MB25410K
DNA methylation and posttranslational histone modifications regulate expression of various genes independently of changes in the DNA sequence. Such epigenetic mechanisms play important roles in controlling cellular functions, including the cell cycle, immunoresponses and signal transduction. On the other hand, epigenetic aberrations are associated with oncogenesis and proliferation of cancer cells, and epigenetic alterations have been identified in many human cancer cells. Furthermore, chemical–biological approaches have uncovered relationships between epigenetic dysregulation and cancer, and several small molecules that modulate epigenetic mechanisms have already been approved for cancer therapy. In this review, we deal with chemical epigenetics relevant to cancer therapy, focusing especially on small molecules that regulate epigenetic mechanisms related to DNA methylation and histone modification.
Co-reporter:Kazuyuki Aizawa, Hidehiko Nakagawa, Kazuya Matsuo, Kodai Kawai, Naoya Ieda, Takayoshi Suzuki, Naoki Miyata
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 8) pp:2340-2343
Publication Date(Web):15 April 2013
DOI:10.1016/j.bmcl.2013.02.062
Recent studies have shown that nitroxyl (HNO) (1HNO/3NO−), which is the one-electron-reduced form of nitric oxide (NO), has unique biological activities, especially in the cardiovascular system, and HNO-releasing agents may have therapeutic potential. Since few HNO donors are available for use under physiological conditions, we synthesized and evaluated a series of Piloty’s acid (PA) derivatives and evaluated their HNO-releasing activity under physiological conditions. N-Hydroxy-2-nitrobenzenesulfonamide (17) was the most efficient HNO donor among our synthesized PA derivatives, including the lead compound, 2-bromo-N-hydroxybenzenesulfonamide (2). The high HNO-releasing activity is suggested to be due to electronic and steric effects. Compound 17 may be a useful tool for biological experiments.
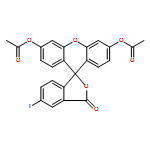
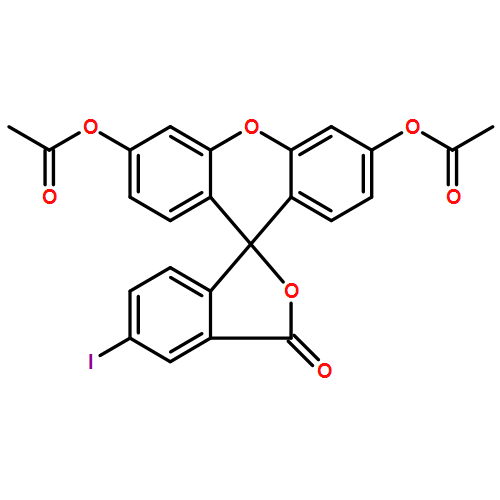
Co-reporter:Daisuke Ogasawara;Dr. Yukihiro Itoh;Dr. Hiroki Tsumoto;Dr. Taeko Kakizawa;Dr. Koshiki Mino;Dr. Kiyoshi Fukuhara; Hidehiko Nakagawa;Dr. Makoto Hasegawa;Dr. Ryuzo Sasaki; Tamio Mizukami; Naoki Miyata; Takayoshi Suzuki
Angewandte Chemie International Edition 2013 Volume 52( Issue 33) pp:8620-8624
Publication Date(Web):
DOI:10.1002/anie.201303999
Co-reporter:Naoya Ieda ; Hidehiko Nakagawa ; Tao Peng ; Dan Yang ; Takayoshi Suzuki
Journal of the American Chemical Society 2012 Volume 134(Issue 5) pp:2563-2568
Publication Date(Web):January 5, 2012
DOI:10.1021/ja206744z
We designed and synthesized a photocontrollable peroxynitrite (ONOO–) generator, P-NAP, which has N-methyl-N-nitrosoaminophenol structure with four methyl groups introduced onto the benzene ring to block reaction of the photodecomposition product with ONOO– and to lower the semiquinoneimine’s redox potential. The semiquinoneimine intermediate generated by photoinduced release of nitric oxide (NO) reduces dissolved molecular oxygen to generate superoxide radical anion (O2•–), which reacts with NO to afford ONOO– under diffusion control (k = 6.7 × 109 M–1 s–1). NO release from P-NAP under UV-A (330–380 nm) irradiation was confirmed by ESR spin trapping. Tyrosine nitration, characteristic of ONOO–, was demonstrated by HPLC analysis of a photoirradiated aqueous solution of P-NAP and N-acetyl-l-tyrosine ethyl ester. ONOO– formation was confirmed with a ONOO–-specific fluorogenic probe, HKGreen-3, and compared with that from 3-(4-morpholinyl)sydnonimine hydrochloride (SIN-1), which is the most widely used ONOO– generator at present. The photoreaction of P-NAP was influenced by superoxide dismutase, indicating that generation of O2•– occurs before ONOO– formation. The quantum yield for formation of duroquinone, the main P-NAP photodecomposition product, was measured as 0.86 ± 0.07 at 334 nm with a potassium ferrioxalate actinometer. Generation of ONOO– from P-NAP in HCT-116 cells upon photoirradiation was successfully imaged with HKGreen-3A. This is the first example of a photocontrollable ONOO– donor applicable to cultured cells.
Co-reporter:Takayoshi Suzuki ; Mohammed Naseer Ahmed Khan ; Hideyuki Sawada ; Erika Imai ; Yukihiro Itoh ; Katsura Yamatsuta ; Natsuko Tokuda ; Jun Takeuchi ; Takuya Seko ; Hidehiko Nakagawa
Journal of Medicinal Chemistry 2012 Volume 55(Issue 12) pp:5760-5773
Publication Date(Web):May 29, 2012
DOI:10.1021/jm3002108
Selective inhibitors of human sirtuin 2 (SIRT2), a deacetylase, are candidate therapeutic agents for neurodegenerative diseases such as Parkinson’s disease and Huntington’s disease as well as potential tools for elucidating the biological functions of SIRT2. On the basis of homology models of SIRT1 and SIRT2, we designed and prepared a series of 2-anilinobenzamide analogues. Enzyme assays using recombinant SIRT1 and SIRT2 revealed that 3′-phenethyloxy-2-anilinobenzamide analogues such as 33a and 33i are potent and selective SIRT2 inhibitors, showing more than 3.5-fold greater SIRT2-inhibitory activity and more than 35-fold greater SIRT2-selectivity compared with AGK2 (3), a previously reported SIRT2-selective inhibitor. Compound 33a also induced a dose-dependent selective increase of α-tubulin acetylation in human colon cancer HCT116 cells, indicating selective inhibition of SIRT2 in the cells. These 3′-phenethyloxy-2-anilinobenzamide derivatives represent an entry into a new class of SIRT2-selective inhibitors.
Co-reporter:Takayoshi Suzuki ; Yosuke Ota ; Masaki Ri ; Masashige Bando ; Aogu Gotoh ; Yukihiro Itoh ; Hiroki Tsumoto ; Prima R. Tatum ; Tamio Mizukami ; Hidehiko Nakagawa ; Shinsuke Iida ; Ryuzo Ueda ; Katsuhiko Shirahige
Journal of Medicinal Chemistry 2012 Volume 55(Issue 22) pp:9562-9575
Publication Date(Web):November 1, 2012
DOI:10.1021/jm300837y
To find HDAC8-selective inhibitors, we designed a library of HDAC inhibitor candidates, each containing a zinc-binding group that coordinates with the active-site zinc ion, linked via a triazole moiety to a capping structure that interacts with residues on the rim of the active site. These compounds were synthesized by using click chemistry. Screening identified HDAC8-selective inhibitors including C149 (IC50 = 0.070 μM), which was more potent than PCI-34058 (6) (IC50 = 0.31 μM), a known HDAC8 inhibitor. Molecular modeling suggested that the phenylthiomethyl group of C149 binds to a unique hydrophobic pocket of HDAC8, and the orientation of the phenylthiomethyl and hydroxamate moieties (fixed by the triazole moiety) is important for the potency and selectivity. The inhibitors caused selective acetylation of cohesin in cells and exerted growth-inhibitory effects on T-cell lymphoma and neuroblastoma cells (GI50 = 3–80 μM). These findings suggest that HDAC8-selective inhibitors have potential as anticancer agents.
Co-reporter:Mamiko Ikeda, Hidehiko Nakagawa, Takayoshi Suzuki, Naoki Miyata
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 5) pp:1949-1952
Publication Date(Web):1 March 2012
DOI:10.1016/j.bmcl.2012.01.042
Nuclear oxidative stress damages genomic DNA and may lead to cell death, leading to aging and aging-related disorders. Though it is important to measure the nuclear oxidative stress separately, there are still little examples that applicable to living cells. We have designed and synthesized three bisbenzimide-nitroxides as probes to selectively visualize nuclear redox changes in terms of fluorescence. Compound 3, containing two radical moieties, showed the largest reduction-induced fluorescence change, with good localization in nuclei. RAW264.7 murine macrophage cells were loaded with compound 3 and then treated with 100 μM hydrogen peroxide for 5 min to show the fluorescence increase. This fluorescence increase was inhibited by pretreatment of 1 mM ascorbic acid. These results show that compound 3 was suitable for nuclear-specific redox imaging in murine macrophages.
Co-reporter:Naoya Ieda, Hidehiko Nakagawa, Taeko Horinouchi, Tao Peng, Dan Yang, Hiroki Tsumoto, Takayoshi Suzuki, Kiyoshi Fukuhara and Naoki Miyata
Chemical Communications 2011 vol. 47(Issue 22) pp:6449-6451
Publication Date(Web):06 May 2011
DOI:10.1039/C1CC11681A
Photocontrollable ONOO− generation from a nitrobenzene derivative was demonstrated. The designed compound released NO in response to photoirradiation, and the resulting semiquinone reduced molecular oxygen to generate O2˙−; reaction of the two generated ONOO−, as confirmed with an ONOO−fluorescent probe, HKGreen-3.
Co-reporter:Takayoshi Suzuki
Journal of Medicinal Chemistry 2011 Volume 54(Issue 24) pp:8236-8250
Publication Date(Web):September 28, 2011
DOI:10.1021/jm201048w
Co-reporter:Daisuke Ogasawara, Takayoshi Suzuki, Koshiki Mino, Rie Ueda, Mohammed Naseer Ahmed Khan, Takuya Matsubara, Koichi Koseki, Makoto Hasegawa, Ryuzo Sasaki, Hidehiko Nakagawa, Tamio Mizukami, Naoki Miyata
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 12) pp:3702-3708
Publication Date(Web):15 June 2011
DOI:10.1016/j.bmc.2010.12.024
Optically active (1S,2R)-NCL-1 and (1R,2S)-NCL-1 were synthesized and evaluated for their lysine-specific demethylase 1 inhibitory activity and cell growth inhibitory activity. In enzyme assays, the (1S,2R)-isomer was approximately four times more potent than the (1R,2S)-isomer. In cell growth inhibition assays, the two isomers showed similar activity in HEK293 cells and SH-SY5Y cells, whereas the (1S,2R)-isomer showed approximately four times more potent activity than the (1R,2S)-isomer in HeLa cells.We synthesized optically active NCL-1, a lysine-specific demethylase 1 inhibitor, and evaluated its biological activity. In enzyme assays, the (1S,2R)-isomer was approximately four times more potent than the (1R,2S)-isomer. In cell-based assays, the two isomers showed similar activity in HEK293 cells and SH-SY5Y cells, whereas the (1S,2R)-isomer showed approximately four times more potent activity than the (1R,2S)-isomer in HeLa cells.
Co-reporter:Taeko Horinouchi;Dr. Hidehiko Nakagawa;Dr. Takayoshi Suzuki;Dr. Kiyoshi Fukuhara;Dr. Naoki Miyata
Chemistry - A European Journal 2011 Volume 17( Issue 17) pp:4809-4813
Publication Date(Web):
DOI:10.1002/chem.201001967
Abstract
We report a novel NO donor (RpNO), containing a 2,6-dimethylnitrobenzene moiety for photocontrollable NO release and a rhodamine moiety for targeting to mitochondria. Photorelease of NO from RpNO in aqueous solution was confirmed by means of ESR analysis. Cellular release of NO from RpNO was confirmed with the aid of DAF-FM DA, an NO-specific fluorescence probe. RpNO was colocalized with MitoTracker Green FM, a mitochondrial stain, in HCT116 colon cancer cells and exhibited photodependent cytotoxicity. Our results indicate that RpNO is an effective NO donor for time-controlled, mitochondria-specific NO treatment.
Co-reporter:Kazuya Matsuo, Hidehiko Nakagawa, Yusuke Adachi, Eri Kameda, Hiroki Tsumoto, Takayoshi Suzuki and Naoki Miyata
Chemical Communications 2010 vol. 46(Issue 21) pp:3788-3790
Publication Date(Web):14 Apr 2010
DOI:10.1039/C001502D
A hydrophilic hetero-Diels–Alder cycloadduct was synthesized as a novel photocontrollable donor of reactive nitrogen species. Production of either nitric oxide (NO) or nitroxyl (HNO) was photoinduced from this compound, depending on the environmental polarity.
Co-reporter:Shohei Hamada ; Takayoshi Suzuki ; Koshiki Mino ; Koichi Koseki ; Felix Oehme ; Ingo Flamme ; Hiroki Ozasa ; Yukihiro Itoh ; Daisuke Ogasawara ; Haruka Komaarashi ; Aiko Kato ; Hiroki Tsumoto ; Hidehiko Nakagawa ; Makoto Hasegawa ; Ryuzo Sasaki ; Tamio Mizukami
Journal of Medicinal Chemistry 2010 Volume 53(Issue 15) pp:5629-5638
Publication Date(Web):July 14, 2010
DOI:10.1021/jm1003655
Selective inhibitors of Jumonji domain-containing protein (JMJD) histone demethylases are candidate anticancer agents as well as potential tools for elucidating the biological functions of JMJDs. On the basis of the crystal structure of JMJD2A and a homology model of JMJD2C, we designed and prepared a series of hydroxamate analogues bearing a tertiary amine. Enzyme assays using JMJD2C, JMJD2A, and prolyl hydroxylases revealed that hydroxamate analogue 8 is a potent and selective JMJD2 inhibitor, showing 500-fold greater JMJD2C-inhibitory activity and more than 9100-fold greater JMJD2C-selectivity compared with the lead compound N-oxalylglycine 2. Compounds 17 and 18, prodrugs of compound 8, each showed synergistic growth inhibition of cancer cells in combination with an inhibitor of lysine-specific demethylase 1 (LSD1). These findings suggest that combination treatment with JMJD2 inhibitors and LSD1 inhibitors may represent a novel strategy for anticancer chemotherapy.
Co-reporter:Takayoshi Suzuki, Rikako Tanaka, Shohei Hamada, Hidehiko Nakagawa, Naoki Miyata
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 3) pp:1124-1127
Publication Date(Web):1 February 2010
DOI:10.1016/j.bmcl.2009.12.016
To identify novel non-nucleoside DNA methyltransferase (DNMT) inhibitors, we designed and synthesized a series of maleimide derivatives. Among this series, compounds 5–8 were found to be more potent DNMT1 inhibitors than RG108, a DNMT1 inhibitor reported previously by Siedlecki et al. The binding mode analysis of compound 5 is also reported.To identify novel non-nucleoside DNA methyltransferase (DNMT) inhibitors, we designed and synthesized a series of maleimide derivatives. Among this series, compounds 5–8 were found to be more potent DNMT1 inhibitors than RG108, a DNMT1 inhibitor reported previously by Siedlecki et al.
Co-reporter:Hiroki Tsumoto, Syo Kawahara, Yuki Fujisawa, Takayoshi Suzuki, Hidehiko Nakagawa, Kohfuku Kohda, Naoki Miyata
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 6) pp:1948-1952
Publication Date(Web):15 March 2010
DOI:10.1016/j.bmcl.2010.01.142
Water-soluble [60]fullerene (C60) derivatives were synthesized to examine their bioactivities. PC12 cells were used as a model of nerve cells and the bioactivities of synthesized C60 derivatives together with some reported ones were tested. Among the compounds tested, C60/(γ-CyD)2, C60-bis(γ-CyD) (5) containing C60-mono(γ-CyD) (5′), and C60/PVP were sufficiently soluble in water and showed an enhancing effect on the neurite outgrowth of NGF-treated PC12 cells.Syntheses of water-soluble C60 derivatives and the examination of their enhancing effect on neurite outgrowth of NGF-treated PC12 cells are reported.
Co-reporter:Dr. Takayoshi Suzuki;Yosuke Ota;Yuki Kasuya;Dr. Motoh Mutsuga;Dr. Yoko Kawamura;Dr. Hiroki Tsumoto;Dr. Hidehiko Nakagawa; M.G. Finn; Naoki Miyata
Angewandte Chemie International Edition 2010 Volume 49( Issue 38) pp:6817-6820
Publication Date(Web):
DOI:10.1002/anie.201002205
Co-reporter:Nobuaki Suzuki ; Takayoshi Suzuki ; Yosuke Ota ; Tatsuya Nakano ; Masaaki Kurihara ; Haruhiro Okuda ; Takao Yamori ; Hiroki Tsumoto ; Hidehiko Nakagawa
Journal of Medicinal Chemistry 2009 Volume 52(Issue 9) pp:2909-2922
Publication Date(Web):April 14, 2009
DOI:10.1021/jm900125m
Guided by the proposed catalytic mechanism of histone deacetylases (HDACs), we designed and synthesized a series of boronic acid-based HDAC inhibitors bearing an α-amino acid moiety. In this series, compounds (S)-18, 20, and 21 showed potent HDAC-inhibitory activity, highlighting the significance of the (S)-amino acid moiety. In cancer cell growth inhibition assays, compounds (S)-18, 20, and 21 exerted strong activity, and the values of the ratio of the concentration causing 50% growth inhibition (GI50) to the concentration causing 50% enzyme inhibition (IC50), i.e., GI50/IC50, were low. The potency of these compounds was similar to that of clinically used suberoylanilide hydroxamic acid (SAHA) (2). The results of Western blot analysis indicated that the cancer cell growth-inhibitory activity of compounds (S)-18, 20, and 21 is the result of HDAC inhibition. A molecular modeling study suggested that the hydrated boronic acid interacts with zinc ion, Tyr residue, and His residue in the active site of HDACs. Our findings indicate that these boronic acid derivatives represent an entry into a new class of HDAC inhibitors.
Co-reporter:Takayoshi Suzuki, Keiko Imai, Erika Imai, Shinsuke Iida, Ryuzo Ueda, Hiroki Tsumoto, Hidehiko Nakagawa, Naoki Miyata
Bioorganic & Medicinal Chemistry 2009 Volume 17(Issue 16) pp:5900-5905
Publication Date(Web):15 August 2009
DOI:10.1016/j.bmc.2009.07.001
A series of 2-anilinobenzamide derivatives were designed, synthesized and evaluated for their SIRT1-inhibitory activity. Among these, compounds 3 and 5 inhibited SIRT1 activity in enzyme assays and suppressed the growth of Daudi and HCT116 cells.A series of 2-anilinobenzamide derivatives were designed and synthesized as SIRT1 inhibitors. In this series, compounds 3 and 5 showed SIRT1 inhibition in vitro and suppressed the growth of Daudi and HCT116 cells.
Co-reporter:Takayoshi Suzuki, Tomomi Asaba, Erika Imai, Hiroki Tsumoto, Hidehiko Nakagawa, Naoki Miyata
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 19) pp:5670-5672
Publication Date(Web):1 October 2009
DOI:10.1016/j.bmcl.2009.08.028
To identify cell-active sirtuin inhibitors containing N-thioacetyl lysine, we synthesized compound 1, which was designed based on the structure of the reported N-ethoxycarbonylacetyl lysine-based sirtuin inhibitor NCS-12k. Compound 1 selectively inhibited SIRT1 in enzyme assays. Compound 1 also caused a dose-dependent increase in p53 acetylation in human colon cancer HCT116 cells, indicating the inhibition of SIRT1 in these cells.To identify cell-active sirtuin inhibitors containing N-thioacetyl lysine, we synthesized compound 1, which was designed based on the structure of the reported N-ethoxycarbonylacetyl lysine-based sirtuin inhibitor NCS-12k. Compound 1 selectively inhibited SIRT1 in enzyme assays. Compound 1 also caused a dose-dependent increase in p53 acetylation in human colon cancer HCT116 cells, indicating the inhibition of SIRT1 in these cells.
Co-reporter:Shohei Hamada, Tae-Dong Kim, Takayoshi Suzuki, Yukihiro Itoh, Hiroki Tsumoto, Hidehiko Nakagawa, Ralf Janknecht, Naoki Miyata
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 10) pp:2852-2855
Publication Date(Web):15 May 2009
DOI:10.1016/j.bmcl.2009.03.098
N-Oxalylglycine (NOG) derivatives were synthesized, and their inhibitory effect on histone lysine demethylase activity was evaluated. NOG and compound 1 inhibited histone lysine demethylases JMJD2A, 2C and 2D in enzyme assays, and their dimethyl ester prodrugs DMOG and 21 exerted histone lysine methylating activity in cellular assays.N-Oxalylglycine (NOG) derivatives were synthesized and their inhibitory effect on histone lysine demethylase activity was evaluated. This study showed that NOG and compound 1 inhibit histone lysine demethylases both in enzyme assays and cellular assays.
Co-reporter:Yusuke Adachi, Hidehiko Nakagawa, Kazuya Matsuo, Takayoshi Suzuki and Naoki Miyata
Chemical Communications 2008 (Issue 41) pp:5149-5151
Publication Date(Web):23 Sep 2008
DOI:10.1039/B811985F
We synthesized hetero-Diels–Alder cycloadducts from acyl nitroso derivatives and 9,10-dimethylanthracene, to be photo-inducible HNO-releasing agents and found that introduction of conjugated nitroaromatic groups effectively enhanced the responsiveness of HNO release to UV-A irradiation; we confirmed photoinduced HNO formation by EPR and GCMS analysis.
Co-reporter:Takayoshi Suzuki ; Sou-ichi Igari ; Akira Hirasawa ; Mie Hata ; Masaji Ishiguro ; Hiroki Fujieda ; Yukihiro Itoh ; Tatsuya Hirano ; Hidehiko Nakagawa ; Michitaka Ogura ; Makoto Makishima ; Gozoh Tsujimoto
Journal of Medicinal Chemistry 2008 Volume 51(Issue 23) pp:7640-7644
Publication Date(Web):November 13, 2008
DOI:10.1021/jm800970b
A weak, nonselective G protein-coupled receptor 120 (GPR120) agonist 10 was found by screening a series of carboxylic acids derived from the peroxisome proliferator-activated receptor γ (PPARγ) agonist 3. Modification based on the homology model of GPR120 led to the first GPR120-selective agonist 12. These results provide a basis for constructing new tools for probing the biology of GPR120 and for developing new candidate therapeutic agents.
Co-reporter:Hiroki Tsumoto, Katsumasa Takahashi, Takayoshi Suzuki, Hidehiko Nakagawa, Kohfuku Kohda, Naoki Miyata
Bioorganic & Medicinal Chemistry Letters 2008 Volume 18(Issue 2) pp:657-660
Publication Date(Web):15 January 2008
DOI:10.1016/j.bmcl.2007.11.065
We report the syntheses of C60-based active esters and the coupling of their C60 moiety to various amines or alcohols. Methano[60]fullerene carboxylic acid was activated by esterification with N-hydroxysuccinimide (NHS) or pentafluorophenol (PFP) and the active esters were isolated. Reactions of the active esters with amines or alcohols proceeded easily to give a variety of compounds having the C60 moiety.We report the synthesis of C60-based active esters and the coupling of their C60 moiety to various amines or alcohols.
Co-reporter:Takayoshi Suzuki, Shinya Hisakawa, Yukihiro Itoh, Sakiko Maruyama, Mineko Kurotaki, Hidehiko Nakagawa, Naoki Miyata
Bioorganic & Medicinal Chemistry Letters 2007 Volume 17(Issue 6) pp:1558-1561
Publication Date(Web):15 March 2007
DOI:10.1016/j.bmcl.2006.12.117
To identify prodrugs of a thiolate histone deacetylase inhibitor NCH-31 that show potent antiproliferative activity and are stable in human plasma, we synthesized several candidate prodrugs of NCH-31. Among these compounds, S-2-methyl-3-phenylpropanoyl compound 2 showed more potent antiproliferative activity and higher stability in human plasma than S-isobutyryl compound NCH-51.To identify prodrugs of a thiolate histone deacetylase inhibitor NCH-31 that show potent antiproliferative activity and are stable in human plasma, we synthesized several S-acyl derivatives of NCH-31. Among these compounds, S-2-methyl-3-phenylpropanoyl compound 2 showed potent antiproliferative activity and high stability in human plasma.
Co-reporter:Hiroki Fujieda, Shinya Usui, Takayoshi Suzuki, Hidehiko Nakagawa, Michitaka Ogura, Makoto Makishima, Naoki Miyata
Bioorganic & Medicinal Chemistry Letters 2007 Volume 17(Issue 15) pp:4351-4357
Publication Date(Web):1 August 2007
DOI:10.1016/j.bmcl.2007.05.017
To find novel PPARδ-selective agonists, we designed and synthesized phenylpropanoic acid derivatives bearing 6-substituted benzothiazoles. Optimization of this series led to the identification of a potent and selective PPARδ agonist 17. Molecular modeling suggested that compound 17 occupies the Y-shaped pocket of PPARδ appropriately.To find novel PPARδ-selective agonists, we designed and synthesized a series of phenylpropanoic acid derivatives bearing a benzothiazole ring. Optimization of this series led to the identification of a potent and selective PPARδ agonist 17.
Co-reporter:Takayoshi Suzuki, Shinya Hisakawa, Yukihiro Itoh, Nobuaki Suzuki, Katsumasa Takahashi, Masatoshi Kawahata, Kentaro Yamaguchi, Hidehiko Nakagawa, Naoki Miyata
Bioorganic & Medicinal Chemistry Letters 2007 Volume 17(Issue 15) pp:4208-4212
Publication Date(Web):1 August 2007
DOI:10.1016/j.bmcl.2007.05.040
Aiming to develop selective anticancer drugs, we designed and synthesized three disulfides bearing a folic acid moiety as candidate folate receptor (FR)-targeted prodrugs of thiolate histone deacetylase inhibitors. Among them, compound 1 displayed growth-inhibitory activity toward folate receptor-positive MCF-7 breast cancer cells. The activity of 1 was significantly reduced by free folic acid, suggesting that cellular uptake of 1 is mediated by FR.Aiming to develop selective anticancer drugs, we designed and synthesized three disulfides bearing a folic acid moiety as candidate folate receptor (FR)-targeted prodrugs of thiolate histone deacetylase inhibitors. One of the folate conjugates showed growth-inhibitory activity toward folate receptor-positive MCF-7 breast cancer cells.
Co-reporter:Takayoshi Suzuki Dr.;Keiko Imai;Hidehiko Nakagawa Dr.
ChemMedChem 2006 Volume 1(Issue 10) pp:
Publication Date(Web):20 SEP 2006
DOI:10.1002/cmdc.200600162
SIRTs, class III histone deacetylases, have been suggested to be associated with certain diseases such as cancer and HIV. Thus, SIRT inhibitors are of interest not only to elucidate the biological functions of the enzyme, but also as potential therapeutic agents. 2-Anilinobenzamide was identified in a nocotinamide- and benzamide-focused compound library as a novel SIRT inhibitor. This compound caused the accumulation of acetylated p53 in HCT116 cells.
Co-reporter:Takayoshi Suzuki Dr.;Keiko Imai;Hidehiko Nakagawa Dr.
ChemMedChem 2006 Volume 1(Issue 10) pp:
Publication Date(Web):6 OCT 2006
DOI:10.1002/cmdc.200690035
Co-reporter:Takayoshi Suzuki, Akiyasu Kouketsu, Azusa Matsuura, Arihiro Kohara, Shin-ichi Ninomiya, Kohfuku Kohda, Naoki Miyata
Bioorganic & Medicinal Chemistry Letters 2004 Volume 14(Issue 12) pp:3313-3317
Publication Date(Web):21 June 2004
DOI:10.1016/j.bmcl.2004.03.063
In order to find novel nonhydroxamate histone deacetylase (HDAC) inhibitors, a series of thiol-based compounds modeled after suberoylanilide hydroxamic acid (SAHA) was synthesized, and their inhibitory effect on HDACs was evaluated. Compound 6, in which the hydroxamic acid of SAHA was replaced by a thiol, was found to be as potent as SAHA, and optimization of this series led to the identification of HDAC inhibitors more potent than SAHA.A series of thiol-based SAHA analogues was designed and synthesized. Compound 6, in which the hydroxamic acid of SAHA was replaced by a thiol, was found to be as potent as SAHA, and optimization of this series led to the identification of HDAC inhibitors (17, 21, 24, and 25) more potent than SAHA.
Co-reporter:Takayoshi Suzuki, Yuki Nagano, Azusa Matsuura, Arihiro Kohara, Shin-ichi Ninomiya, Kohfuku Kohda, Naoki Miyata
Bioorganic & Medicinal Chemistry Letters 2003 Volume 13(Issue 24) pp:4321-4326
Publication Date(Web):15 December 2003
DOI:10.1016/j.bmcl.2003.09.048
In order to find novel non-hydroxamate histone deacetylase (HDAC) inhibitors, a series of compounds modeled after suberoylanilide hydroxamic acid (SAHA) were designed and synthesized as (i) substrate (acetyl lysine) analogues (compounds 3–7), (ii) analogues bearing various functional groups expected to chelate zinc ion (compounds 8–15), and (iii) analogues bearing nucleophilic functional groups which could bind covalently to HDACs (compounds 16–18). In this series, semicarbazide 8b and bromoacetamides 18b,c were found to be potent HDAC inhibitors for non-hydroxamates.A series of SAHA-based non-hydroxamates was designed and synthesized, and semicarbazide 8b and bromoacetamides 18b,c were found to be potent HDAC inhibitors for non-hydroxamates. The binding mode analyses by computer calculation of 8b and 18c are also reported.
Co-reporter:Ikuo Nakanishi, Kei Ohkubo, Shunsuke Fujita, Shunichi Fukuzumi, Toshifumi Konishi, Mamoru Fujitsuka, Osamu Ito and Naoki Miyata
Organic & Biomolecular Chemistry 2002 (Issue 11) pp:1829-1833
Publication Date(Web):26 Sep 2002
DOI:10.1039/B206887G
Visible-light irradiation of poly(vinylpyrrolidone)
(PVP)-solubilized C60 in water in the presence of NADH (dihydronicotinamide adenine dinucleotide) and molecular oxygen (O2) results in formation of superoxide anion (O2˙−). Formation of O2˙− having a characteristic g‖ value of 2.18 was evidenced by the direct observation with use of a low-temperature EPR technique at 77 K. Photoinduced O2˙− formation was also observed for an N-methyl-2-pyrrolidone (NMP) solution of C60 and 1-benzyl-1,4-dihydronicotinamide (BNAH) in the presence of O2, whereas C60 radical anion (C60˙−) was formed in the absence of O2 under otherwise the same experimental conditions. These results suggest that C602− formed in the photoinduced electron-transfer reduction of C60 by BNAH acts as an electron donor to O2 to give O2˙− in NMP.
Co-reporter:Mamiko Ikeda, Hidehiko Nakagawa, Shizuka Ban, Hiroki Tsumoto, ... Naoki Miyata
Free Radical Biology and Medicine (1 December 2010) Volume 49(Issue 11) pp:1792-1797
Publication Date(Web):1 December 2010
DOI:10.1016/j.freeradbiomed.2010.09.009
Oxidative stress in nuclei is known to induce either oxidative modification of DNA bases or single/double-strand breaks, which may lead to carcinogenesis. To evaluate the redox status in nuclei in living cells, we designed a novel nucleus-localizing redox spin probe, F-DisT, which contains a fluorescein fluorophore linked to a DNA minor-groove-binding moiety. Nuclear distribution of the probe was easily confirmed by colocalization with a nuclear stain, Hoechst 33342, in confocal microscopy. Measurement of oxidative stress with F-DisT in a murine macrophage cell line exposed to endotoxin (lipopolysaccharide) showed a remarkable increase in the ESR signal decay rate. This increase was significantly inhibited by Nω-nitro-l-arginine (nitric oxide synthase inhibitor) and diphenyleneiodonium chloride (NADPH oxidase inhibitor). These results indicate that nitric oxide and superoxide contribute to oxidative stress in nuclei. Similar studies in membrane or mitochondria using respective organelle-specific spin probes indicated that the redox microenvironments in these organelles are markedly different from that in nuclei. Thus, subcellular redox microenvironments show marked variability in endotoxin-stimulated living cells.
Co-reporter:Rie Ueda ; Takayoshi Suzuki ; Koshiki Mino ; Hiroki Tsumoto ; Hidehiko Nakagawa ; Makoto Hasegawa ; Ryuzo Sasaki ; Tamio Mizukami
Journal of the American Chemical Society () pp:
Publication Date(Web):November 16, 2009
DOI:10.1021/ja907055q
Lysine specific demethylase 1 (LSD1) plays a key role in the regulation of gene expression by removing the methyl groups from methylated Lys4 of histone H3 (H3K4). Here we report the identification of the first small-molecule LSD1-selective inhibitors. These inhibitors show in vivo H3K4-methylating activity and antiproliferative activity and should be useful as lead structures for anticancer drugs and as tools for studying the biological roles of LSD1.
Co-reporter:Yusuke Adachi;Hidehiko Nakagawa;Kazuya Matsuo;Takayoshi Suzuki
Chemical Communications 2008(Issue 41) pp:
Publication Date(Web):2008/10/27
DOI:10.1039/B811985F
We synthesized hetero-Diels–Alder cycloadducts from acyl nitroso derivatives and 9,10-dimethylanthracene, to be photo-inducible HNO-releasing agents and found that introduction of conjugated nitroaromatic groups effectively enhanced the responsiveness of HNO release to UV-A irradiation; we confirmed photoinduced HNO formation by EPR and GCMS analysis.
Co-reporter:Naoya Ieda, Hidehiko Nakagawa, Taeko Horinouchi, Tao Peng, Dan Yang, Hiroki Tsumoto, Takayoshi Suzuki, Kiyoshi Fukuhara and Naoki Miyata
Chemical Communications 2011 - vol. 47(Issue 22) pp:NaN6451-6451
Publication Date(Web):2011/05/06
DOI:10.1039/C1CC11681A
Photocontrollable ONOO− generation from a nitrobenzene derivative was demonstrated. The designed compound released NO in response to photoirradiation, and the resulting semiquinone reduced molecular oxygen to generate O2˙−; reaction of the two generated ONOO−, as confirmed with an ONOO−fluorescent probe, HKGreen-3.
Co-reporter:Kazuya Matsuo, Hidehiko Nakagawa, Yusuke Adachi, Eri Kameda, Hiroki Tsumoto, Takayoshi Suzuki and Naoki Miyata
Chemical Communications 2010 - vol. 46(Issue 21) pp:NaN3790-3790
Publication Date(Web):2010/04/14
DOI:10.1039/C001502D
A hydrophilic hetero-Diels–Alder cycloadduct was synthesized as a novel photocontrollable donor of reactive nitrogen species. Production of either nitric oxide (NO) or nitroxyl (HNO) was photoinduced from this compound, depending on the environmental polarity.





![Glucitol, 1-deoxy-1-[(dithiocarboxy)methylamino]-](http://img.cochemist.com/ccimg/874200/874112-84-2.png)
![Glucitol, 1-deoxy-1-[(dithiocarboxy)methylamino]-](http://img.cochemist.com/ccimg/874200/874112-84-2_b.png)
![METHANONE, [3-(1-METHYLETHENYL)PHENYL]PHENYL-](http://img.cochemist.com/ccimg/856900/856893-07-7.png)
![METHANONE, [3-(1-METHYLETHENYL)PHENYL]PHENYL-](http://img.cochemist.com/ccimg/856900/856893-07-7_b.png)

















![Spiro[isobenzofuran-1(3H),9'-[9H]xanthen]-3-one,3',6'-bis(acetyloxy)-4-amino-2',7'-difluoro-5-(methylamino)-](/data/chemimg/410800/254109-22-3.png)
![Spiro[isobenzofuran-1(3H),9'-[9H]xanthen]-3-one,3',6'-bis(acetyloxy)-4-amino-2',7'-difluoro-5-(methylamino)-](/data/chemimg/410800/254109-22-3_b.png)










![Benzene, 1,1'-[thiobis(methylene)]bis[2-nitro-](http://img.cochemist.com/ccimg/99400/99366-73-1.png)
![Benzene, 1,1'-[thiobis(methylene)]bis[2-nitro-](http://img.cochemist.com/ccimg/99400/99366-73-1_b.png)


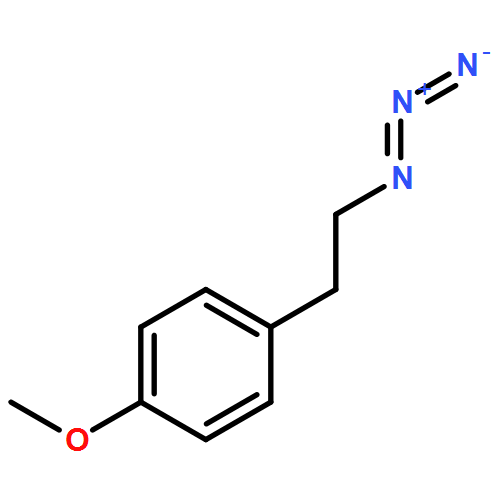
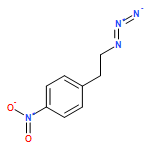
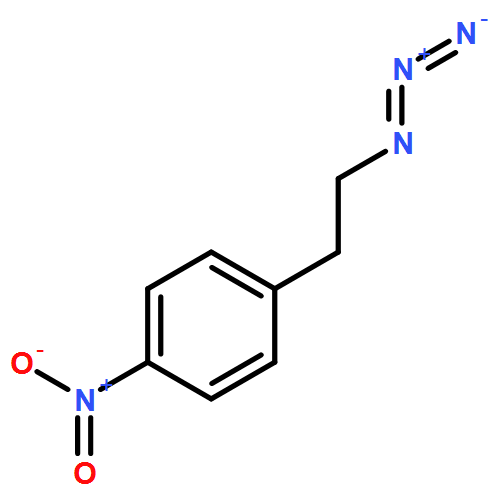
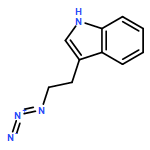
![Tricyclo[3.3.1.13,7]decane, 1-(azidomethyl)-](http://img.cochemist.com/ccimg/63600/63534-36-1.png)
![Tricyclo[3.3.1.13,7]decane, 1-(azidomethyl)-](http://img.cochemist.com/ccimg/63600/63534-36-1_b.png)









![Benzene, [(1E)-2-azidoethenyl]-](/data/chemimg/3722800/18756-03-1.png)
![Benzene, [(1E)-2-azidoethenyl]-](/data/chemimg/3722800/18756-03-1_b.png)



![PHOSPHONIC ACID, [(3,5-DIMETHYL-4-NITROPHENYL)METHYL]-, DIETHYL ESTER](http://img.cochemist.com/ccimg/864600/864513-22-4.png)
![PHOSPHONIC ACID, [(3,5-DIMETHYL-4-NITROPHENYL)METHYL]-, DIETHYL ESTER](http://img.cochemist.com/ccimg/864600/864513-22-4_b.png)