Co-reporter:Gayan B. Wijeratne, Shabnam Hematian, Maxime A. Siegler, and Kenneth D. Karlin
Journal of the American Chemical Society September 27, 2017 Volume 139(Issue 38) pp:13276-13276
Publication Date(Web):August 18, 2017
DOI:10.1021/jacs.7b07808
A copper complex, [CuI(tmpa)(MeCN)]+, effectively reductively couples NO(g) at RT in methanol (MeOH), giving a structurally characterized hyponitrito-dicopper(II) adduct. Hydrogen-bonding from MeOH is critical for the hyponitrite complex formation and stabilization. This complex exhibits the reverse redox process in aprotic solvents, giving CuI + NO(g), leading to CuI-mediated NO(g)-disproportionation. The relationship of this chemistry to biological iron and/or copper mediated NO(g) reductive coupling to give N2O(g) is discussed.
Co-reporter:Isaac Garcia-Bosch, Ryan E. Cowley, Daniel E. Díaz, Ryan L. Peterson, Edward I. Solomon, and Kenneth D. Karlin
Journal of the American Chemical Society March 1, 2017 Volume 139(Issue 8) pp:3186-3186
Publication Date(Web):February 14, 2017
DOI:10.1021/jacs.6b12990
Copper-dependent metalloenzymes are widespread throughout metabolic pathways, coupling the reduction of O2 with the oxidation of organic substrates. Small-molecule synthetic analogs are useful platforms to generate L/Cu/O2 species that reproduce the structural, spectroscopic, and reactive properties of some copper-/O2-dependent enzymes. Landmark studies have shown that the conversion between dicopper(II)-peroxo species (L2CuII2(O22–) either side-on peroxo, SP, or end-on trans-peroxo, TP) and dicopper(III)-bis(μ-oxo) (L2CuIII2(O2–)2: O) can be controlled through ligand design, reaction conditions (temperature, solvent, and counteranion), or substrate coordination. We recently published ( J. Am. Chem. Soc. 2012, 134, 8513, DOI: 10.1021/ja300674m) the crystal structure of an unusual SP species [(MeAN)2CuII2(O22–)]2+ (SPMeAN, MeAN: N-methyl-N,N-bis[3-(dimethylamino)propyl]amine) that featured an elongated O–O bond but did not lead to O–O cleavage or reactivity toward external substrates. Herein, we report that SPMeAN can be activated to generate OMeAN and perform the oxidation of external substrates by two complementary strategies: (i) coordination of substituted sodium phenolates to form the substrate-bound OMeAN-RPhO- species that leads to ortho-hydroxylation in a tyrosinase-like fashion and (ii) addition of stoichiometric amounts (1 or 2 equiv) of Lewis acids (LA’s) to form an unprecedented series of O-type species (OMeAN-LA) able to oxidize C–H and O–H bonds. Spectroscopic, computational, and mechanistic studies emphasize the unique plasticity of the SPMeAN core, which combines the assembly of exogenous reagents in the primary (phenolates) and secondary (Lewis acids association to the MeAN ligand) coordination spheres with O–O cleavage. These findings are reminiscent of the strategy followed by several metalloproteins and highlight the possible implication of O-type species in copper-/dioxygen-dependent enzymes such as tyrosinase (Ty) and particulate methane monooxygenase (pMMO).
Co-reporter:Andrew W. Schaefer, Matthew T. Kieber-Emmons, Suzanne M. Adam, Kenneth D. Karlin, and Edward I. Solomon
Journal of the American Chemical Society June 14, 2017 Volume 139(Issue 23) pp:7958-7958
Publication Date(Web):May 18, 2017
DOI:10.1021/jacs.7b03292
This study evaluates the reaction of a biomimetic heme–peroxo–copper complex, {[(DCHIm)(F8)FeIII]–(O22–)–[CuII(AN)]}+ (1), with a phenolic substrate, involving a net H-atom abstraction to cleave the bridging peroxo O–O bond that produces FeIV═O, CuII—OH, and phenoxyl radical moieties, analogous to the chemistry carried out in heme–copper oxidases (HCOs). A 3D potential energy surface generated for this reaction reveals two possible reaction pathways: one involves nearly complete proton transfer (PT) from the phenol to the peroxo ligand before the barrier; the other involves O–O homolysis, where the phenol remains H-bonding to the peroxo OCu in the transition state (TS) and transfers the H+ after the barrier. In both mechanisms, electron transfer (ET) from phenol occurs after the PT (and after the barrier); therefore, only the interaction with the H+ is involved in lowering the O–O cleavage barrier. The relative barriers depend on covalency (which governs ET from Fe), and therefore vary with DFT functional. However, as these mechanisms differ by the amount of PT at the TS, kinetic isotope experiments were conducted to determine which mechanism is active. It is found that the phenolic proton exhibits a secondary kinetic isotope effect, consistent with the calculations for the H-bonded O–O homolysis mechanism. The consequences of these findings are discussed in relation to O–O cleavage in HCOs, supporting a model in which a peroxo intermediate serves as the active H+ acceptor, and both the H+ and e– required for O–O cleavage derive from the cross-linked Tyr residue present at the active site.
Co-reporter:David A. Quist;Daniel E. Diaz
JBIC Journal of Biological Inorganic Chemistry 2017 Volume 22( Issue 2-3) pp:
Publication Date(Web):
DOI:10.1007/s00775-016-1415-2
Co-reporter:Pankaj Kumar; Yong-Min Lee; Lianrui Hu; Jianwei Chen; Young Jun Park; Jiannian Yao; Hui Chen; Kenneth D. Karlin;Wonwoo Nam
Journal of the American Chemical Society 2016 Volume 138(Issue 24) pp:7753-7762
Publication Date(Web):May 24, 2016
DOI:10.1021/jacs.6b04040
Metal–nitrosyl complexes are key intermediates involved in many biological and physiological processes of nitric oxide (NO) activation by metalloproteins. In this study, we report the reactivities of mononuclear cobalt(III)–nitrosyl complexes bearing N-tetramethylated cyclam (TMC) ligands, [(14-TMC)CoIII(NO)]2+ and [(12-TMC)CoIII(NO)]2+, in NO-transfer and dioxygenation reactions. The Co(III)–nitrosyl complex bearing 14-TMC ligand, [(14-TMC)CoIII(NO)]2+, transfers the bound nitrosyl ligand to [(12-TMC)CoII]2+ via a dissociative pathway, {[(14-TMC)CoIII(NO)]2+ → {(14-TMC)Co···NO}2+}, thus affording [(12-TMC)CoIII(NO)]2+ and [(14-TMC)CoII]2+ as products. The dissociation of NO from the [(14-TMC)CoIII(NO)]2+ complex prior to NO-transfer is supported experimentally and theoretically. In contrast, the reverse reaction, which is the NO-transfer from [(12-TMC)CoIII(NO)]2+ to [(14-TMC)CoII]2+, does not occur. In addition to the NO-transfer reaction, dioxygenation of [(14-TMC)CoIII(NO)]2+ by O2 produces [(14-TMC)CoII(NO3)]+, which possesses an O,O-chelated nitrato ligand and where, based on an experiment using 18O-labeled O2, two of the three O-atoms in the [(14-TMC)CoII(NO3)]+ product derive from O2. The dioxygenation reaction is proposed to occur via a dissociative pathway, as proposed in the NO-transfer reaction, and via the formation of a Co(II)–peroxynitrite intermediate, based on the observation of phenol ring nitration. In contrast, [(12-TMC)CoIII(NO)]2+ does not react with O2. Thus, the present results demonstrate unambiguously that the NO-transfer/dioxygenation reactivity of the cobalt(III)–nitrosyl complexes bearing TMC ligands is significantly influenced by the ring size of the TMC ligands and/or the spin state of the cobalt ion.
Co-reporter:Suzanne M. Adam, Isaac Garcia-Bosch, Andrew W. Schaefer, Savita K. Sharma, Maxime A. Siegler, Edward I. Solomon, and Kenneth D. Karlin
Journal of the American Chemical Society 2016 Volume 139(Issue 1) pp:472-481
Publication Date(Web):December 28, 2016
DOI:10.1021/jacs.6b11322
The 4H+/4e– reduction of O2 to water, a key fuel-cell reaction also carried out in biology by oxidase enzymes, includes the critical O–O bond reductive cleavage step. Mechanistic investigations on active-site model compounds, which are synthesized by rational design to incorporate systematic variations, can focus on and resolve answers to fundamental questions, including protonation and/or H-bonding aspects, which accompany electron transfer. Here, we describe the nature and comparative reactivity of two low-spin heme–peroxo–Cu complexes, LS-4DCHIm, [(DCHIm)F8FeIII-(O22–)-CuII(DCHIm)4]+, and LS-3DCHIm, [(DCHIm)F8FeIII-(O22–)-CuII(DCHIm)3]+ (F8 = tetrakis(2,6-difluorophenyl)-porphyrinate; DCHIm = 1,5-dicyclohexylimidazole), toward different proton (4-nitrophenol and [DMF·H+](CF3SO3–)) (DMF = dimethyl-formamide) or electron (decamethylferrocene (Fc*)) sources. Spectroscopic reactivity studies show that differences in structure and electronic properties of LS-3DCHIm and LS-4DCHIm lead to significant differences in behavior. LS-3DCHIm is resistant to reduction, is unreactive toward weakly acidic 4-NO2–phenol, and stronger acids cleave the metal–O bonds, releasing H2O2. By contrast, LS-4DCHIm forms an adduct with 4-NO2–phenol, which includes an H-bond to the peroxo O-atom distal to Fe (resonance Raman (rR) spectroscopy and DFT). With addition of Fc* (2 equiv overall required), O–O reductive cleavage occurs, giving water, Fe(III), and Cu(II) products; however, a kinetic study reveals a one-electron rate-determining process, ket = 1.6 M–1 s–1 (−90 °C). The intermediacy of a high-valent [(DCHIm)F8FeIV═O] species is thus implied, and separate experiments show that one-electron reduction-protonation of [(DCHIm)F8FeIV═O] occurs faster (ket2 = 5.0 M–1 s–1), consistent with the overall postulated mechanism. The importance of the H-bonding interaction as a prerequisite for reductive cleavage is highlighted.
Co-reporter:Rui Cao, Lee Taylor Elrod, Ryan L. Lehane, Eunsuk Kim, and Kenneth D. Karlin
Journal of the American Chemical Society 2016 Volume 138(Issue 49) pp:16148-16158
Publication Date(Web):December 2, 2016
DOI:10.1021/jacs.6b10689
Co-reporter:Rui Cao; Claudio Saracini; Jake W. Ginsbach; Matthew T. Kieber-Emmons; Maxime A. Siegler; Edward I. Solomon; Shunichi Fukuzumi
Journal of the American Chemical Society 2016 Volume 138(Issue 22) pp:7055-7066
Publication Date(Web):May 26, 2016
DOI:10.1021/jacs.6b02404
Oxygenation of [Cu2(UN-O–)(DMF)]2+ (1), a structurally characterized dicopper Robin–Day class I mixed-valent Cu(II)Cu(I) complex, with UN-O– as a binucleating ligand and where dimethylformamide (DMF) binds to the Cu(II) ion, leads to a superoxo-dicopper(II) species [CuII2(UN-O–)(O2•–)]2+ (2). The formation kinetics provide that kon = 9 × 10–2 M–1 s–1 (−80 °C), ΔH‡ = 31.1 kJ mol–1 and ΔS‡ = −99.4 J K–1 mol–1 (from −60 to −90 °C data). Complex 2 can be reversibly reduced to the peroxide species [CuII2(UN-O–)(O22–)]+ (3), using varying outer-sphere ferrocene or ferrocenium redox reagents. A Nernstian analysis could be performed by utilizing a monodiphenylamine substituted ferrocenium salt to oxidize 3, leading to an equilibrium mixture with Ket = 5.3 (−80 °C); a standard reduction potential for the superoxo–peroxo pair is calculated to be E° = +130 mV vs SCE. A literature survey shows that this value falls into the range of biologically relevant redox reagents, e.g., cytochrome c and an organic solvent solubilized ascorbate anion. Using mixed-isotope resonance Raman (rRaman) spectroscopic characterization, accompanied by DFT calculations, it is shown that the superoxo complex consists of a mixture of μ-1,2- (21,2) and μ-1,1- (21,1) isomers, which are in rapid equilibrium. The electron transfer process involves only the μ-1,2-superoxo complex [CuII2(UN-O–)(μ-1,2-O2•–)]2+ (21,2) and μ-1,2-peroxo structures [CuII2(UN-O–)(O22–)]+ (3) having a small bond reorganization energy of 0.4 eV (λin). A stopped-flow kinetic study results reveal an outer-sphere electron transfer process with a total reorganization energy (λ) of 1.1 eV between 21,2 and 3 calculated in the context of Marcus theory.
Co-reporter:Mihoko Yamada, Kenneth D. Karlin and Shunichi Fukuzumi
Chemical Science 2016 vol. 7(Issue 4) pp:2856-2863
Publication Date(Web):05 Jan 2016
DOI:10.1039/C5SC04312C
Benzene was hydroxylated with hydrogen peroxide (H2O2) in the presence of catalytic amounts of copper complexes in acetone to yield phenol at 298 K. At higher temperatures, phenol was further hydroxylated with H2O2 by catalysis of copper complexes to yield p-benzoquinone. The kinetic study revealed that the rate was proportional to concentrations of benzene and H2O2, but to the square root of the concentration of a copper(II) complex ([Cu(tmpa)]2+: tmpa = tris(2-pyridylmethyl)amine). The addition of a spin trapping reagent resulted in formation of a spin adduct of hydroperoxyl radical (HO2˙), as observed by EPR spectroscopy, inhibiting phenol formation. HO2˙ produced by the reaction of [Cu(tmpa)]2+ with H2O2 acts as a chain carrier for the radical chain reactions for formation of phenol. When [Cu(tmpa)]2+ was incorporated into mesoporous silica–alumina (Al-MCM-41) by a cation exchange reaction, the selectivity for production of phenol was much enhanced by prevention of hydroxylation of phenol, which was not adsorbed to Al-MCM-41. The high durability with a turnover number of 4320 for the hydroxylation of benzene to phenol with H2O2 was achieved using [Cu(tmpa)]2+ incorporated into Al-MCM-41 as an efficient and selective catalyst.
Peter M.H. Kroneck, Martha E. Sosa Torres (Guest editors), “Sustaining Life on Planet Earth: Metalloenzymes Mastering Dioxygen and Other Chewy Gases”, A. Sigel, H. Sigel, R.K.O. Sigel (Series editors), Metal Ions in Life Sciences, vol. 15, Springer International Publishing AG, Switzerland, 2015, 329 pp [ISSN 1559-0836; ISSN 1868-0402 (electronic); ISBN 978-3-319-12414-8; ISBN 978-3-319-12415-5 (eBook), http://dx.doi.org/10.1007/978-3-319-12415-5.
Co-reporter:Kenneth D. Karlin
Inorganica Chimica Acta 2016 Volume 451() pp:207-208
Publication Date(Web):1 September 2016
DOI:10.1016/j.ica.2016.07.035
Co-reporter:Shabnam Hematian, Isaac Garcia-Bosch, and Kenneth D. Karlin
Accounts of Chemical Research 2015 Volume 48(Issue 8) pp:2462
Publication Date(Web):August 5, 2015
DOI:10.1021/acs.accounts.5b00265
Our long-time niche in synthetic biological inorganic chemistry has been to design ligands and generate coordination complexes of copper or iron ions or both, those reacting with dioxygen (O2) or nitrogen oxides (e.g., nitric oxide (NO(g)) and nitrite (NO2–)) or both. As inspiration for this work, we turn to mitochondrial cytochrome c oxidase, which is responsible for dioxygen consumption and is also the predominant target for NO(g) and nitrite within mitochondria. In this Account, we highlight recent advances in studying synthetic heme/Cu complexes in two respects. First, there is the design, synthesis, and characterization of new O2 adducts whose further study will add insights into O2 reductive cleavage chemistry. Second, we describe how related heme/Cu constructs reduce nitrite ion to NO(g) or the reverse, oxidize NO(g) to nitrite. The reactions of nitrogen oxides occur as part of CcO’s function, which is intimately tied to cellular O2 balance.We had first discovered that reduced heme/Cu compounds react with O2 giving μ-oxo heme-FeIII–O–CuII(L) products; their properties are discussed. The O-atom is derived from dioxygen, and interrogations of these systems led to the construction and characterization of three distinctive classes of heme–peroxo complexes, two high-spin and one low-spin species.Recent investigations include a new approach to the synthesis of low-spin heme–peroxo–Cu complexes, employing a “naked” synthon, where the copper ligand denticity and geometric types can be varied. The result is a collection of such complexes; spectroscopic and structural features (by DFT calculations) are described. Some of these compounds are reactive toward reductants/protons effecting subsequent O–O cleavage. This points to how subtle improvements in ligand environment lead to a desired local structure and resulting optimized reactivity, as known to occur at enzyme active sites.The other sector of research is focused on heme/Cu assemblies mediating the redox interplay between nitrite and NO(g). In the nitrite reductase chemistry, the cupric center serves as a Lewis acid, while the heme is the redox active center providing the electron. The orientation of nitrite in approaching the ferrous heme center and N-atom binding are important. Also, detailed spectroscopic and kinetic studies of the NO(g) oxidase chemistry, in excellent agreement with theoretical calculations, reveal the intermediates and key mechanistic steps. Thus, we suggest that both chemical and biochemical heme/Cu-mediated nitrite reductase and NO(g) oxidase chemistry require N-atom binding to a ferrous heme along with cupric ion O-atom coordination, proceeding via a three-membered O–Fe–N chelate ring transition state. These important mechanistic features of heme/Cu systems interconverting NO(g) and nitrite are discussed for the first time.
Co-reporter:Isaac Garcia-Bosch; Suzanne M. Adam; Andrew W. Schaefer; Savita K. Sharma; Ryan L. Peterson; Edward I. Solomon
Journal of the American Chemical Society 2015 Volume 137(Issue 3) pp:1032-1035
Publication Date(Web):January 16, 2015
DOI:10.1021/ja5115198
Here we describe a new approach for the generation of heme-peroxo-Cu compounds, using a “naked” complex synthon, [(F8)FeIII-(O22–)-CuII(MeTHF)3]+ (MeTHF = 2-methyltetrahydrofuran; F8 = tetrakis(2,6-difluorophenyl)porphyrinate). Addition of varying ligands (L) for Cu allows the generation and spectroscopic characterization of a family of high- and low-spin FeIII-(O22–)-CuII(L) complexes. These possess markedly varying CuII coordination geometries, leading to tunable Fe-O, O-O, and Cu-O bond strengths. DFT calculations accompanied by vibrational data correlations give detailed structural insights.
Co-reporter:Sunghee Kim; Jake W. Ginsbach; Jung Yoon Lee; Ryan L. Peterson; Jeffrey J. Liu; Maxime A. Siegler; Amy A. Sarjeant; Edward I. Solomon
Journal of the American Chemical Society 2015 Volume 137(Issue 8) pp:2867-2874
Publication Date(Web):February 23, 2015
DOI:10.1021/ja508371q
Copper(II) hydroperoxide species are significant intermediates in processes such as fuel cells and (bio)chemical oxidations, all involving stepwise reduction of molecular oxygen. We previously reported a CuII-OOH species that performs oxidative N-dealkylation on a dibenzylamino group that is appended to the 6-position of a pyridyl donor of a tripodal tetradentate ligand. To obtain insights into the mechanism of this process, reaction kinetics and products were determined employing ligand substrates with various para-substituent dibenzyl pairs (-H,-H; -H,-Cl; -H,-OMe, and -Cl,-OMe), or with partially or fully deuterated dibenzyl N-(CH2Ph)2 moieties. A series of ligand–copper(II) bis-perchlorate complexes were synthesized, characterized, and the X-ray structures of the -H,-OMe analogue were determined. The corresponding metastable CuII-OOH species were generated by addition of H2O2/base in acetone at −90 °C. These convert (t1/2 ≈ 53 s) to oxidatively N-dealkylated products, producing para-substituted benzaldehydes. Based on the experimental observations and supporting DFT calculations, a reaction mechanism involving dibenzylamine H-atom abstraction or electron-transfer oxidation by the CuII-OOH entity could be ruled out. It is concluded that the chemistry proceeds by rate limiting Cu-O homolytic cleavage of the CuII-(OOH) species, followed by site-specific copper Fenton chemistry. As a process of broad interest in copper as well as iron oxidative (bio)chemistries, a detailed computational analysis was performed, indicating that a CuIOOH species undergoes O–O homolytic cleavage to yield a hydroxyl radical and CuIIOH rather than heterolytic cleavage to yield water and a CuII-O•– species.
Co-reporter:Saya Kakuda; Clarence J. Rolle; Kei Ohkubo; Maxime A. Siegler; Kenneth D. Karlin;Shunichi Fukuzumi
Journal of the American Chemical Society 2015 Volume 137(Issue 9) pp:3330-3337
Publication Date(Web):February 7, 2015
DOI:10.1021/ja512584r
Mononuclear copper complexes, [(tmpa)CuII(CH3CN)](ClO4)2 (1, tmpa = tris(2-pyridylmethyl)amine) and [(BzQ)CuII(H2O)2](ClO4)2 (2, BzQ = bis(2-quinolinylmethyl)benzylamine)], act as efficient catalysts for the selective two-electron reduction of O2 by ferrocene derivatives in the presence of scandium triflate (Sc(OTf)3) in acetone, whereas 1 catalyzes the four-electron reduction of O2 by the same reductant in the presence of Brønsted acids such as triflic acid. Following formation of the peroxo-bridged dicopper(II) complex [(tmpa)CuII(O2)CuII(tmpa)]2+, the two-electron reduced product of O2 with Sc3+ is observed to be scandium peroxide ([ScIII(O22–)]+). In the presence of 3 equiv of hexamethylphosphoric triamide (HMPA), [ScIII(O22–)]+ was oxidized by [Fe(bpy)3]3+ (bpy = 2,2-bipyridine) to the known superoxide species [(HMPA)3ScIII(O2•–)]2+ as detected by EPR spectroscopy. A kinetic study revealed that the rate-determining step of the catalytic cycle for the two-electron reduction of O2 with 1 is electron transfer from Fc* to 1 to give a cuprous complex which is highly reactive toward O2, whereas the rate-determining step with 2 is changed to the reaction of the cuprous complex with O2 following electron transfer from ferrocene derivatives to 2. The explanation for the change in catalytic O2-reaction stoichiometry from four-electron with Brønsted acids to two-electron reduction in the presence of Sc3+ and also for the change in the rate-determining step is clarified based on a kinetics interrogation of the overall catalytic cycle as well as each step of the catalytic cycle with study of the observed effects of Sc3+ on copper–oxygen intermediates.
Co-reporter:Pankaj Kumar; Yong-Min Lee; Young Jun Park; Maxime A. Siegler; Kenneth D. Karlin;Wonwoo Nam
Journal of the American Chemical Society 2015 Volume 137(Issue 13) pp:4284-4287
Publication Date(Web):March 20, 2015
DOI:10.1021/ja513234b
New CoIII–nitrosyl complexes bearing N-tetramethylated cyclam (TMC) ligands, [(12-TMC)CoIII(NO)]2+ (1) and [(13-TMC)CoIII(NO)]2+ (2), were synthesized via [(TMC)CoII(CH3CN)]2+ + NO(g) reactions. Spectroscopic and structural characterization showed that these compounds bind the nitrosyl moiety in a bent end-on fashion. Complexes 1 and 2 reacted with KO2/2.2.2-cryptand to produce [(12-TMC)CoII(NO2)]+ (3) and [(13-TMC)CoII(NO2)]+ (4), respectively; these possess O,O′-chelated nitrito ligands. Mechanistic studies using 18O-labeled superoxide (18O2•–) showed that one O atom in the nitrito ligand is derived from superoxide and the O2 produced comes from the other superoxide O atom. Evidence supporting the formation of a Co–peroxynitrite intermediate is also presented.
Co-reporter:Shabnam Hematian; Isabell Kenkel; Tatyana E. Shubina; Maximilian Dürr; Jeffrey J. Liu; Maxime A. Siegler; Ivana Ivanovic-Burmazovic
Journal of the American Chemical Society 2015 Volume 137(Issue 20) pp:6602-6615
Publication Date(Web):May 14, 2015
DOI:10.1021/jacs.5b02174
While nitric oxide (NO, nitrogen monoxide) is a critically important signaling agent, its cellular concentrations must be tightly controlled, generally through its oxidative conversion to nitrite (NO2–) where it is held in reserve to be reconverted as needed. In part, this reaction is mediated by the binuclear heme a3/CuB active site of cytochrome c oxidase. In this report, the oxidation of NO(g) to nitrite is shown to occur efficiently in new synthetic μ-oxo heme-FeIII–O–CuII(L) constructs (L being a tridentate or tetradentate pyridyl/alkylamino ligand), and spectroscopic and kinetic investigations provide detailed mechanistic insights. Two new X-ray structures of μ-oxo complexes have been determined and compared to literature analogs. All μ-oxo complexes react with 2 mol equiv NO(g) to give 1:1 mixtures of discrete [(L)CuII(NO2–)]+ plus ferrous heme-nitrosyl compounds; when the first NO(g) equiv reduces the heme center and itself is oxidized to nitrite, the second equiv of NO(g) traps the ferrous heme thus formed. For one μ-oxo heme-FeIII–O–CuII(L) compound, the reaction with NO(g) reveals an intermediate species (“intermediate”), formally a bis-NO adduct, [(NO)(porphyrinate)FeII–(NO2–)–CuII(L)]+ (λmax = 433 nm), confirmed by cryo-spray ionization mass spectrometry and EPR spectroscopy, along with the observation that cooling a 1:1 mixture of [(L)CuII(NO2–)]+ and heme-FeII(NO) to −125 °C leads to association and generation of the key 433 nm UV–vis feature. Kinetic-thermodynamic parameters obtained from low-temperature stopped-flow measurements are in excellent agreement with DFT calculations carried out which describe the sequential addition of NO(g) to the μ-oxo complex.
Co-reporter:Sunghee Kim; Jung Yoon Lee; Ryan E. Cowley; Jake W. Ginsbach; Maxime A. Siegler; Edward I. Solomon
Journal of the American Chemical Society 2015 Volume 137(Issue 8) pp:2796-2799
Publication Date(Web):February 20, 2015
DOI:10.1021/ja511504n
Previous efforts to synthesize a cupric superoxide complex possessing a thioether donor have resulted in the formation of an end-on trans-peroxo-dicopper(II) species, [{(Ligand)CuII}2(μ-1,2-O22–)]2+. Redesign/modification of previous N3S tetradentate ligands has now allowed for the stabilization of the monomeric, superoxide product possessing a S(thioether) ligation, [(DMAN3S)CuII(O2•–)]+ (2S), as characterized by UV–vis and resonance Raman spectroscopies. This complex mimics the putative CuII(O2•–) active species of the copper monooxygenase PHM and exhibits enhanced reactivity toward both O–H and C–H substrates in comparison to close analogues [(L)CuII(O2•–)]+, where L contains only nitrogen donor atoms. Also, comparisons of [(L)CuII/I]n+ compound reduction potentials (L = various N4 vs DMAN3S ligands) provide evidence that DMAN3S is a weaker donor to copper ion than is found for any N4 ligand-complex.
Co-reporter:Sudipta Chatterjee; Kushal Sengupta; Shabnam Hematian; Kenneth D. Karlin;Abhishek Dey
Journal of the American Chemical Society 2015 Volume 137(Issue 40) pp:12897-12905
Publication Date(Web):September 30, 2015
DOI:10.1021/jacs.5b06513
A synthetic heme–Cu CcO model complex shows selective and highly efficient electrocatalytic 4e–/4H+ O2-reduction to H2O with a large catalytic rate (>105 M–1 s–1). While the heme-Cu model (FeCu) shows almost exclusive 4e–/4H+ reduction of O2 to H2O (detected using ring disk electrochemistry and rotating ring disk electrochemistry), when imidazole is bound to the heme (Fe(Im)Cu), this same selective O2-reduction to water occurs only under slow electron fluxes. Surface enhanced resonance Raman spectroscopy coupled to dynamic electrochemistry data suggests the formation of a bridging peroxide intermediate during O2-reduction by both complexes under steady state reaction conditions, indicating that O–O bond heterolysis is likely to be the rate-determining step (RDS) at the mass transfer limited region. The O–O vibrational frequencies at 819 cm–1 in 16O2 (759 cm–1 in 18O2) for the FeCu complex and at 847 cm–1 (786 cm–1) for the Fe(Im)Cu complex, indicate the formation of side-on and end-on bridging Fe-peroxo-Cu intermediates, respectively, during O2-reduction in an aqueous environment. These data suggest that side-on bridging peroxide intermediates are involved in fast and selective O2-reduction in these synthetic complexes. The greater amount of H2O2 production by the imidazole bound complex under fast electron transfer is due to 1e–/1H+ O2-reduction by the distal Cu where O2 binding to the water bound low spin FeII complex is inhibited.
Co-reporter:Claudio Saracini; Kei Ohkubo; Tomoyoshi Suenobu; Gerald J. Meyer; Kenneth D. Karlin;Shunichi Fukuzumi
Journal of the American Chemical Society 2015 Volume 137(Issue 50) pp:15865-15874
Publication Date(Web):December 11, 2015
DOI:10.1021/jacs.5b10177
Photoexcitation of end-on trans-μ-1,2-peroxodicopper(II) complex [(tmpa)2CuII2(O2)]2+ (1) (λmax = 525 and 600 nm) and side-on μ-η2:η2-peroxodicopper(II) complexes [(N5)CuII2(O2)]2+ (2) and [(N3)CuII2(O2)]2+ (3) at −80 °C in acetone led to one-photon two-electron peroxide-to-dioxygen oxidation chemistry (O22– + hν → O2 + 2e–). Interestingly, light excitation of 2 and 3 (having side-on μ-η2:η2-peroxo ligation) led to release of dioxygen, while photoexcitation of 1 (having an end-on trans-1,2-peroxo geometry) did not, even though spectroscopic studies revealed that both reactions proceeded through previously unknown mixed-valent superoxide species: [CuII(O2•–)CuI]2+ (λmax = 685–740 nm). For 1, this intermediate underwent further fast intramolecular electron transfer to yield an “O2-caged” dicopper(I) adduct, CuI2–O2, and a barrierless stepwise back electron transfer to regenerate 1 occurred. Femtosecond laser excitation of 2 and 3 under the same conditions still led to [CuII(O2•–)CuI]2+ intermediates that, instead, underwent O2 release with a quantum yield of 0.14 ± 0.1 for 3. Such remarkable differences in reaction pathways likely result from the well-known ligand-derived stability of 2 and 3 vs 1 indicated by ligand–CuII/I redox potentials; (N5)CuI and (N3)CuI complexes are far more stable than (tmpa)CuI species. The fast CuI2/O2 rebinding kinetics was also measured after photoexcitation of 2 and 3, with the results closely tracking those known for the dicopper proteins hemocyanin and tyrosinase, for which the synthetic dicopper(I) precursors [(N5)CuI2]2+ and [(N3)CuI2]2+ and their dioxygen adducts serve as models. The biological relevance of the present findings is discussed, including the potential impact on the solar water splitting process.
Co-reporter:Ga Young Park ; Jung Yoon Lee ; Richard A. Himes ; Gnana S. Thomas ; Ninian J. Blackburn
Journal of the American Chemical Society 2014 Volume 136(Issue 36) pp:12532-12535
Publication Date(Web):August 29, 2014
DOI:10.1021/ja505098v
Oxygen-activating copper proteins may possess His-Xaa-His chelating sequences at their active sites and additionally exhibit imidiazole group δN vs εN tautomeric preferences. As shown here, such variations strongly affect copper ion’s coordination geometry, redox behavior, and oxidative reactivity. Copper(I) complexes bound to either δ-HGH or ε-HGH tripeptides were synthesized and characterized. Structural investigations using X-ray absorption spectroscopy, density functional theory calculations, and solution conductivity measurements reveal that δ-HGH forms the CuI dimer complex [{CuI(δ-HGH)}2]2+ (1) while ε-HGH binds CuI to give the monomeric complex [CuI(ε-HGH)]+ (2). Only 2 exhibits any reactivity, forming a strong CO adduct, [CuI(ε-HGH)(CO)]+, with properties closely matching those of the copper monooxygenase PHM. Also, 2 is reactive toward O2 or H2O2, giving a new type of O2-adduct or CuII–OOH complex, respectively.
Co-reporter:Sunghee Kim ; Jake W. Ginsbach ; A. Imtiaz Billah ; Maxime A. Siegler ; Cathy D. Moore ; Edward I. Solomon
Journal of the American Chemical Society 2014 Volume 136(Issue 22) pp:8063-8071
Publication Date(Web):May 22, 2014
DOI:10.1021/ja502974c
Current interest in copper/dioxygen reactivity includes the influence of thioether sulfur ligation, as it concerns the formation, structures, and properties of derived copper-dioxygen complexes. Here, we report on the chemistry of {L-CuI}2-(O2) species L = DMMESE, DMMESP, and DMMESDP, which are N3S(thioether)-based ligands varied in the nature of a substituent on the S atom, along with a related N3O(ether) (EOE) ligand. CuI and CuII complexes have been synthesized and crystallographically characterized. Copper(I) complexes are dimeric in the solid state, [{L-CuI}2](B(C6F5)4)2, however are shown by diffusion-ordered NMR spectroscopy to be mononuclear in solution. Copper(II) complexes with a general formulation [L-CuII(X)]n+ {X = ClO4–, n = 1, or X = H2O, n = 2} exhibit distorted square pyramidal coordination geometries and progressively weaker axial thioether ligation across the series. Oxygenation (−130 °C) of {(DMMESE)CuI}+ results in the formation of a trans-μ-1,2-peroxodicopper(II) species [{(DMMESE)CuII}2(μ-1,2-O22–)]2+ (1P). Weakening the Cu–S bond via a change to the thioether donor found in DMMESP leads to the initial formation of [{(DMMESP)CuII}2(μ-1,2-O22–)]2+ (2P) that subsequently isomerizes to a bis-μ-oxodicopper(III) complex, [{(DMMESP)CuIII}2(μ-O2–)2]2+ (2O), with 2P and 2O in equilibrium (Keq = [2O]/[2P] = 2.6 at −130 °C). Formulations for these Cu/O2 adducts were confirmed by resonance Raman (rR) spectroscopy. This solution mixture is sensitive to the addition of methylsulfonate, which shifts the equilibrium toward the bis-μ-oxo isomer. Further weakening of the Cu–S bond in DMMESDP or substitution with an ether donor in DMMEOE leads to only a bis-μ-oxo species (3O and 4O, respectively). Reactivity studies indicate that the bis-μ-oxodicopper(III) species (2O, 3O) and not the trans-peroxo isomers (1P and 2P) are responsible for the observed ligand sulfoxidation. Our findings concerning the existence of the 2P/2O equilibrium contrast with previously established ligand-CuI/O2 reactivity and possible implications are discussed.
Co-reporter:Jung Yoon Lee ; Ryan L. Peterson ; Kei Ohkubo ; Isaac Garcia-Bosch ; Richard A. Himes ; Julia Woertink ; Cathy D. Moore ; Edward I. Solomon ; Shunichi Fukuzumi
Journal of the American Chemical Society 2014 Volume 136(Issue 28) pp:9925-9937
Publication Date(Web):June 22, 2014
DOI:10.1021/ja503105b
To obtain mechanistic insights into the inherent reactivity patterns for copper(I)–O2 adducts, a new cupric–superoxo complex [(DMM-tmpa)CuII(O2•–)]+ (2) [DMM-tmpa = tris((4-methoxy-3,5-dimethylpyridin-2-yl)methyl)amine] has been synthesized and studied in phenol oxidation–oxygenation reactions. Compound 2 is characterized by UV–vis, resonance Raman, and EPR spectroscopies. Its reactions with a series of para-substituted 2,6-di-tert-butylphenols (p-X-DTBPs) afford 2,6-di-tert-butyl-1,4-benzoquinone (DTBQ) in up to 50% yields. Significant deuterium kinetic isotope effects and a positive correlation of second-order rate constants (k2) compared to rate constants for p-X-DTBPs plus cumylperoxyl radical reactions indicate a mechanism that involves rate-limiting hydrogen atom transfer (HAT). A weak correlation of (kBT/e) ln k2 versus Eox of p-X-DTBP indicates that the HAT reactions proceed via a partial transfer of charge rather than a complete transfer of charge in the electron transfer/proton transfer pathway. Product analyses, 18O-labeling experiments, and separate reactivity employing the 2,4,6-tri-tert-butylphenoxyl radical provide further mechanistic insights. After initial HAT, a second molar equiv of 2 couples to the phenoxyl radical initially formed, giving a CuII–OO–(ArO′) intermediate, which proceeds in the case of p-OR-DTBP substrates via a two-electron oxidation reaction involving hydrolysis steps which liberate H2O2 and the corresponding alcohol. By contrast, four-electron oxygenation (O–O cleavage) mainly occurs for p-R-DTBP which gives 18O-labeled DTBQ and elimination of the R group.
Co-reporter:Claudio Saracini ; Dimitrios G. Liakos ; Jhon E. Zapata Rivera ; Frank Neese ; Gerald J. Meyer
Journal of the American Chemical Society 2014 Volume 136(Issue 4) pp:1260-1263
Publication Date(Web):January 15, 2014
DOI:10.1021/ja4115314
Irradiation of the copper(II)–superoxide synthetic complexes [(TMG3tren)CuII(O2)]+ (1) and [(PV-TMPA)CuII(O2)]+ (2) with visible light resulted in direct photogeneration of O2 gas at low temperature (from −40 °C to −70 °C for 1 and from −125 to −135 °C for 2) in 2-methyltetrahydrofuran (MeTHF) solvent. The yield of O2 release was wavelength dependent: λexc = 436 nm, ϕ = 0.29 (for 1), ϕ = 0.11 (for 2), and λexc = 683 nm, ϕ = 0.035 (for 1), ϕ = 0.078 (for 2), which was followed by fast O2-recombination with [(TMG3tren)CuI]+ (3) and [(PV-TMPA)CuI]+ (4). Enthalpic barriers for O2 rebinding to the copper(I) center (∼10 kJ mol–1) and for O2 dissociation from the superoxide compound 1 (45 kJ mol–1) were determined. TD-DFT studies, carried out for 1, support the experimental results confirming the dissociative character of the excited states formed upon blue- or red-light laser excitation.
Co-reporter:Sunghee Kim, Maxime A. Siegler and Kenneth D. Karlin
Chemical Communications 2014 vol. 50(Issue 22) pp:2844-2846
Publication Date(Web):09 Dec 2013
DOI:10.1039/C3CC47942K
New peroxynitrite–copper chemistry ensues via addition of nitric oxide (˙NO(g)) to a CuII–hydroperoxo species. In characterizing the system, the ligand–Cu(I) complex was shown to effect a seldom observed ˙NO(g) reductive coupling reaction. Biological implications are discussed.
Co-reporter:Atsutoshi Yokoyama, Jung Eun Han, Kenneth D. Karlin and Wonwoo Nam
Chemical Communications 2014 vol. 50(Issue 14) pp:1742-1744
Publication Date(Web):13 Dec 2013
DOI:10.1039/C3CC48782B
Reaction of a nonheme iron(III)-peroxo complex, [FeIII(14-TMC)(O2)]+, with NO+, a transformation which is essentially isoelectronic with that for nitric oxide dioxygenases [Fe(III)(O2˙−) + NO], affords an iron(IV)-oxo complex, [FeIV(14-TMC)(O)]2+, and nitrogen dioxide (NO2), followed by conversion to an iron(III)-nitrato complex, [FeIII(14-TMC)(NO3)(F)]+.
Co-reporter:Dr. Matthew T. Kieber-Emmons;Jake W. Ginsbach;Dr. Patrick K. Wick;Dr. Heather R. Lucas;Dr. Matthew E. Helton;Dr. Baldo Lucchese; Masatatsu Suzuki; Andreas D. Zuberbühler; Kenneth D. Karlin; Edward I. Solomon
Angewandte Chemie International Edition 2014 Volume 53( Issue 19) pp:4935-4939
Publication Date(Web):
DOI:10.1002/anie.201402166
Abstract
Synthesis of small-molecule Cu2O2 adducts has provided insight into the related biological systems and their reactivity patterns including the interconversion of the CuII2(μ-η2:η2-peroxo) and CuIII2(μ-oxo)2 isomers. In this study, absorption spectroscopy, kinetics, and resonance Raman data show that the oxygenated product of [(BQPA)CuI]+ initially yields an “end-on peroxo” species, that subsequently converts to the thermodynamically more stable “bis-μ-oxo” isomer (Keq=3.2 at −90 °C). Calibration of density functional theory calculations to these experimental data suggest that the electrophilic reactivity previously ascribed to end-on peroxo species is in fact a result of an accessible bis-μ-oxo isomer, an electrophilic Cu2O2 isomer in contrast to the nucleophilic reactivity of binuclear CuII end-on peroxo species. This study is the first report of the interconversion of an end-on peroxo to bis-μ-oxo species in transition metal-dioxygen chemistry.
Co-reporter:Dr. Matthew T. Kieber-Emmons;Jake W. Ginsbach;Dr. Patrick K. Wick;Dr. Heather R. Lucas;Dr. Matthew E. Helton;Dr. Baldo Lucchese; Masatatsu Suzuki; Andreas D. Zuberbühler; Kenneth D. Karlin; Edward I. Solomon
Angewandte Chemie 2014 Volume 126( Issue 19) pp:5035-5039
Publication Date(Web):
DOI:10.1002/ange.201402166
Abstract
Synthesis of small-molecule Cu2O2 adducts has provided insight into the related biological systems and their reactivity patterns including the interconversion of the CuII2(μ-η2:η2-peroxo) and CuIII2(μ-oxo)2 isomers. In this study, absorption spectroscopy, kinetics, and resonance Raman data show that the oxygenated product of [(BQPA)CuI]+ initially yields an “end-on peroxo” species, that subsequently converts to the thermodynamically more stable “bis-μ-oxo” isomer (Keq=3.2 at −90 °C). Calibration of density functional theory calculations to these experimental data suggest that the electrophilic reactivity previously ascribed to end-on peroxo species is in fact a result of an accessible bis-μ-oxo isomer, an electrophilic Cu2O2 isomer in contrast to the nucleophilic reactivity of binuclear CuII end-on peroxo species. This study is the first report of the interconversion of an end-on peroxo to bis-μ-oxo species in transition metal-dioxygen chemistry.
Co-reporter:Ryan L. Peterson ; Jake W. Ginsbach ; Ryan E. Cowley ; Munzarin F. Qayyum ; Richard A. Himes ; Maxime A. Siegler ; Cathy D. Moore ; Britt Hedman ; Keith O. Hodgson ; Shunichi Fukuzumi ; Edward I. Solomon
Journal of the American Chemical Society 2013 Volume 135(Issue 44) pp:16454-16467
Publication Date(Web):October 28, 2013
DOI:10.1021/ja4065377
The protonation–reduction of a dioxygen adduct with [LCuI][B(C6F5)4], cupric superoxo complex [LCuII(O2•–)]+ (1) (L = TMG3tren (1,1,1-tris[2-[N2-(1,1,3,3-tetramethylguanidino)]ethyl]amine)) has been investigated. Trifluoroacetic acid (HOAcF) reversibly associates with the superoxo ligand in ([LCuII(O2•–)]+) in a 1:1 adduct [LCuII(O2•–)(HOAcF)]+ (2), as characterized by UV–visible, resonance Raman (rR), nuclear magnetic resonance (NMR), and X-ray absorption (XAS) spectroscopies, along with density functional theory (DFT) calculations. Chemical studies reveal that for the binding of HOAcF with 1 to give 2, Keq = 1.2 × 105 M–1 (−130 °C) and ΔHo = −6.9(7) kcal/mol, ΔSo = −26(4) cal mol–1 K–1). Vibrational (rR) data reveal a significant increase (29 cm–1) in vO–O (= 1149 cm–1) compared to that known for [LCuII(O2•–)]+ (1). Along with results obtained from XAS and DFT calculations, hydrogen bonding of HOAcF to a superoxo O-atom in 2 is established. Results from NMR spectroscopy of 2 at −120 °C in 2-methyltetrahydrofuran are also consistent with 1/HOAcF = 1:1 formulation of 2 and with this complex possessing a triplet (S = 1) ground state electronic configuration, as previously determined for 1. The pre-equilibrium acid association to 1 is followed by outer-sphere electron-transfer reduction of 2 by decamethylferrocene (Me10Fc) or octamethylferrocene (Me8Fc), leading to the products H2O2, the corresponding ferrocenium salt, and [LCuII(OAcF)]+. Second-order rate constants for electron transfer (ket) were determined to be 1365 M–1 s–1 (Me10Fc) and 225 M–1 s–1 (Me8Fc) at −80 °C. The (bio)chemical relevance of the proton-triggered reduction of the metal-bound dioxygen-derived fragment is discussed.
Co-reporter:Shunichi Fukuzumi, Kenneth D. Karlin
Coordination Chemistry Reviews 2013 Volume 257(Issue 1) pp:187-195
Publication Date(Web):1 January 2013
DOI:10.1016/j.ccr.2012.05.031
The kinetics and thermodynamics of formation of Cu(II)-superoxo (Cu–O2) complexes by the reaction of Cu(I) complexes with dioxygen (O2) and the reduction of Cu(II)-superoxo complexes to dinuclear Cu-peroxo complexes are discussed. In the former case, electron transfer from a Cu(I) complex to O2 occurs concomitantly with binding of O2−to the corresponding Cu(II) species. This is defined as an inner-sphere Cu(II) ion-coupled electron transfer process. Electron transfer from another Cu(I) complex to preformed Cu(II)-superoxo complexes also occurs concomitantly with binding of the Cu(II)-peroxo species with the Cu(II) species to produce the dinuclear Cu-peroxo (Cu2–O2) complexes. The kinetics and thermodynamics of outer-sphere electron-transfer reduction of Cu2–O2 complexes are also been discussed in light of the Marcus theory of outer-sphere electron transfer.Graphical abstractIn terms of Marcus theory of electron-transfer, the kinetics and thermodynamics of O2-reaction with copper(I) complexes are overviewed and evaluated, including for the reactions leading to the formation of copper(II)-superoxo products. Dinuclear species of three common types may then form from copper(II)-superoxo complex reaction with another ligand-copper(I) reductant. All of these reactions occur via an inner-sphere Cu(II) ion-coupled electron transfer process. A case of copper(II) superoxo complex reduction by hydride reagents is also presented. Of interest in catalytic dioxygen reduction and other chemistries, the kinetics and thermodynamics of outer-sphere electron-transfer reduction of peroxodicopper(II) or bis-μ-oxodicopper(III) adducts are also discussed.Highlights► Dioxygen binding to copper(I) complexes gives superoxo-copper(II), peroxodicopper(II) or bis-μ-oxodicopper(III) adducts. ► Electron-transfer from copper(I) occurs concomitant with superoxide anion binding to copper(II) rather than by stepwise electron-transfer followed by ligation. ► The kinetics and thermodynamics of electron-transfer reduction of various Cu2–O2 species are discussed in terms of Marcus theory. ► Electron-transfer reduction of Cu2–O2 complexes is the fastest for bis-μ-oxodicopper(III) complexes and for at least one case, electron-transfer reduction of a μ–η2:η2-peroxodicopper(II) complex occurs directly and without prior conversion to an isomeric bis-μ-oxodicopper(III) species.
Co-reporter:Dipanwita Das ; Yong-Min Lee ; Kei Ohkubo ; Wonwoo Nam ; Kenneth D. Karlin ;Shunichi Fukuzumi
Journal of the American Chemical Society 2013 Volume 135(Issue 7) pp:2825-2834
Publication Date(Web):February 8, 2013
DOI:10.1021/ja312523u
Selective two-electron plus two-proton (2e–/2H+) reduction of O2 to hydrogen peroxide by ferrocene (Fc) or 1,1′-dimethylferrocene (Me2Fc) in the presence of perchloric acid is catalyzed efficiently by a mononuclear copper(II) complex, [CuII(tepa)]2+ (1; tepa = tris[2-(2-pyridyl)ethyl]amine) in acetone. The E1/2 value for [CuII(tepa)]2+ as measured by cyclic voltammetry is 0.07 V vs Fc/Fc+ in acetone, being significantly positive, which makes it possible to use relatively weak one-electron reductants such as Fc and Me2Fc for the overall two-electron reduction of O2. Fast electron transfer from Fc or Me2Fc to 1 affords the corresponding CuI complex [CuI(tepa)]+ (2), which reacts at low temperature (193 K) with O2, however only in the presence of HClO4, to afford the hydroperoxo complex [CuII(tepa)(OOH)]+ (3). A detailed kinetic study on the homogeneous catalytic system reveals the rate-determining step to be the O2-binding process in the presence of HClO4 at lower temperature as well as at room temperature. The O2-binding kinetics in the presence of HClO4 were studied, demonstrating that the rate of formation of the hydroperoxo complex 3 as well as the overall catalytic reaction remained virtually the same with changing temperature. The apparent lack of activation energy for the catalytic two-electron reduction of O2 is shown to result from the existence of a pre-equilibrium between 2 and O2 prior to the formation of the hydroperoxo complex 3. No further reduction of [CuII(tepa)(OOH)]+ (3) by Fc or Me2Fc occurred, and instead 3 is protonated by HClO4 to yield H2O2 accompanied by regeneration of 1, thus completing the catalytic cycle for the two-electron reduction of O2 by Fc or Me2Fc.
Co-reporter:Dipanwita Das ; Yong-Min Lee ; Kei Ohkubo ; Wonwoo Nam ; Kenneth D. Karlin ;Shunichi Fukuzumi
Journal of the American Chemical Society 2013 Volume 135(Issue 10) pp:4018-4026
Publication Date(Web):February 26, 2013
DOI:10.1021/ja312256u
Catalytic four-electron reduction of O2 by ferrocene (Fc) and 1,1′-dimethylferrocene (Me2Fc) occurs efficiently with a dinuclear copper(II) complex [CuII2(XYLO)(OH)]2+ (1), where XYLO is a m-xylene-linked bis[(2-(2-pyridyl)ethyl)amine] dinucleating ligand with copper-bridging phenolate moiety], in the presence of perchloric acid (HClO4) in acetone at 298 K. The hydroxide and phenoxo group in [CuII2(XYLO)(OH)]2+ (1) undergo protonation with HClO4 to produce [CuII2(XYLOH)]4+ (2) where the two copper centers become independent and the reduction potential shifts from −0.68 V vs SCE in the absence of HClO4 to 0.47 V; this makes possible the use of relatively weak one-electron reductants such as Fc and Me2Fc, significantly reducing the effective overpotential in the catalytic O2-reduction reaction. The mechanism of the reaction has been clarified on the basis of kinetic studies on the overall catalytic reaction as well as each step in the catalytic cycle and also by low-temperature detection of intermediates. The O2-binding to the fully reduced complex [CuI2(XYLOH)]2+ (3) results in the reversible formation of the hydroperoxo complex ([CuII2(XYLO)(OOH)]2+) (4), followed by proton-coupled electron-transfer (PCET) reduction to complete the overall O2-to-2H2O catalytic conversion.
Co-reporter:Saya Kakuda ; Ryan L. Peterson ; Kei Ohkubo ; Kenneth D. Karlin ;Shunichi Fukuzumi
Journal of the American Chemical Society 2013 Volume 135(Issue 17) pp:6513-6522
Publication Date(Web):March 20, 2013
DOI:10.1021/ja3125977
A copper complex, [(PV-tmpa)CuII](ClO4)2 (1) [PV-tmpa = bis(pyrid-2-ylmethyl){[6-(pivalamido)pyrid-2-yl]methyl}amine], acts as a more efficient catalyst for the four-electron reduction of O2 by decamethylferrocene (Fc*) in the presence of trifluoroacetic acid (CF3COOH) in acetone as compared with the corresponding copper complex without a pivalamido group, [(tmpa)CuII](ClO4)2 (2) (tmpa = tris(2-pyridylmethyl)amine). The rate constant (kobs) of formation of decamethylferrocenium ion (Fc*+) in the catalytic four-electron reduction of O2 by Fc* in the presence of a large excess CF3COOH and O2 obeyed first-order kinetics. The kobs value was proportional to the concentration of catalyst 1 or 2, whereas the kobs value remained constant irrespective of the concentration of CF3COOH or O2. This indicates that electron transfer from Fc* to 1 or 2 is the rate-determining step in the catalytic cycle of the four-electron reduction of O2 by Fc* in the presence of CF3COOH. The second-order catalytic rate constant (kcat) for 1 is 4 times larger than the corresponding value determined for 2. With the pivalamido group in 1 compared to 2, the CuII/CuI potentials are –0.23 and –0.05 V vs SCE, respectively. However, during catalytic turnover, the CF3COO– anion present readily binds to 2 shifting the resulting complex’s redox potential to –0.35 V. The pivalamido group in 1 is found to inhibit anion binding. The overall effect is to make 1 easier to reduce (relative to 2) during catalysis, accounting for the relative kcat values observed. 1 is also an excellent catalyst for the two-electron two-proton reduction of H2O2 to water and is also more efficient than is 2. For both complexes, reaction rates are greater than for the overall four-electron O2-reduction to water, an important asset in the design of catalysts for the latter.
Co-reporter:Atsutoshi Yokoyama ; Kyung-Bin Cho ; Kenneth D. Karlin ;Wonwoo Nam
Journal of the American Chemical Society 2013 Volume 135(Issue 40) pp:14900-14903
Publication Date(Web):September 25, 2013
DOI:10.1021/ja405891n
The reaction of an end-on Cr(III)-superoxo complex bearing a 14-membered tetraazamacrocyclic TMC ligand, [CrIII(14-TMC)(O2)(Cl)]+, with nitric oxide (NO) resulted in the generation of a stable Cr(IV)-oxo species, [CrIV(14-TMC)(O)(Cl)]+, via the formation of a Cr(III)-peroxynitrite intermediate and homolytic O–O bond cleavage of the peroxynitrite ligand. Evidence for the latter comes from electron paramagnetic resonance spectroscopy, computational chemistry and the observation of phenol nitration chemistry. The Cr(IV)-oxo complex does not react with nitrogen dioxide (NO2), but reacts with NO to afford a Cr(III)-nitrito complex, [CrIII(14-TMC)(NO2)(Cl)]+. The Cr(IV)-oxo and Cr(III)-nitrito complexes were also characterized spectroscopically and/or structurally.
Co-reporter:Isaac Garcia-Bosch ; Savita K. Sharma
Journal of the American Chemical Society 2013 Volume 135(Issue 44) pp:16248-16251
Publication Date(Web):October 22, 2013
DOI:10.1021/ja405739m
The selective oxidation of the α-position of two heme-FeIII tetraarylporphryinate complexes occurs when water(hydroxide) attacks their oxidized Cmpd I-type equivalents, high-valent FeIV═O π-cation radical species ((P+•)FeIV═O). Stepwise intermediate formation occurs, as detected by UV–vis spectroscopic monitoring or mass spectrometric interrogation, being iron(III) isoporphyrins, iron(III) benzoyl-biliverdins, and the final verdoheme-like products. Heme oxygenase (HO) enzymes could proceed through heterolytic cleavage of an iron(III)-hydroperoxo intermediate to form a transient Cmpd I-type species.
Co-reporter:Jake W. Ginsbach ; Ryan L. Peterson ; Ryan E. Cowley ; Kenneth D. Karlin ;Edward I. Solomon
Inorganic Chemistry 2013 Volume 52(Issue 22) pp:12872-12874
Publication Date(Web):October 28, 2013
DOI:10.1021/ic402357u
The geometry of mononuclear copper(II) superoxide complexes has been shown to determine their ground state where side-on bonding leads to a singlet ground state and end-on complexes have triplet ground states. In an apparent contrast to this trend, the recently synthesized (HIPT3tren)CuIIO2•– (1) was proposed to have an end-on geometry and a singlet ground state. However, reexamination of 1 with resonance Raman, magnetic circular dichroism, and 2H NMR spectroscopies indicate that 1 is, in fact, an end-on superoxide species with a triplet ground state that results from the single CuIIO2•– bonding interaction being weaker than the spin-pairing energy.
Co-reporter:Yuqi Li, Savita K. Sharma, Kenneth D. Karlin
Polyhedron 2013 Volume 58() pp:190-196
Publication Date(Web):13 July 2013
DOI:10.1016/j.poly.2012.11.011
Inspired by the chemistry relevant to dioxygen storage, transport and activation by metalloproteins, in particular for heme/copper oxidases, and carbon monoxide binding to metal-containing active sites as a probe or surrogate for dioxygen binding, a series of heme derived dioxygen and CO complexes have been designed, synthesized, and characterized with respect to their physical properties and reactivity. The focus of this study is in the description and comparison of three types heme–superoxo and heme–CO adducts. The starting point is in the characterization of the reduced heme complexes, [(F8)FeII], [(PPy)FeII] and [(PIm)FeII], where F8, PPy and PIm are iron(II)–porphyrinates and where PPy and PIm possess a covalently tethered axial base pyridyl or imidazolyl group, respectively. The spin-state properties of these complexes vary with solvent. The low temperature reactions between O2 and these reduced porphyrin FeII complexes yield distinctive low spin heme–superoxo adducts. The dioxygen binding properties for all three complexes are shown to be reversible, via alternate argon or O2 bubbling. Carbon monoxide binds to the reduced heme–FeII precursors to form low spin heme–CO adducts. The implications for future investigations of these heme O2 and CO adducts are discussed.Graphical abstractInspired by the chemistry relevant to dioxygen processing by metalloproteins, in particular for heme/copper oxidases, a series of heme derived dioxygen and CO complexes have been designed, synthesized, and characterized with respect to their physical properties and reactivity. The focus of this study is the description and comparison of three types of heme–superoxo and heme–CO adducts. Where the porphyrinates are one without an axial base covalently attached, while the others possess a covalently tethered axial base pyridyl or imidazolyl group.Highlights► New pyridyl or imidazolyl tethered heme–FeII, heme–FeIII–superoxo and heme–FeII–CO complexes have been generated and characterized. ► The new heme–Fe–superoxo and heme–Fe–CO complexes are determined to be six coordinate low spin complexes by using 1H or 2H NMR spectroscopy. ► Dioxygen (O2) binding to reduced base tethered heme–FeII complexes is reversible.
Co-reporter:Shunichi Fukuzumi ; Laleh Tahsini ; Yong-Min Lee ; Kei Ohkubo ; Wonwoo Nam
Journal of the American Chemical Society 2012 Volume 134(Issue 16) pp:7025-7035
Publication Date(Web):March 30, 2012
DOI:10.1021/ja211656g
The selective two-electron reduction of O2 by one-electron reductants such as decamethylferrocene (Fc*) and octamethylferrocene (Me8Fc) is efficiently catalyzed by a binuclear Cu(II) complex [CuII2(LO)(OH)]2+ (D1) {LO is a binucleating ligand with copper-bridging phenolate moiety} in the presence of trifluoroacetic acid (HOTF) in acetone. The protonation of the hydroxide group of [CuII2(LO)(OH)]2+ with HOTF to produce [CuII2(LO)(OTF)]2+ (D1-OTF) makes it possible for this to be reduced by 2 equiv of Fc* via a two-step electron-transfer sequence. Reactions of the fully reduced complex [CuI2(LO)]+ (D3) with O2 in the presence of HOTF led to the low-temperature detection of the absorption spectra due to the peroxo complex [CuII2(LO)(OO)] (D) and the protonated hydroperoxo complex [CuII2(LO)(OOH)]2+ (D4). No further Fc* reduction of D4 occurs, and it is instead further protonated by HOTF to yield H2O2 accompanied by regeneration of [CuII2(LO)(OTF)]2+ (D1-OTF), thus completing the catalytic cycle for the two-electron reduction of O2 by Fc*. Kinetic studies on the formation of Fc*+ under catalytic conditions as well as for separate examination of the electron transfer from Fc* to D1-OTF reveal there are two important reaction pathways operating. One is a rate-determining second reduction of D1-OTF, thus electron transfer from Fc* to a mixed-valent intermediate [CuIICuI(LO)]2+ (D2), which leads to [CuI2(LO)]+ that is coupled with O2 binding to produce [CuII2(LO)(OO)]+ (D). The other involves direct reaction of O2 with the mixed-valent compound D2 followed by rapid Fc* reduction of a putative superoxo-dicopper(II) species thus formed, producing D.
Co-reporter:Ga Young Park ; Munzarin F. Qayyum ; Julia Woertink ; Keith O. Hodgson ; Britt Hedman ; Amy A. Narducci Sarjeant ; Edward I. Solomon
Journal of the American Chemical Society 2012 Volume 134(Issue 20) pp:8513-8524
Publication Date(Web):May 9, 2012
DOI:10.1021/ja300674m
Certain side-on peroxo-dicopper(II) species with particularly low νO–O (710–730 cm–1) have been found in equilibrium with their bis-μ-oxo-dicopper(III) isomer. An issue is whether such side-on peroxo bridges are further activated for O–O cleavage. In a previous study (Liang, H.-C., et al. J. Am. Chem. Soc.2002, 124, 4170), we showed that oxygenation of the three-coordinate complex [CuI(MeAN)]+ (MeAN = N-methyl-N,N-bis[3-(dimethylamino)propyl]amine) leads to a low-temperature stable [{CuII(MeAN)}2(μ-η2:η2-O22–)]2+ peroxo species with low νO–O (721 cm–1), as characterized by UV–vis absorption and resonance Raman (rR) spectroscopies. Here, this complex has been crystallized as its SbF6– salt, and an X-ray structure indicates the presence of an unusually long O–O bond (1.540(5) Å) consistent with the low νO–O. Extended X-ray absorption fine structure and rR spectroscopic and reactivity studies indicate the exclusive formation of [{CuII(MeAN)}2(μ-η2:η2-O22–)]2+ without any bis-μ-oxo-dicopper(III) isomer present. This is the first structure of a side-on peroxo-dicopper(II) species with a significantly long and weak O–O bond. DFT calculations show that the weak O–O bond results from strong σ donation from the MeAN ligand to Cu that is compensated by a decrease in the extent of peroxo to Cu charge transfer. Importantly, the weak O–O bond does not reflect an increase in backbonding into the σ* orbital of the peroxide. Thus, although the O–O bond is unusually weak, this structure is not further activated for reductive cleavage to form a reactive bis-μ-oxo dicopper(III) species. These results highlight the necessity of understanding electronic structure changes associated with spectral changes for correlations to reactivity.
Co-reporter:Atsutoshi Yokoyama ; Jung Eun Han ; Jaeheung Cho ; Minoru Kubo ; Takashi Ogura ; Maxime A. Siegler ; Kenneth D. Karlin ;Wonwoo Nam
Journal of the American Chemical Society 2012 Volume 134(Issue 37) pp:15269-15272
Publication Date(Web):September 5, 2012
DOI:10.1021/ja307384e
The O2 and NO reactivity of a Cr(II) complex bearing a 12-membered tetraazamacrocyclic N-tetramethylated cyclam (TMC) ligand, [CrII(12-TMC)(Cl)]+ (1), and the NO reactivity of its peroxo derivative, [CrIV(12-TMC)(O2)(Cl)]+ (2), are described. By contrast to the previously reported Cr(III)–superoxo complex, [CrIII(14-TMC)(O2)(Cl)]+, the Cr(IV)–peroxo complex 2 is formed in the reaction of 1 and O2. Full spectroscopic and X-ray analysis revealed that 2 possesses side-on η2-peroxo ligation. The quantitative reaction of 2 with NO affords a reduction in Cr oxidation state, producing a Cr(III)–nitrato complex, [CrIII(12-TMC)(NO3)(Cl)]+ (3). The latter is suggested to form via a Cr(III)–peroxynitrite intermediate. [CrII(12-TMC)(NO)(Cl)]+ (4), a Cr(II)–nitrosyl complex derived from 1 and NO, could also be synthesized; however, it does not react with O2.
Co-reporter:Shabnam Hematian ; Maxime A. Siegler
Journal of the American Chemical Society 2012 Volume 134(Issue 46) pp:18912-18915
Publication Date(Web):November 6, 2012
DOI:10.1021/ja3083818
The hemea3/CuB active site of cytochrome c oxidase is responsible for cellular nitrite reduction to nitric oxide; the same center can return NO to the nitrite pool via oxidative chemistry. Here, we show that a partially reduced heme/Cu assembly reduces NO2– ion, producing nitric oxide. The heme serves as the reductant, but the CuII ion is also required. In turn, a μ-oxo heme-FeIII–O–CuII complex facilitates NO oxidation to nitrite; the final products are the reduced heme and CuII–nitrito complexes.
Co-reporter:Shunichi Fukuzumi, Yusuke Yamada, Kenneth D. Karlin
Electrochimica Acta 2012 Volume 82() pp:493-511
Publication Date(Web):1 November 2012
DOI:10.1016/j.electacta.2012.03.132
This review describes homogeneous and heterogeneous catalytic reduction of dioxygen with metal complexes focusing on the catalytic two-electron reduction of dioxygen to produce hydrogen peroxide. Whether two-electron reduction of dioxygen to produce hydrogen peroxide or four-electron O2-reduction to produce water occurs depends on the types of metals and ligands that are utilized. Those factors controlling the two processes are discussed in terms of metal–oxygen intermediates involved in the catalysis. Metal complexes acting as catalysts for selective two-electron reduction of oxygen can be utilized as metal complex-modified electrodes in the electrocatalytic reduction to produce hydrogen peroxide. Hydrogen peroxide thus produced can be used as a fuel in a hydrogen peroxide fuel cell. A hydrogen peroxide fuel cell can be operated with a one-compartment structure without a membrane, which is certainly more promising for the development of low-cost fuel cells as compared with two compartment hydrogen fuel cells that require membranes. Hydrogen peroxide is regarded as an environmentally benign energy carrier because it can be produced by the electrocatalytic two-electron reduction of O2, which is abundant in air, using solar cells; the hydrogen peroxide thus produced could then be readily stored and then used as needed to generate electricity through the use of hydrogen peroxide fuel cells.
Co-reporter:Sunghee Kim, Claudio Saracini, Maxime A. Siegler, Natalia Drichko, and Kenneth D. Karlin
Inorganic Chemistry 2012 Volume 51(Issue 23) pp:12603-12605
Publication Date(Web):November 15, 2012
DOI:10.1021/ic302071e
At −90 °C in acetone, a stable hydroperoxo complex [(BA)CuIIOOH]+ (2) (BA, a tetradentate N4 ligand possessing a pendant −N(H)CH2C6H5 group) is generated by reacting [(BA)CuII(CH3COCH3)]2+ with only 1 equiv of H2O2/Et3N. The exceptional stability of 2 is ascribed to internal H-bonding. Species 2 is also generated in a manner not previously known in copper chemistry, by adding 1.5 equiv of H2O2 (no base) to the cuprous complex [(BA)CuI]+. The broad implications for this finding are discussed. Species 2 slowly converts to a μ-1,2-peroxodicopper(II) analogue (3) characterized by UV–vis and resonance Raman spectroscopies. Unlike a close analogue not possessing internal H-bonding, 2 affords no oxidative reactivity with internal or external substrates. However, 2 can be protonated to release H2O2, but only with HClO4, while 1 equiv Et3N restores 2.
Co-reporter:Yusuke Yamada, Kazuki Maeda, Kei Ohkubo, Kenneth D. Karlin and Shunichi Fukuzumi
Physical Chemistry Chemical Physics 2012 vol. 14(Issue 27) pp:9654-9659
Publication Date(Web):14 May 2012
DOI:10.1039/C2CP41207A
The catalytic durability of an organic photocatalyst, 9-mesityl-10-methyl acridinium ion (Acr+–Mes), has been dramatically improved by the addition of [{tris(2-pyridylmethyl)amine}CuII](ClO4)2 ([(tmpa)CuII]2+) in the photocatalytic oxygenation of p-xylene by molecular oxygen in acetonitrile. Such an improvement is not observed by the addition of Cu(ClO4)2 in the absence of organic ligands. The addition of [(tmpa)Cu]2+ in the reaction solution resulted in more than an 11 times higher turnover number (TON) compared with the TON obtained without [(tmpa)CuII]2+. In the photocatalytic oxygenation, a stoichiometric amount of H2O2 formation was observed in the absence of [(tmpa)CuII]2+, however, much less H2O2 formation was observed in the presence of [(tmpa)CuII]2+. The photocatalytic mechanism was investigated by laser flash photolysis measurements in order to detect intermediates. The reaction of O2˙− with [(tmpa)CuII]2+ monitored by UV-vis spectroscopy in propionitrile at 203 K suggested formation of [{(tmpa)CuII}2O2]2+, a transformation which is crucial for the overall 4-electron reduction of molecular O2 to water, and a key in the observed improvement in the catalytic durability of Acr+–Mes.
Co-reporter:Reza A. Ghiladi, Arnold L. Rheingold, Maxime A. Siegler, Kenneth D. Karlin
Inorganica Chimica Acta 2012 Volume 389() pp:131-137
Publication Date(Web):1 July 2012
DOI:10.1016/j.ica.2012.02.020
This report describes our approach towards modeling the copper cluster active sites of nitrous oxide reductase and the multicopper oxidases/oxygenases. We have synthesized two mesitylene-based trinucleating ligands, MesPY1 and MesPY2, which employ bis(2-picolyl)amine (PY1) and bis(2-pyridylethyl)amine (PY2) tridentate copper chelates, respectively. Addition of cuprous salts to these ligands leads to the isolation of tricopper(I) complexes [(Mes-PY1)CuI3(CH3CN)3](ClO4)3·0.25Et2O (1) and [(Mes-PY2)CuI3](PF6)3 (3). Each of the three copper centers in 1 is most likely four-coordinate, with ligated acetonitrile as the fourth ligand; by contrast, the copper centers in 3 are three-coordinate, as determined by X-ray crystallography. The synthesis of [(Mes-PY1)CuII3(CH3CN)2(CH3OH)2](ClO4)6·(CH3OH) (2) was accomplished by addition of three equivalents of the copper(II) salt, Cu(ClO4)2·6H2O, to the ligand. The structure of 2 shows that two of the copper centers are tetracoordinate (with MeCN solvent ligation), but have additional weak axial (fifth ligand) interactions with the perchlorate anions; the third copper is unique in that it is coordinated by two MeOH solvent molecules, making it overall five-coordinate. For complexes 2 and 3, one copper ion center is located on the opposite side of the mesitylene plane as the other two. These observations, although in the solid state, must be taken into account for future studies where intramolecular tricopper(I)/O2 (or other small molecules of interest) interactions in solution are desirable.Graphical abstractSynthetic bioinorganic modeling of copper ion clusters is important due to their functioning in both dioxygen activation and nitrogen oxide reduction. Here mesitylene based trinucleating ligands have been designed, synthesized and both copper(I) and copper(II) complexes generated. These are [(Mes-PY1)CuI3(CH3CN)3]3+ (1), [(Mes-PY1)CuII3(CH3CN)2(CH3OH)2]6+ (2) and [(Mes-PY2)CuI3]3+ (3), where bis(2-picolyl)amine (PY1) and bis(2-pyridylethyl)amine (PY2) are the tridentate chelates involved. X-ray structures of 2 and 3 are described.Highlights► Mesitylene can be derivatized to give trinucleating ligands for copper ion complexes. ► Trinuclear copper(I) and copper(II) complexes have been synthesized. ► X-ray structures of two tricopper complexes are reported.
Co-reporter:Kenneth D. Karlin, Christiana Xin Zhang, Arnold L. Rheingold, Benedikt Galliker, Susan Kaderli, Andreas D. Zuberbühler
Inorganica Chimica Acta 2012 Volume 389() pp:138-150
Publication Date(Web):1 July 2012
DOI:10.1016/j.ica.2012.01.042
Copper–dioxygen interactions are of intrinsic importance in a wide range of biological and industrial processes. Here, we present detailed kinetic/thermodynamic studies on the O2-binding and arene hydroxylation reactions of a series of xylyl-bridged binuclear copper(I) complexes, where the effects of ligand electronic and structural elements on these reactions are investigated. Ligand 4-pyridyl substituents influence the reversible formation of side-on bound μ-η2:η2-peroxodicopper(II) complexes, with stronger donors leading to more rapid formation and greater thermodynamic stability of product complexes [CuII2(RXYL)(O22−)]2+. An interaction of the latter with the xylyl π-system is indicated. Subsequent peroxo electrophilic attack on the arene leads to C–H activation and oxygenation with hydroxylated products [CuII2(RXYLO–)(−OH)]2+ being formed. A related unsymmetrical binucleating ligand was also employed. Its corresponding O2-adduct [CuII2(UN)(O22−)]2+ is more stable, but primarily because the subsequent decay by hydroxylation is in a relative sense slower. The study emphasizes how ligand electronic effects can and do influence and tune copper(I)–dioxygen complex formation and subsequent reactivity.Graphical abstractBinuclear copper(I) complexes react with dioxygen to give adducts, which effect arene hydroxylation reactions. The kinetics and thermodynamics are studied and presented.Highlights► Low-temperature stopped-flow kinetics provide insights into copper(I)–O2 chemistry. ► Dicopper(I)–O2 reactions leads to Cu2O2 formation and arene substrate hydroxylation. ► Ligand 4-pyridyl substituents tune copper(I)–O2 kinetics–thermodynamics. ► An unsymmetrical binucleating ligand generates altered CuI2–O2 reactivity behavior.
Co-reporter:Dr. Laleh Tahsini;Dr. Hiroaki Kotani;Dr. Yong-Min Lee;Dr. Jaeheung Cho;Dr. Wonwoo Nam;Dr. Kenneth D. Karlin;Dr. Shunichi Fukuzumi
Chemistry - A European Journal 2012 Volume 18( Issue 4) pp:1084-1093
Publication Date(Web):
DOI:10.1002/chem.201103215
Abstract
The four-electron reduction of dioxygen by decamethylferrocene (Fc*) to water is efficiently catalyzed by a binuclear copper(II) complex (1) and a mononuclear copper(II) complex (2) in the presence of trifluoroacetic acid in acetone at 298 K. Fast electron transfer from Fc* to 1 and 2 affords the corresponding CuI complexes, which react at low temperature (193 K) with dioxygen to afford the η2:η2-peroxo dicopper(II) (3) and bis-μ-oxo dicopper(III) (4) intermediates, respectively. The rate constants for electron transfer from Fc* and octamethylferrocene (Me8Fc) to 1 as well as electron transfer from Fc* and Me8Fc to 3 were determined at various temperatures, leading to activation enthalpies and entropies. The activation entropies of electron transfer from Fc* and Me8Fc to 1 were determined to be close to zero, as expected for outer-sphere electron-transfer reactions without formation of any intermediates. For electron transfer from Fc* and Me8Fc to 3, the activation entropies were also found to be close to zero. Such agreement indicates that the η2:η2-peroxo complex (3) is directly reduced by Fc* rather than via the conversion to the corresponding bis-μ-oxo complex, followed by the electron-transfer reduction by Fc* leading to the four-electron reduction of dioxygen to water. The bis-μ-oxo species (4) is reduced by Fc* with a much faster rate than the η2:η2-peroxo complex (3), but this also leads to the four-electron reduction of dioxygen to water.
Co-reporter:Dr. Matthew T. Kieber-Emmons;Munzarin F. Qayyum;Yuqi Li;Dr. Zakaria Halime; Keith O. Hodgson; Britt Hedman; Kenneth D. Karlin; Edward I. Solomon
Angewandte Chemie 2012 Volume 124( Issue 1) pp:172-176
Publication Date(Web):
DOI:10.1002/ange.201104080
Co-reporter:Dr. Matthew T. Kieber-Emmons;Munzarin F. Qayyum;Yuqi Li;Dr. Zakaria Halime; Keith O. Hodgson; Britt Hedman; Kenneth D. Karlin; Edward I. Solomon
Angewandte Chemie International Edition 2012 Volume 51( Issue 1) pp:168-172
Publication Date(Web):
DOI:10.1002/anie.201104080
Co-reporter:Akinori Itoh;Shunichi Fukuzumi;Kei Ohkubo;Kaoru Doi;Tomoyoshi Suenobu;Yusuke Yamada
PNAS 2012 Volume 109 (Issue 39 ) pp:
Publication Date(Web):2012-09-25
DOI:10.1073/pnas.1119994109
A simple donor-acceptor linked dyad, 9-mesityl-10-methylacridinium ion (Acr+-Mes) was incorporated into nanosized mesoporous silica-alumina to form a composite, which in acetonitrile is highly dispersed.
In this medium, upon visible light irradiation, the formation of an extremely long-lived electron-transfer state (Acr•-Mes•+) was confirmed by EPR and laser flash photolysis spectroscopic methods. The composite of Acr+-Mes-incorporated mesoporous silica-alumina with an added copper complex [(tmpa)CuII] (tmpa = tris(2-pyridylmethyl)amine) acts as an efficient and robust photocatalyst for the selective oxygenation of p-xylene by molecular oxygen to produce p-tolualdehyde and hydrogen peroxide. Thus, incorporation of Acr+-Mes into nanosized mesoporous silica-alumina combined with an O2-reduction catalyst ([(tmpa)CuII]2+) provides a promising method in the development of efficient and robust organic photocatalysts for substrate oxygenation
by dioxygen, the ultimate environmentally benign oxidant.
Co-reporter:Ryan L. Peterson ; Richard A. Himes ; Hiroaki Kotani ; Tomoyoshi Suenobu ; Li Tian ; Maxime A. Siegler ; Edward I. Solomon ; Shunichi Fukuzumi
Journal of the American Chemical Society 2011 Volume 133(Issue 6) pp:1702-1705
Publication Date(Web):January 25, 2011
DOI:10.1021/ja110466q
The new cupric superoxo complex [LCuII(O2•−)]+, which possesses particularly strong O−O and Cu−O bonding, is capable of intermolecular C−H activation of the NADH analogue 1-benzyl-1,4-dihydronicotinamide (BNAH). Kinetic studies indicated a first-order dependence on both the Cu complex and BNAH with a deuterium kinetic isotope effect (KIE) of 12.1, similar to that observed for certain copper monooxygenases.
Co-reporter:Zakaria Halime;Hiroaki Kotani;Yuqi Li;Shunichi Fukuzumi
PNAS 2011 Volume 108 (Issue 34 ) pp:
Publication Date(Web):2011-08-23
DOI:10.1073/pnas.1104698108
An efficient and selective four-electron plus four-proton (4e-/4H+) reduction of O2 to water by decamethylferrocene and trifluoroacetic acid can be catalyzed by a synthetic analog of the heme a3/CuB site in cytochrome c oxidase (6LFeCu) or its Cu-free version (6LFe) in acetone. A detailed mechanistic-kinetic study on the homogeneous catalytic system reveals spectroscopically detectable
intermediates and that the rate-determining step changes from the O2-binding process at 25 °C room temperature (RT) to the O-O bond cleavage of a newly observed FeIII-OOH species at lower temperature (-60 °C). At RT, the rate of O2-binding to 6LFeCu is significantly faster than that for 6LFe, whereas the rates of the O-O bond cleavage of the FeIII-OOH species observed (-60 °C) with either the 6LFeCu or 6LFe catalyst are nearly the same. Thus, the role of the Cu ion is to assist the heme and lead to faster O2-binding at RT. However, the proximate Cu ion has no effect on the O-O bond cleavage of the FeIII-OOH species at low temperature.
Co-reporter:Shunichi Fukuzumi ; Hiroaki Kotani ; Heather R. Lucas ; Kaoru Doi ; Tomoyoshi Suenobu ; Ryan L. Peterson
Journal of the American Chemical Society 2010 Volume 132(Issue 20) pp:6874-6875
Publication Date(Web):May 5, 2010
DOI:10.1021/ja100538x
A mononuclear CuII complex acts as an efficient catalyst for four-electron reduction of O2 to H2O. Its reduction by a ferrocene derivative (Fc*) and reaction with O2 leads to the formation of a peroxodicopper(II) complex; this is subsequently reduced by Fc* in the presence of protons to regenerate the CuII complex.
Co-reporter:Ga Young Park, Yunho Lee, Dong-Heon Lee, Julia S. Woertink, Amy A. Narducci Sarjeant, Edward I. Solomon and Kenneth D. Karlin
Chemical Communications 2010 vol. 46(Issue 1) pp:91-93
Publication Date(Web):04 Nov 2009
DOI:10.1039/B918616F
[(ANS)CuI(CH3CN)]+ reacts with O2 giving [{(ANS)CuII}2(μ-η2:η2-O22−)]2+, νO–O = 731 cm−1, shown to possess S-thioether ligation, based on comparisons with analogues having all N-ligands or a –S(Ph) group. The finding is a rare occurrence and new for side-on O22− binding.
Co-reporter:Jun Wang ; Mark P. Schopfer ; Simona C. Puiu ; Amy A. N. Sarjeant
Inorganic Chemistry 2010 Volume 49(Issue 4) pp:1404-1419
Publication Date(Web):December 23, 2009
DOI:10.1021/ic901431r
The interactions of nitrogen monoxide (•NO; nitric oxide) with transition metal centers continue to be of great interest, in part due to their importance in biochemical processes. Here, we describe •NO(g) reductive coupling chemistry of possible relevance to that process (i.e., nitric oxide reductase (NOR) biochemistry), which occurs at the heme/Cu active site of cytochrome c oxidases (CcOs). In this report, heme/Cu/•NO(g) activity is studied using 1:1 ratios of heme and copper complex components, (F8)Fe (F8 = tetrakis(2,6-difluorophenyl)porphyrinate(2-)) and [(tmpa)CuI(MeCN)]+ (TMPA = tris(2-pyridylmethyl)amine). The starting point for heme chemistry is the mononitrosyl complex (F8)Fe(NO) (λmax = 399 (Soret), 541 nm in acetone). Variable-temperature 1H and 2H NMR spectra reveal a broad peak at δ = 6.05 ppm (pyrrole) at room temperature (RT), which gives rise to asymmetrically split pyrrole peaks at 9.12 and 8.54 ppm at −80 °C. A new heme dinitrosyl species, (F8)Fe(NO)2, obtained by bubbling (F8)Fe(NO) with •NO(g) at −80 °C, could be reversibly formed, as monitored by UV−vis (λmax = 426 (Soret), 538 nm in acetone), EPR (silent), and NMR spectroscopies; that is, the mono-NO complex was regenerated upon warming to RT. (F8)Fe(NO)2 reacts with [(tmpa)CuI(MeCN)]+ and 2 equiv of acid to give [(F8)FeIII]+, [(tmpa)CuII(solvent)]2+, and N2O(g), fitting the stoichiometric •NO(g) reductive coupling reaction: 2•NO(g) + FeII + CuI + 2H+ → N2O(g) + FeIII + CuII + H2O, equivalent to one enzyme turnover. Control reaction chemistry shows that both iron and copper centers are required for the NOR-type chemistry observed and that, if acid is not present, half the •NO is trapped as a (F8)Fe(NO) complex, while the remaining nitrogen monoxide undergoes copper complex promoted disproportionation chemistry. As part of this study, [(F8)FeIII]SbF6 was synthesized and characterized by X-ray crystallography, along with EPR (77 K: g = 5.84 and 6.12 in CH2Cl2 and THF, respectively) and variable-temperature NMR spectroscopies. These structural and physical properties suggest that at RT this complex consists of an admixture of high and intermediate spin states.
Co-reporter:Zakaria Halime ; Matthew T. Kieber-Emmons ; Munzarin F. Qayyum ; Biplab Mondal ; Thirumanavelan Gandhi ; Simona C. Puiu ; Eduardo E. Chufán ; Amy A. N. Sarjeant ; Keith O. Hodgson ; Britt Hedman ; Edward I. Solomon
Inorganic Chemistry 2010 Volume 49(Issue 8) pp:3629-3645
Publication Date(Web):April 12, 2010
DOI:10.1021/ic9020993
The nature of the ligand is an important aspect of controlling the structure and reactivity in coordination chemistry. In connection with our study of heme−copper−oxygen reactivity relevant to cytochrome c oxidase dioxygen-reduction chemistry, we compare the molecular and electronic structures of two high-spin heme−peroxo−copper [FeIIIO22−CuII]+ complexes containing N4 tetradentate (1) or N3 tridentate (2) copper ligands. Combining previously reported and new resonance Raman and EXAFS data coupled to density functional theory calculations, we report a geometric structure and more complete electronic description of the high-spin heme−peroxo−copper complexes 1 and 2, which establish μ-(O22−) side-on to the FeIII and end-on to CuII (μ-η2:η1) binding for the complex 1 but side-on/side-on (μ-η2:η2) μ-peroxo coordination for the complex 2. We also compare and summarize the differences and similarities of these two complexes in their reactivity toward CO, PPh3, acid, and phenols. The comparison of a new X-ray structure of μ-oxo complex 2a with the previously reported 1a X-ray structure, two thermal decomposition products respectively of 2 and 1, reveals a considerable difference in the Fe−O−Cu angle between the two μ-oxo complexes (∠Fe−O−Cu = 178.2° in 1a and ∠Fe−O−Cu = 149.5° in 2a). The reaction of 2 with 1 equiv of an exogenous nitrogen-donor axial base leads to the formation of a distinctive low-temperature-stable, low-spin heme−dioxygen−copper complex (2b), but under the same conditions, the addition of an axial base to 1 leads to the dissociation of the heme−peroxo−copper assembly and the release of O2. 2b reacts with phenols performing H-atom (e− + H+) abstraction resulting in O−O bond cleavage and the formation of high-valent ferryl [FeIV=O] complex (2c). The nature of 2c was confirmed by a comparison of its spectroscopic features and reactivity with those of an independently prepared ferryl complex. The phenoxyl radical generated by the H-atom abstraction was either (1) directly detected by electron paramagnetic resonance spectroscopy using phenols that produce stable radicals or (2) indirectly detected by the coupling product of two phenoxyl radicals.
Co-reporter:Mark P. Schopfer ; Jun Wang
Inorganic Chemistry 2010 Volume 49(Issue 14) pp:6267-6282
Publication Date(Web):July 12, 2010
DOI:10.1021/ic100033y
The focus of this Forum Article highlights work from our own laboratories and those of others in the area of biochemical and biologically inspired inorganic chemistry dealing with nitric oxide [nitrogen monoxide, •NO(g)] and its biological roles and reactions. The latter focus is on (i) oxidation of •NO(g) to nitrate by nitric oxide dioxygenases (NODs) and (ii) reductive coupling of two molecules of •NO(g) to give N2O(g). In the former case, NODs are described, and the highlighting of possible peroxynitrite/heme intermediates and the consequences of this are given by a discussion of recent works with myoglobin and a synthetic heme model system for NOD action. Summaries of recent copper complex chemistries with •NO(g) and O2(g), leading to peroxynitrite species, are given. The coverage of biological reductive coupling of •NO(g) deals with bacterial nitric oxide reductases (NORs) with heme/nonheme diiron active sites and on heme/copper oxidases such as cytochrome c oxidase, which can mediate the same chemistry. Recently designed protein and synthetic model compounds (heme/nonheme/diiron or heme/copper) as functional mimics are discussed in some detail. We also highlight examples from the chemical literature, not necessarily involving biologically relevant metal ions, that describe the oxidation of •NO(g) to nitrate (or nitrite) and possible peroxynitrite intermediates or reductive coupling of •NO(g) to give nitrous oxide.
Co-reporter:Yunho Lee, Dong-Heon Lee, Ga Young Park, Heather R. Lucas, Amy A. Narducci Sarjeant, Matthew T. Kieber-Emmons, Michael A. Vance, Ashley E. Milligan, Edward I. Solomon, and Kenneth D. Karlin
Inorganic Chemistry 2010 Volume 49(Issue 19) pp:8873-8885
Publication Date(Web):September 7, 2010
DOI:10.1021/ic101041m
To better understand the effect of thioether coordination in copper-O2 chemistry, the tetradentate N3S ligand LASM (2-(methylthio)-N,N-bis((pyridin-2-yl)methyl)benzenamine) and related alkylether ligand LEOE (2-ethoxy-N,N-bis((pyridin-2-yl)methyl)ethanamine) have been studied. The corresponding copper(I) complexes, [(LASM)CuI]+ (1a) and [(LEOE)CuI]+ (3a), were studied as were the related compound [(LESE)CuI]+ (2a, LESE = (2-ethylthio-N,N-bis((pyridin-2-yl)methyl)ethanamine). The X-ray structure of 1a and its solution conductivity reveal a monomeric molecular structure possessing thioether coordination which persists in solution. In contrast, the C−O stretching frequencies of the derivative Cu(I)-CO complexes reveal that for these complexes, the modulated ligand arms, whether arylthioether, alkylthioether, or ether, are not coordinated to the cuprous ion. Electrochemical data for 1a and 2a in CH3CN and N,N-dimethylformamide (DMF) show the thioanisole moiety to be a poor electron donor compared to alkylthioether (1a is ∼200 mV more positive than 2a). The structures of [(LASM)CuII(CH3OH)]2+ (1c) and [(LESE)CuII(CH3OH)]2+ (2c) have also been obtained and indicate nearly identical copper coordination environments. Oxygenation of 1a at reduced temperature gives a characteristic deep blue intermediate [{(LASM)CuII}2(O22−)]2+ (1bP) with absorption features at 442 (1,500 M−1 cm−1), 530 (8,600 M−1 cm−1), and 605 nm (10,400 M−1 cm−1); these values compare well to the ligand-to-metal charge-transfer (LMCT) transitions previously reported for [{(LESE)CuII}2(O22−)]2+ (2bP). Resonance Raman data for [{(LASM)CuII}2(O22−)]2+ (1bP) support the formation of μ-1,2-peroxo species ν(O−O) = 828 cm−1(Δ(18O2) = 48), νsym(Cu−O) = 547 cm−1 (Δ(18O2) = 23), and νasym(Cu−O) = 497 cm−1 (Δ(18O2) = 22) and suggest the LASM ligand is a poorer electron donor to copper than is LESE. In contrast, the oxygenation of [(LEOE)CuI]+ (3a), possessing an ether donor as an analogue of the thioether in LESE, led to the formation of a bis(μ-oxo) species [{(LEOE)CuIII}2(O2−)2]2+ (3bO; 380 nm, ε ∼ 10,000 M−1 cm−1). This result provides further support for the sulfur influence in 1bP and 2bP, in particular coordination of the sulfur to the Cu. Thermal decomposition of 1bP is accompanied by ligand sulfoxidation. The structure of [{(LEOE)CuII(Cl)}2]+ (3c) generated from the reductive dehalogenation of organic chlorides suggests that the ether moiety is weakly bound to the cupric ion. A detailed discussion of the spectroscopic and structural characteristics of 1bP, 2bP, and 3bO is presented.
Co-reporter:Heather R. Lucas ; Gerald J. Meyer
Journal of the American Chemical Society 2009 Volume 131(Issue 39) pp:13924-13925
Publication Date(Web):September 8, 2009
DOI:10.1021/ja906172c
Intermolecular nitrogen monoxide (·NO) and carbon monoxide (CO) transfer from iron to copper and back, a phenomenon not previously observed, has been accomplished by employing transient-absorbance laser flash photolysis methods. A 1:1 heme/copper component system consisting of a six-coordinate ferrous species, F8FeII(CO)(DCIM) or F8FeII(NO)(thf) [F8 = tetrakis(2,6-difluorophenyl)porphyrinate(2−); DCIM = 1,5-dicyclohexylimidazole; thf = tetrahydrofuran], and two ligand−copper(I) complexes, one with tridentate [BzL = (benzyl)bis(2-pyridylmethyl)amine] and one with tetradentate coordination [PyL = tris(2-pyridylmethyl)amine], was utilized. The results suggest a lower affinity for NO versus CO binding to copper(I) and a higher rate for NO versus CO binding to heme. In fact, the latter event has been observed in cytochrome c oxidase aa3.
Co-reporter:Mark P. Schopfer, Biplab Mondal, Dong-Heon Lee, Amy A. N. Sarjeant and Kenneth D. Karlin
Journal of the American Chemical Society 2009 Volume 131(Issue 32) pp:11304-11305
Publication Date(Web):July 23, 2009
DOI:10.1021/ja904832j
An oxy-heme complex, the heme-superoxo species (tetrahydrofuran)(F8)FeIII-(O2•−) (2) (F8 = an ortho-difluoro substituted tetraarylporphyrinate), reacts with nitrogen monoxide (•NO; nitric oxide) to produce a nitrato-iron(III) compound (F8)FeIII-(NO3−) (3) (X-ray). The chemistry mimics the action of •NO Dioxygenases (NODs), microbial and mammalian heme proteins which facilitate •NO detoxification/homeostasis. A peroxynitrite intermediate complex is implicated; if 2,4-di-tert-butylphenol is added prior to •NO reaction with 2, o-nitration occurs giving 2,4-di-tert-butyl-6-nitrophenol. The iron product is (F8)FeIII-(OH) (4). The results suggest that heme/O2/•NO chemistry may lead to peroxynitrite leakage and/or exogenous substrate oxidative/nitrative reactivity.
Co-reporter:Debabrata Maiti ; Julia S. Woertink ; Reza A. Ghiladi ; Edward I. Solomon
Inorganic Chemistry 2009 Volume 48(Issue 17) pp:8342-8356
Publication Date(Web):August 10, 2009
DOI:10.1021/ic900975y
Our continuing efforts into developing copper coordination chemistry relevant to dioxygen-processing copper proteins has led us to design and synthesize a cyclotriveratrylene (CTV)-based trinucleating ligand, CTV-TMPA, which employs tetradentate tris(2-pyridylmethyl)-amine chelates (TMPA) for their copper ion binding sites. Binding of three copper ions per CTV-TMPA unit was established by various chemical and spectroscopic methods such as UV−vis and resonance Raman (rR) spectroscopies. The following complexes were observed: A tricopper(I) complex [(CTV-TMPA)CuI3]3+ (1), a CO adduct [(CTV-TMPA)CuI3(CO)3]3+ (1-CO; ν(C═O) = 2094 cm−1), a triphenylphosphine adduct [(CTV-TMPA)CuI3(PPh3)3]3+ (1-PPh3), a tricopper(II) complex [(CTV-TMPA)CuII3]3+ (1-Ox), and its tris-monochloride or tris-monobromide adducts. Also, introduction of dioxygen to the −80 °C solutions of 1 leads to O2-adducts, the first example of a synthetic copper complex which can stabilize a mononuclear CuII-superoxo and dinuclear peroxo species simultaneously within one complex {[Cu] = 1.53 mM in THF: (μ-1,2-peroxo complex, λmax = 543 (ε 9650) nm): ν(O−O) = 825 ((Δ18O2) = −47) cm−1; ν(Cu−O) = 506 ((Δ18O2) = −26) cm−1: (superoxo complex, λmax = 427 (ε 3150) nm): ν(O−O) = 1129 ((Δ18O2) = −60) cm−1; ν(Cu−O) = 463 ((Δ18O2) = −27) cm−1}. Elemental sulfur reacts reversibly with 1 leading to a (proposed) hexanuclear species [{(CTV-TMPA)CuII3}2(μ-1,2-S22−)3]6+ (1-S) {λmax = 544 (ε 7270) nm }, possessing one dicopper(II)-disulfide structural type: {THF solvent) ν(S−S) = 489 ((Δ34S) = −10) cm−1; ν(Cu−S) = 307 ((Δ34S) = −5) cm−1}. Derivation of spectroscopic, structural, and chemical conclusions were aided by the study of a close mononuclear analogue with one pyridyl group of the TMPA parent possessing a 6-CH2OCH3 substituent, this being part of the CTV-TMPA architecture.
Co-reporter:Yunho Lee ; Ga Young Park ; Heather R. Lucas ; Peter L. Vajda ; Kaliappan Kamaraj ; Michael A. Vance ; Ashley E. Milligan ; Julia S. Woertink ; Maxime A. Siegler ; Amy A. Narducci Sarjeant ; Lev N. Zakharov ; Arnold L. Rheingold ⊗; Edward I. Solomon
Inorganic Chemistry 2009 Volume 48(Issue 23) pp:11297-11309
Publication Date(Web):November 3, 2009
DOI:10.1021/ic9017695
Cuprous and cupric complexes with the new imidazolyl containing tripodal tetradentate ligands {LMIm, (1H-imidazol-4-yl)-N,N-bis((pyridin-2-yl)methyl)methanamine, and LEIm, 2-(1H-imidazol-4-yl)-N,N-bis((pyridin-2-yl)methyl)ethanamine}, have been investigated to probe differences in their chemistry, especially in copper(I)−dioxygen chemistry, compared to that already known for the pyridyl analogue TMPA, tris(2-pyridyl)methyl)amine. Infrared (IR) stretching frequencies obtained from carbon monoxide adducts of [(LMIm)CuI]+ (1a) and [(LEIm)CuI]+ (2a) show that the imidazolyl donor is stronger than its pyridyl analogue. Electrochemical data suggest differences in the binding constant of CuII to LEIm compared to TMPA and LMIm, reflecting geometric changes. Oxygenation of [(LMIm)CuI]+ (1a) in 2-methyltetrahydrofuran (MeTHF) solvent at −128 °C leads to an intensely purple colored species with a UV−vis spectrum characteristic of an end-on bound peroxodicopper(II) complex [{(LMIm)CuII}2(μ-1,2-O22−)]2+ (1bP) {λmax = 528 nm}, very similar to the previously well characterized complex [{(TMPA)CuII}2(μ-1,2-O22−)]2+ {λmax = 520 nm (ε = 12 000 M−1 cm−1), in MeTHF; resonance Raman (rR) spectroscopy: ν(O−O) = 832 (Δ(18O2) = −44) cm−1}. In the low-temperature oxygenation of 2a, benchtop (−128 °C) and stopped-flow (−90 °C) experiments reveal the formation of an initial superoxo-Cu(II) species [(LEIm)CuII(O2•−)]+ (2bS), λmax = 431 nm in THF) . This converts to the low-temperature stable peroxo complex [{(LEIm)CuII}2(μ-1,2-O22−)]2+ (2bP) {rR spectroscopy: ν(O−O) = 822 (Δ(18O2) = −46) cm−1}. Complex 2bP possess distinctly reduced Cu−O and O−O stretching frequencies and a red-shifted UV−vis feature {to λmax = 535 nm (ε = 11 000 M−1 cm−1)} compared to the TMPA analogue due to a distortion from trigonal bipyramidal (TBP) to a square pyramidal ligand field. This distortion is supported by the structural characterization of related ligand−copper(II) complexes: A stable tetramer cluster complex [(μ2-LEIm−)4(CuII)4]4+, obtained from thermal decomposition of 2bP (with formation of H2O2), also exhibits a distorted square pyramidal Cu(II) ion geometry as does the copper(II) complex [(LEIm)CuII(CH3CN)]2+ (2c), characterized by X-ray crystallography and solution electron paramagnetic resonance (EPR) spectroscopy.
Co-reporter:Richard A. Himes
PNAS 2009 Volume 106 (Issue 45 ) pp:18877-18878
Publication Date(Web):2009-11-10
DOI:10.1073/pnas.0911413106
Co-reporter:Ga Young Park;Subramanian Deepalatha
JBIC Journal of Biological Inorganic Chemistry 2009 Volume 14( Issue 8) pp:
Publication Date(Web):2009 November
DOI:10.1007/s00775-009-0575-8
Reaction of nitrogen monoxide with a copper(I) complex possessing a tridentate alkylamine ligand gives a Cu(I)–(·NO) adduct, which when exposed to dioxygen generates a peroxynitrite (O=NOO−)–Cu(II) species. This undergoes thermal transformation to produce a copper(II) nitrito (NO2–) complex and 0.5 mol equiv O2. In the presence of a substituted phenol, the peroxynitrite complex effects oxidative coupling, whereas addition of chloride ion to dissociate the peroxynitrite moiety instead leads to phenol ortho nitration. Discussions include the structures (including electronic description) of the copper–nitrosyl and copper–peroxynitrite complexes and the formation of the latter, based on density functional theory calculations and accompanying spectroscopic data.
Co-reporter:Debabrata Maiti ; Julia S. Woertink ; Amy A. Narducci Sarjeant ; Edward I. Solomon
Inorganic Chemistry 2008 Volume 47(Issue 9) pp:3787-3800
Publication Date(Web):April 9, 2008
DOI:10.1021/ic702437c
The preference for the formation of a particular Cu2O2 isomer coming from (ligand)-CuI/O2 reactivity can be regulated with the steric demands of a TMPA (tris(2-pyridylmethyl)amine) derived ligand possessing 6-pyridyl substituents on one of the three donor groups of the tripodal tetradentate ligand. When this substituent is an −XHR group (X = N or C) the traditional CuI/O2 adduct forms a (μ-1,2)peroxodicopper(II) species (A). However, when the substituent is the slightly bulkier XR2 moiety {aryl or NR2 (R ≠ H)}, a bis(μ-oxo)dicopper(III) structure (C) is favored. The reactivity of one of the bis(μ-oxo)dicopper(III) species, [{(6tbp)CuIII}2(O2−)2]2+ (7-O2) (6tbp = (6-tBu-phenyl-2-pyridylmethyl)bis(2-pyridylmethyl)amine), was probed, and for the first time, exogenous toluene or ethylbenzene hydrocarbon oxygenation reactions were observed. Typical monooxygenase chemistry occurred: the benzaldehyde product includes an 18-O atom for toluene/7-18O2 reactivity, and a H-atom abstraction by 7-O2 is apparent from study of its reactions with ArOH substrates, as well as the determination of kH/kD ≈ 7 in the toluene oxygenation (i.e., PhCH3 vs PhCD3 substrates). Proposed courses of reaction are presented, including the possible involvement of PhCH2OO• and its subsequent reaction with copper(I) complex, the latter derived from dynamic solution behavior of 7-O2. External TMPA ligand exchange for copper in 7-O2 and O−O bond (re)formation chemistry, along with the ability to protonate 7-O2 and release of H2O2 indicate the presence of an equilibrium between [{(6tbp)CuIII}2(O2−)2]2+ (7-O2) and a (μ-1,2)peroxodicopper(II) form.
Co-reporter:Debabrata Maiti ; Amy A. Narducci Sarjeant
Inorganic Chemistry 2008 Volume 47(Issue 19) pp:8736-8747
Publication Date(Web):September 11, 2008
DOI:10.1021/ic800617m
A substantial oxidative N-debenzylation reaction along with PhCH═O formation occurs from a hydroperoxo−copper(II) complex that has a dibenzylamino substrate (—N(CH2Ph)2 appended as a substituent on one pyridyl group of its tripodal tetradentate TMPA (also TPA, (2-pyridylmethyl)amine)) ligand framework. During the course of the (LN(CH2Ph)2)CuII(−OOH) reactivity, the formation of a substrate and a −OOH-derived (an oxygen atom) alkoxo CuII(−OR) complex occurs. The observation that the same CuII(−OR) species occurs from CuΙ/PhIO chemistry suggests the possibility that a copper−oxo (cupryl) reactive intermediate forms during the alkoxo species formation; new ESI-MS data provide further support for this high-valent intermediate. A net H atom abstraction chemistry is proposed on the basis of the kinetic isotope effect studies provided here and the previously published study for a closely related CuII(−OOH) species incorporating dimethylamine (—N(CH3)2) as the internal substrate;(27) the CuΙ/PhIO reactivity with similar isotope effect results provides further support. The reactivity of these chemical systems closely resembles the proposed oxidative N-dealkylation mechanisms that are effected by the copper monooxygenases, dopamine β-monooxygenase (DβM) and peptidylglycine-α-hydroxylating monooxygenase (PHM).
Co-reporter:Eduardo E. Chufán, Simona C. Puiu and Kenneth D. Karlin
Accounts of Chemical Research 2007 Volume 40(Issue 7) pp:563
Publication Date(Web):June 6, 2007
DOI:10.1021/ar700031t
This Account focuses on our recent developments in synthetic heme/copper/O2 chemistry, potentially relevant to the mechanism of action of heme–copper oxidases (e.g., cytochrome c oxidase) and to dioxygen activation chemistry. Methods for the generation of O2 adducts, which are high-spin heme(FeIII)–peroxo–CuII complexes, are described, along with a detailed structural/electronic characterization of one example. The coordination mode of the O2-derived heme–Cu bridging group depends upon the copper–ligand environment, resulting in μ-(O22–) side-on to FeIII and end-on to CuII (μ-η2:η1) binding for cases having N4 tetradentate ligands but side-on/side-on (μ-η2:η2) μ-peroxo coordination with tridentate copper chelates. The dynamics of the generation of FeIII–(O22–)–CuII complexes are known in some cases, including the initial formation of a short-lived superoxo (heme)FeIII(O2•–) intermediate. Complexes with cross-linked imidazole–phenol “cofactors” adjacent to the copper centers have also been described. Essential investigations of heme–copper-mediated reductive O–O bond cleavage chemistry are ongoing.
Co-reporter:Yunho Lee, Dong-Heon Lee, Amy A. Narducci Sarjeant, Kenneth D. Karlin
Journal of Inorganic Biochemistry 2007 Volume 101(11–12) pp:1845-1858
Publication Date(Web):November 2007
DOI:10.1016/j.jinorgbio.2007.06.016
In order to better understand copper mediated oxidative chemistry via ligand–CuI/O2 reactivity employing S-donor ligands for copper, O2-reactivity studies of the copper(I) complexes (1 and 2, Chart 2) have been carried out with a tridentate N2S thiol ligand (1-(N-methyl-N-(2-(pyridin-2-yl)ethyl)amino)propane-2-thiol; LSH) or its oxidized disulfide form (LSS). Reactions of [LSHCuI]+ (1) and [LSS(CuI)2(X)2]2+ (2) with O2 give ∼90% and ∼70% yields of [LSO3CuII(MeOH)2]+ (3), respectively, where LSO3 is S-oxygenated sulfonate; 3 was characterized by electrospray ionization (ESI) mass spectrometry and X-ray crystallography. Mimicking TyrCys galactose oxidase cofactor biogenesis, a new C–S bond is formed (within new thioether moiety LSPhOH) from cuprous complex (both 1 and 2) dioxygen reactivity in the presence of 2,4-tBu2-phenolate. In addition, the disulfide ligand (LSS) reacts with 2 equiv. cupric ion salts and the phenolate to efficiently give the cross-linked product LSPhOH in high yield (>90%) under anaerobic conditions. Separately, complex [LSPhOCuII(ClO4)] (4), possessing the cross-linked LSPhOH, was characterized by ESI mass spectrometry and X-ray crystallography.
Co-reporter:Debabrata Maiti;Dong-Heon Lee Dr.;Katya Gaoutchenova Dr.;Christian Würtele;MaxC. Holthausen Dr.;AmyA. NarducciSarjeant Dr.;Jörg Sundermeyer Dr.;Siegfried Schindler Dr.;KennethD. Karlin
Angewandte Chemie International Edition 2007 Volume 47( Issue 1) pp:82-85
Publication Date(Web):
DOI:10.1002/anie.200704389
Co-reporter:Debabrata Maiti;Dong-Heon Lee Dr.;Katya Gaoutchenova Dr.;Christian Würtele;MaxC. Holthausen Dr.;AmyA. NarducciSarjeant Dr.;Jörg Sundermeyer Dr.;Siegfried Schindler Dr.;KennethD. Karlin
Angewandte Chemie 2007 Volume 120( Issue 1) pp:88-91
Publication Date(Web):
DOI:10.1002/ange.200704389
Co-reporter:Yunho Lee, Amy A. Narducci Sarjeant and Kenneth D. Karlin
Chemical Communications 2006 (Issue 6) pp:621-623
Publication Date(Web):21 Dec 2005
DOI:10.1039/B513768C
A copper(I) complex with new N2S thiol ligand transforms to a multicopper(I) cluster, [(LS−)6CuI13(S2−)2]3+ (1); its X-ray structure exhibiting μ4-sulfido and μ3-thiolato coordination is presented and compared to other cuprous thiolato/sulfido clusters including that observed in the copper enzyme nitrous oxide reductase.
Co-reporter:Matthew E. Helton, Debabrata Maiti, Lev N. Zakharov, Arnold L. Rheingold, John A. Porco Jr.,Kenneth D. Karlin
Angewandte Chemie International Edition 2006 45(7) pp:
Publication Date(Web):
DOI:10.1002/anie.200503216
Co-reporter:Matthew E. Helton Dr.;Debabrata Maiti;Lev N. Zakharov;Arnold L. Rheingold ;John A. Porco Jr.
Angewandte Chemie 2006 Volume 118(Issue 7) pp:
Publication Date(Web):30 DEC 2005
DOI:10.1002/ange.200503216
Side-on: Elementarer Schwefel (S8) reagiert mit dem Kupfer(I)-Komplex des dreizähnigen Chelatliganden (MePY2) zu dem μ-η2:η2-Disulfido-verbrückten Kupfer(II)-Komplex [{Cu(MePY2)}2(S2)]2+, der röntgenstrukturanalytisch charakterisiert (siehe Struktur; S gelb, Cu grün, N blau, C grau) und auf seine Reaktivität gegenüber PPh3, RNC, CO, O2, PhCH2Br und einem vierzähnigen Chelatliganden untersucht wurde.
Co-reporter:Eunsuk Kim;Matthew E. Helton;Ian M. Wasser;Shen Lu;Hong-wei Huang;Pierre Moënne-Loccoz;Christopher D. Incarvito;Arnold L. Rheingold;Marcus Honecker;Susan Kaderli;Andreas D. Zuberbühler;
Proceedings of the National Academy of Sciences 2003 100(7) pp:3623-3628
Publication Date(Web):March 24, 2003
DOI:10.1073/pnas.0737180100
The O2-reaction chemistry of 1:1 mixtures of (F8)FeII (1; F8 = tetrakis(2,6-diflurorophenyl)porphyrinate) and [(LMe2N)CuI]+ (2; LMe2N = N,N-bis{2-[2-(N′,N′-4-dimethylamino)pyridyl]ethyl}methylamine) is described, to model aspects of the chemistry occurring in cytochrome c oxidase. Spectroscopic investigations, along with stopped-flow kinetics, reveal that low-temperature oxygenation of 1/2 leads
to rapid formation of a heme-superoxo species (F8)FeIII-(O) (3), whether or not 2 is present. Complex 3 subsequently reacts with 2 to form [(F8)FeIII–(O)–CuII(LMe2N)]+ (4), which thermally converts to [(F8)FeIII–(O)–CuII(LMe2N)]+ (5), which has an unusually bent (Fe–O–Cu) bond moiety. Tridentate chelation, compared with tetradentate, is shown to dramatically
lower the ν(O–O) values observed in 4 and give rise to the novel structural features in 5.
Co-reporter:Jacqueline K Barton, Kenneth D Karlin
Current Opinion in Chemical Biology 2001 Volume 5(Issue 2) pp:165-167
Publication Date(Web):1 April 2001
DOI:10.1016/S1367-5931(00)00186-1
Co-reporter:Christiana Xin Zhang, Hong-Chang Liang, Eun-il Kim, Qing-Fen Gan, Zoltán Tyeklár, Kin-Chung Lam, Arnold L. Rheingold, Susan Kaderli, Andreas D. Zuberbühler and Kenneth D. Karlin
Chemical Communications 2001 (Issue 7) pp:631-632
Publication Date(Web):14 Mar 2001
DOI:10.1039/B009053K
Reaction of dioxygen with a dinuclear copper(I)
complex of a new binucleating ligand is described, wherein a
peroxo–dicopper(II) (Cu2–O2)
intermediate leads to an oxo-transfer reaction to give an N-oxide
of an N-benzyl internal ligand substrate; additionally observed
regioselective oxidative N-dealkylation chemistry occurs.
Co-reporter:Heather R. Lucas ; Gerald J. Meyer
Journal of the American Chemical Society () pp:
Publication Date(Web):August 20, 2010
DOI:10.1021/ja104107q
The kinetics, thermodynamics, and coordination dynamics are reported for O2 and CO 1:1 binding to a series of pseudo-tetradentate ligand−copper(I) complexes (DLCuI) to give CuI/O2 and CuI/CO product species. Members of the DLCuI series possess an identical tridentate core structure where the cuprous ion binds to the bispicolylamine (L) fragment. DL also contains a fourth variable N-donor moiety {D = benzyl (Bz); pyridyl (Py); imidazolyl (Im); dimethylamino (NMe2); (tert-butylphenyl)pyridyl (TBP); quinolyl (Q)}. The structural characteristics of DLCuI−CO and DLCuI are detailed, with X-ray crystal structures reported for TBPLCuI−CO, BzLCuI−CO, and QLCuI. Infrared studies (solution and solid-state) confirm that DLCuI−CO possess the same four-coordinate core structure in solution with the variable D moiety “dangling”, i.e., not coordinated to the copper(I) ion. Other trends observed for the present series appear to derive from the degree to which the D-group interacts with the cuprous ion center. Electrochemical studies reveal close similarities of behavior for ImLCuI and NMe2LCuI (as well as for TBPLCuI and QLCuI), which relate to the O2 binding kinetics and thermodynamics. Equilibrium CO binding data (KCO, ΔH°, ΔS°) were obtained by conducting UV−visible spectrophotometric CO titrations, while CO binding kinetics and thermodynamics (kCO, ΔH⧧, ΔS⧧) were measured through variable-temperature (193−293 K) transient absorbance laser flash photolysis experiments, λex = 355 nm. Carbon monoxide dissociation rate constants (k−CO) and corresponding activation parameters (ΔH⧧, ΔS⧧) have also been obtained. CO binding to DLCuI follows an associative mechanism, with the increased donation from D leading to higher kCO values. Unlike observations from previous work, the KCO values increased as the kCO and k−CO values declined; the latter decreased at a faster rate. By using the “flash-and-trap” method (λex = 355 nm, 188−218 K), the kinetics and thermodynamics (kO2, ΔH⧧, ΔS⧧) for O2 binding to NMe2LCuI and ImLCuI were measured and compared to those for PyLCuI. A surprising change in the O2 binding mechanism was deduced from the thermodynamic ΔS⧧ values observed, associative for PyLCuI but dissociative for NMe2LCuI and ImLCuI; these results are interpreted as arising from a difference in the timing of electron transfer from copper(I) to O2 as this molecule coordinates and a tetrahydrofuran (THF) solvent molecule dissociates. The change in mechanism was not simply related to alterations in DLCuII/I geometries or the order in which O2 and THF coordinate. The equilibrium O2 binding constant (KO2, ΔH°, ΔS°) and O2 dissociation rate constants (k−O2, ΔH⧧, ΔS⧧) were also determined. Overall the results demonstrate that subtle changes in the coordination environment, as occur over time through evolution in nature or through controlled ligand design in synthetic systems, dictate to a critically detailed level the observed chemistry in terms of reaction kinetics, structure, and reactivity, and thus function. Results reported here are also compared to relevant copper and/or iron biological systems and analogous synthetic ligand−copper systems.
Co-reporter:Sunghee Kim, Maxime A. Siegler and Kenneth D. Karlin
Chemical Communications 2014 - vol. 50(Issue 22) pp:NaN2846-2846
Publication Date(Web):2013/12/09
DOI:10.1039/C3CC47942K
New peroxynitrite–copper chemistry ensues via addition of nitric oxide (˙NO(g)) to a CuII–hydroperoxo species. In characterizing the system, the ligand–Cu(I) complex was shown to effect a seldom observed ˙NO(g) reductive coupling reaction. Biological implications are discussed.
Co-reporter:Ga Young Park, Yunho Lee, Dong-Heon Lee, Julia S. Woertink, Amy A. Narducci Sarjeant, Edward I. Solomon and Kenneth D. Karlin
Chemical Communications 2010 - vol. 46(Issue 1) pp:NaN93-93
Publication Date(Web):2009/11/04
DOI:10.1039/B918616F
[(ANS)CuI(CH3CN)]+ reacts with O2 giving [{(ANS)CuII}2(μ-η2:η2-O22−)]2+, νO–O = 731 cm−1, shown to possess S-thioether ligation, based on comparisons with analogues having all N-ligands or a –S(Ph) group. The finding is a rare occurrence and new for side-on O22− binding.
Co-reporter:Atsutoshi Yokoyama, Jung Eun Han, Kenneth D. Karlin and Wonwoo Nam
Chemical Communications 2014 - vol. 50(Issue 14) pp:NaN1744-1744
Publication Date(Web):2013/12/13
DOI:10.1039/C3CC48782B
Reaction of a nonheme iron(III)-peroxo complex, [FeIII(14-TMC)(O2)]+, with NO+, a transformation which is essentially isoelectronic with that for nitric oxide dioxygenases [Fe(III)(O2˙−) + NO], affords an iron(IV)-oxo complex, [FeIV(14-TMC)(O)]2+, and nitrogen dioxide (NO2), followed by conversion to an iron(III)-nitrato complex, [FeIII(14-TMC)(NO3)(F)]+.
Co-reporter:Yusuke Yamada, Kazuki Maeda, Kei Ohkubo, Kenneth D. Karlin and Shunichi Fukuzumi
Physical Chemistry Chemical Physics 2012 - vol. 14(Issue 27) pp:NaN9659-9659
Publication Date(Web):2012/05/14
DOI:10.1039/C2CP41207A
The catalytic durability of an organic photocatalyst, 9-mesityl-10-methyl acridinium ion (Acr+–Mes), has been dramatically improved by the addition of [{tris(2-pyridylmethyl)amine}CuII](ClO4)2 ([(tmpa)CuII]2+) in the photocatalytic oxygenation of p-xylene by molecular oxygen in acetonitrile. Such an improvement is not observed by the addition of Cu(ClO4)2 in the absence of organic ligands. The addition of [(tmpa)Cu]2+ in the reaction solution resulted in more than an 11 times higher turnover number (TON) compared with the TON obtained without [(tmpa)CuII]2+. In the photocatalytic oxygenation, a stoichiometric amount of H2O2 formation was observed in the absence of [(tmpa)CuII]2+, however, much less H2O2 formation was observed in the presence of [(tmpa)CuII]2+. The photocatalytic mechanism was investigated by laser flash photolysis measurements in order to detect intermediates. The reaction of O2˙− with [(tmpa)CuII]2+ monitored by UV-vis spectroscopy in propionitrile at 203 K suggested formation of [{(tmpa)CuII}2O2]2+, a transformation which is crucial for the overall 4-electron reduction of molecular O2 to water, and a key in the observed improvement in the catalytic durability of Acr+–Mes.
Co-reporter:Mihoko Yamada, Kenneth D. Karlin and Shunichi Fukuzumi
Chemical Science (2010-Present) 2016 - vol. 7(Issue 4) pp:NaN2863-2863
Publication Date(Web):2016/01/05
DOI:10.1039/C5SC04312C
Benzene was hydroxylated with hydrogen peroxide (H2O2) in the presence of catalytic amounts of copper complexes in acetone to yield phenol at 298 K. At higher temperatures, phenol was further hydroxylated with H2O2 by catalysis of copper complexes to yield p-benzoquinone. The kinetic study revealed that the rate was proportional to concentrations of benzene and H2O2, but to the square root of the concentration of a copper(II) complex ([Cu(tmpa)]2+: tmpa = tris(2-pyridylmethyl)amine). The addition of a spin trapping reagent resulted in formation of a spin adduct of hydroperoxyl radical (HO2˙), as observed by EPR spectroscopy, inhibiting phenol formation. HO2˙ produced by the reaction of [Cu(tmpa)]2+ with H2O2 acts as a chain carrier for the radical chain reactions for formation of phenol. When [Cu(tmpa)]2+ was incorporated into mesoporous silica–alumina (Al-MCM-41) by a cation exchange reaction, the selectivity for production of phenol was much enhanced by prevention of hydroxylation of phenol, which was not adsorbed to Al-MCM-41. The high durability with a turnover number of 4320 for the hydroxylation of benzene to phenol with H2O2 was achieved using [Cu(tmpa)]2+ incorporated into Al-MCM-41 as an efficient and selective catalyst.

![Iron, chloro[5,10,15,20-tetrakis(4-methoxyphenyl)-21H,23H-porphinato(2-)-κN21,κN22,κN23,κN24]-, (SP-5-12)-](/data/chemimg/27600/36995-20-7.png)
![Iron, chloro[5,10,15,20-tetrakis(4-methoxyphenyl)-21H,23H-porphinato(2-)-κN21,κN22,κN23,κN24]-, (SP-5-12)-](/data/chemimg/27600/36995-20-7_b.png)
![1,5-Pentanediamine, N,N,N',N'-tetrakis[2-(2-pyridinyl)ethyl]-](http://img.cochemist.com/ccimg/98300/98218-52-1.png)
![1,5-Pentanediamine, N,N,N',N'-tetrakis[2-(2-pyridinyl)ethyl]-](http://img.cochemist.com/ccimg/98300/98218-52-1_b.png)
![1,3-Propanediamine, N,N,N',N'-tetrakis[2-(2-pyridinyl)ethyl]-](http://img.cochemist.com/ccimg/89000/88917-40-2.png)
![1,3-Propanediamine, N,N,N',N'-tetrakis[2-(2-pyridinyl)ethyl]-](http://img.cochemist.com/ccimg/89000/88917-40-2_b.png)
![2-[(2-methylphenyl)methylsulfanyl]ethanamine](http://img.cochemist.com/ccimg/60200/60116-43-0.png)
![2-[(2-methylphenyl)methylsulfanyl]ethanamine](http://img.cochemist.com/ccimg/60200/60116-43-0_b.png)


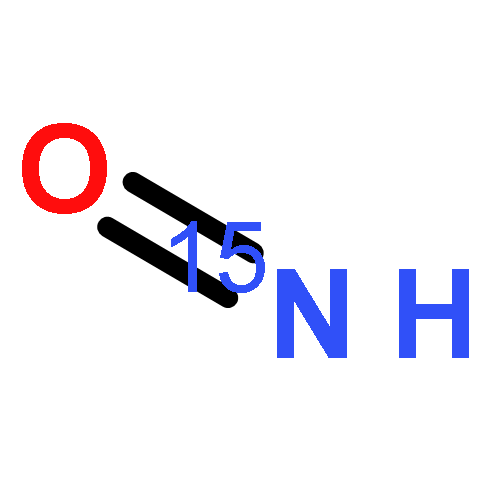


























































































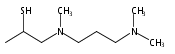
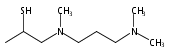
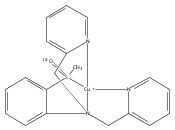
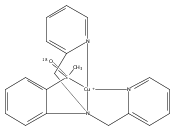
























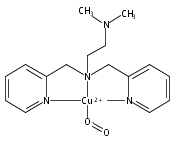

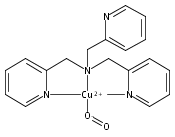









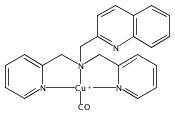
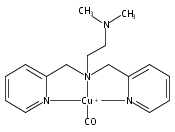
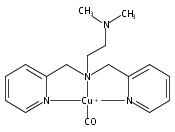
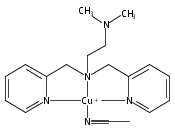
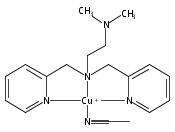

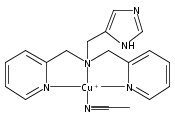













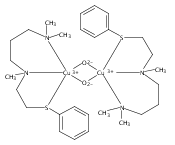
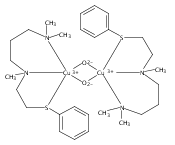
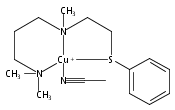
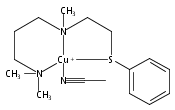


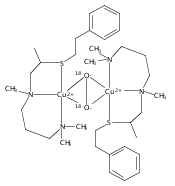
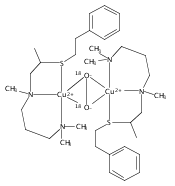

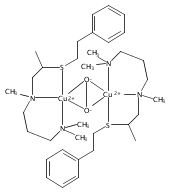

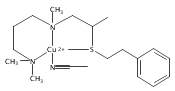






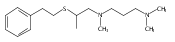
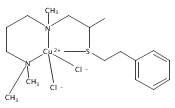
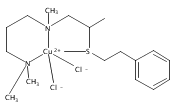


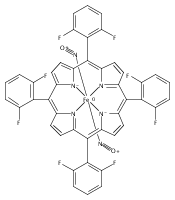











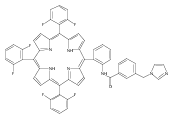
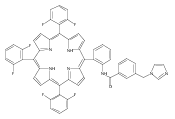


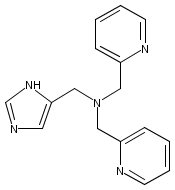





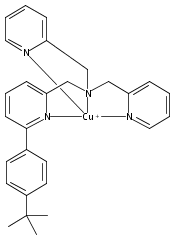











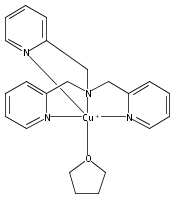
![Propanamide,N-[6-[[bis(2-pyridinylmethyl)amino]methyl]-2-pyridinyl]-2,2-dimethyl-](http://img.cochemist.com/ccimg/351500/351415-88-8.png)
![Propanamide,N-[6-[[bis(2-pyridinylmethyl)amino]methyl]-2-pyridinyl]-2,2-dimethyl-](http://img.cochemist.com/ccimg/351500/351415-88-8_b.png)





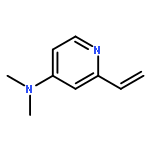
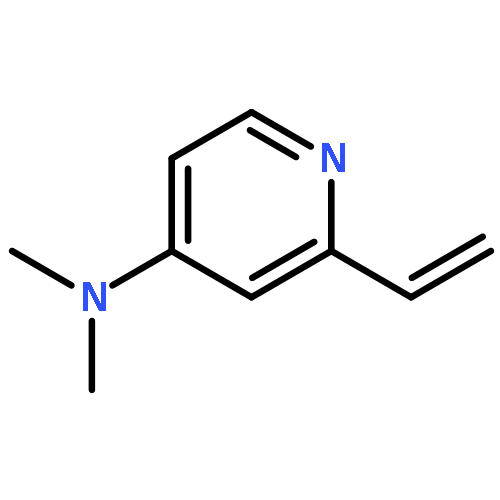


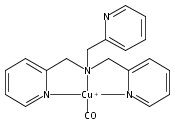
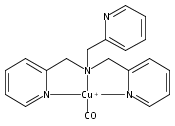



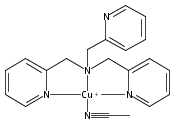
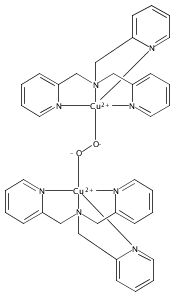
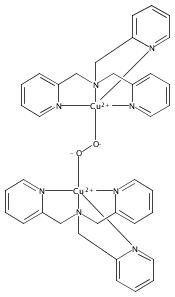
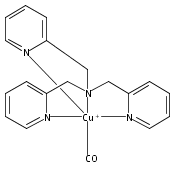



![Propanamide, N-[6-(bromomethyl)-2-pyridinyl]-2,2-dimethyl-](http://img.cochemist.com/ccimg/111500/111477-43-1.png)
![Propanamide, N-[6-(bromomethyl)-2-pyridinyl]-2,2-dimethyl-](http://img.cochemist.com/ccimg/111500/111477-43-1_b.png)




![2-Pyridineethanamine, N-[2-(2-pyridinyl)ethyl]-](http://img.cochemist.com/ccimg/15500/15496-36-3.png)
![2-Pyridineethanamine, N-[2-(2-pyridinyl)ethyl]-](http://img.cochemist.com/ccimg/15500/15496-36-3_b.png)


















![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8.png)
![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8_b.png)