Co-reporter:Maris A. Cinelli, Huiying Li, Georges Chreifi, Thomas L. Poulos, and Richard B. Silverman
Journal of Medicinal Chemistry May 11, 2017 Volume 60(Issue 9) pp:3958-3958
Publication Date(Web):April 19, 2017
DOI:10.1021/acs.jmedchem.7b00259
Neuronal nitric oxide synthase (nNOS) inhibition is a promising strategy to treat neurodegenerative disorders, but the development of nNOS inhibitors is often hindered by poor pharmacokinetics. We previously developed a class of membrane-permeable 2-aminoquinoline inhibitors and later rearranged the scaffold to decrease off-target binding. However, the resulting compounds had decreased permeability, low human nNOS activity, and low selectivity versus human eNOS. In this study, 5-substituted phenyl ether-linked aminoquinolines and derivatives were synthesized and assayed against purified NOS isoforms. 5-Cyano compounds are especially potent and selective rat and human nNOS inhibitors. Activity and selectivity are mediated by the binding of the cyano group to a new auxiliary pocket in nNOS. Potency was enhanced by methylation of the quinoline and by introduction of simple chiral moieties, resulting in a combination of hydrophobic and auxiliary pocket effects that yielded high (∼500-fold) n/e selectivity. Importantly, the Caco-2 assay also revealed improved membrane permeability over previous compounds.
Co-reporter:Anthony V. Pensa, Maris A. Cinelli, Huiying Li, Georges Chreifi, Paramita Mukherjee, Linda J. Roman, Pavel Martásek, Thomas L. Poulos, and Richard B. Silverman
Journal of Medicinal Chemistry August 24, 2017 Volume 60(Issue 16) pp:7146-7146
Publication Date(Web):August 4, 2017
DOI:10.1021/acs.jmedchem.7b00835
Neuronal nitric oxide synthase (nNOS) is a target for development of antineurodegenerative agents. Most nNOS inhibitors mimic l-arginine and have poor bioavailability. 2-Aminoquinolines showed promise as bioavailable nNOS inhibitors but suffered from low human nNOS inhibition, low selectivity versus human eNOS, and significant binding to other CNS targets. We aimed to improve human nNOS potency and selectivity and reduce off-target binding by (a) truncating the original scaffold or (b) introducing a hydrophilic group to interrupt the lipophilic, promiscuous pharmacophore and promote interaction with human nNOS-specific His342. We synthesized both truncated and polar 2-aminoquinoline derivatives and assayed them against recombinant NOS enzymes. Although aniline and pyridine derivatives interact with His342, benzonitriles conferred the best rat and human nNOS inhibition. Both introduction of a hydrophobic substituent next to the cyano group and aminoquinoline methylation considerably improved isoform selectivity. Most importantly, these modifications preserved Caco-2 permeability and reduced off-target CNS binding.
Co-reporter:Heng-Yen Wang; Yajuan Qin; Huiying Li; Linda J. Roman; Pavel Martásek; Thomas L. Poulos
Journal of Medicinal Chemistry 2016 Volume 59(Issue 10) pp:4913-4925
Publication Date(Web):April 6, 2016
DOI:10.1021/acs.jmedchem.6b00273
Neuronal nitric oxide synthase (nNOS) is an important therapeutic target for the treatment of various neurodegenerative disorders. A major challenge in the design of nNOS inhibitors focuses on potency in humans and selectivity over other NOS isoforms. Here we report potent and selective human nNOS inhibitors based on the 2-aminopyridine scaffold with a central pyridine linker. Compound 14j, the most promising inhibitor in this study, exhibits excellent potency for rat nNOS (Ki = 16 nM) with 828-fold n/e and 118-fold n/i selectivity with a Ki value of 13 nM against human nNOS with 1761-fold human n/e selectivity. Compound 14j also displayed good metabolic stability in human liver microsomes, low plasma protein binding, and minimal binding to cytochromes P450 (CYPs), although it had little to no Caco-2 permeability.
Co-reporter:Jianbin Zheng, Long Chen, Michael Schwake, Richard B. Silverman, and Dimitri Krainc
Journal of Medicinal Chemistry 2016 Volume 59(Issue 18) pp:8508-8520
Publication Date(Web):September 6, 2016
DOI:10.1021/acs.jmedchem.6b00930
Gaucher’s disease is a common genetic disease caused by mutations in the β-glucocerebrosidase (GBA1) gene that have been also linked to increased risk of Parkinson’s disease and Lewy body dementia. Stabilization of misfolded mutant β-glucocerebrosidase (GCase) represents an important therapeutic strategy in synucleinopathies. Here we report a novel class of GCase quinazoline inhibitors, obtained in a high throughput screening, with moderate potency against wild-type GCase. Rational design and a SAR study of this class of compounds led to a new series of quinazoline derivatives with single-digit nanomolar potency. These compounds were shown to selectively stabilize GCase when compared to other lysosomal enzymes and to increase N370S mutant GCase protein concentration and activity in cell assays. To the best of our knowledge, these molecules are the most potent noniminosugar GCase modulators to date that may prove useful for future mechanistic studies and therapeutic approaches in Gaucher’s and Parkinson’s diseases.
Co-reporter:Jeffrey K. Holden, Matthew C. Lewis, Maris A. Cinelli, Ziad Abdullatif, Anthony V. Pensa, Richard B. Silverman, and Thomas L. Poulos
Biochemistry 2016 Volume 55(Issue 39) pp:5587
Publication Date(Web):September 8, 2016
DOI:10.1021/acs.biochem.6b00786
Nitric oxide is produced in Gram-positive pathogens Bacillus anthracis and Staphylococcus aureus by the bacterial isoform of nitric oxide synthase (NOS). Inhibition of bacterial nitric oxide synthase (bNOS) has been identified as a promising antibacterial strategy for targeting methicillin-resistant S. aureus [Holden, J. K., et al. (2015) Chem. Biol. 22, 785–779]. One class of NOS inhibitors that demonstrates antimicrobial efficacy utilizes an aminoquinoline scaffold. Here we report on a variety of aminoquinolines that target the bacterial NOS active site, in part, by binding to a hydrophobic patch that is unique to bNOS. Through mutagenesis and crystallographic studies, our findings demonstrate that aminoquinolines are an excellent scaffold for further aiding in the development of bNOS specific inhibitors.
Co-reporter:Hyunbeom Lee; Emma H. Doud; Rui Wu; Ruslan Sanishvili; Jose I. Juncosa; Dali Liu; Neil L. Kelleher
Journal of the American Chemical Society 2015 Volume 137(Issue 7) pp:2628-2640
Publication Date(Web):January 23, 2015
DOI:10.1021/ja512299n
γ-Aminobutyric acid aminotransferase (GABA-AT) is a pyridoxal 5′-phosphate (PLP)-dependent enzyme that degrades GABA, the principal inhibitory neurotransmitter in mammalian cells. When the concentration of GABA falls below a threshold level, convulsions can occur. Inhibition of GABA-AT raises GABA levels in the brain, which can terminate seizures as well as have potential therapeutic applications in treating other neurological disorders, including drug addiction. Among the analogues that we previously developed, (1S,3S)-3-amino-4-difluoromethylene-1-cyclopentanoic acid (CPP-115) showed 187 times greater potency than that of vigabatrin, a known inactivator of GABA-AT and approved drug (Sabril) for the treatment of infantile spasms and refractory adult epilepsy. Recently, CPP-115 was shown to have no adverse effects in a Phase I clinical trial. Here we report a novel inactivation mechanism for CPP-115, a mechanism-based inactivator that undergoes GABA-AT-catalyzed hydrolysis of the difluoromethylene group to a carboxylic acid with concomitant loss of two fluoride ions and coenzyme conversion to pyridoxamine 5′-phosphate (PMP). The partition ratio for CPP-115 with GABA-AT is about 2000, releasing cyclopentanone-2,4-dicarboxylate (22) and two other precursors of this compound (20 and 21). Time-dependent inactivation occurs by a conformational change induced by the formation of the aldimine of 4-aminocyclopentane-1,3-dicarboxylic acid and PMP (20), which disrupts an electrostatic interaction between Glu270 and Arg445 to form an electrostatic interaction between Arg445 and the newly formed carboxylate produced by hydrolysis of the difluoromethylene group in CPP-115, resulting in a noncovalent, tightly bound complex. This represents a novel mechanism for inactivation of GABA-AT and a new approach for the design of mechanism-based inactivators in general.
Co-reporter:Hoang V. Le; Dustin D. Hawker; Rui Wu; Emma Doud; Julia Widom; Ruslan Sanishvili; Dali Liu; Neil L. Kelleher
Journal of the American Chemical Society 2015 Volume 137(Issue 13) pp:4525-4533
Publication Date(Web):March 17, 2015
DOI:10.1021/jacs.5b01155
Low levels of γ-aminobutyric acid (GABA), one of two major neurotransmitters that regulate brain neuronal activity, are associated with many neurological disorders, such as epilepsy, Parkinson’s disease, Alzheimer’s disease, Huntington’s disease, and cocaine addiction. One of the main methods to raise the GABA level in human brain is to use small molecules that cross the blood–brain barrier and inhibit the activity of γ-aminobutyric acid aminotransferase (GABA-AT), the enzyme that degrades GABA. We have designed a series of conformationally restricted tetrahydrothiophene-based GABA analogues with a properly positioned leaving group that could facilitate a ring-opening mechanism, leading to inactivation of GABA-AT. One compound in the series is 8 times more efficient an inactivator of GABA-AT than vigabatrin, the only FDA-approved inactivator of GABA-AT. Our mechanistic studies show that the compound inactivates GABA-AT by a new mechanism. The metabolite resulting from inactivation does not covalently bind to amino acid residues of GABA-AT but stays in the active site via H-bonding interactions with Arg-192, a π–π interaction with Phe-189, and a weak nonbonded S···O═C interaction with Glu-270, thereby inactivating the enzyme.
Co-reporter:Wei Tang; Huiying Li; Emma H. Doud; Yunqiu Chen; Stephanie Choing; Carla Plaza; Neil L. Kelleher; Thomas L. Poulos
Journal of the American Chemical Society 2015 Volume 137(Issue 18) pp:5980-5989
Publication Date(Web):April 15, 2015
DOI:10.1021/jacs.5b01202
Nitric oxide synthase (NOS) catalyzes the conversion of l-arginine to l-citrulline and the second messenger nitric oxide. Three mechanistic pathways are proposed for the inactivation of neuronal NOS (nNOS) by (S)-2-amino-5-(2-(methylthio)acetimidamido)pentanoic acid (1): sulfide oxidation, oxidative dethiolation, and oxidative demethylation. Four possible intermediates were synthesized. All compounds were assayed with nNOS, their IC50, KI, and kinact values were obtained, and their crystal structures were determined. The identification and characterization of the products formed during inactivation provide evidence for the details of the inactivation mechanism. On the basis of these studies, the most probable mechanism for the inactivation of nNOS involves oxidative demethylation with the resulting thiol coordinating to the cofactor heme iron. Although nNOS is a heme-containing enzyme, this is the first example of a NOS that catalyzes an S-demethylation reaction; the novel mechanism of inactivation described here could be applied to the design of inactivators of other heme-dependent enzymes.
Co-reporter:Soosung Kang; Huiying Li; Wei Tang; Pavel Martásek; Linda J. Roman; Thomas L. Poulos
Journal of Medicinal Chemistry 2015 Volume 58(Issue 14) pp:5548-5560
Publication Date(Web):June 29, 2015
DOI:10.1021/acs.jmedchem.5b00573
We have analyzed a recently obtained crystal structure of human neuronal nitric oxide synthase (nNOS) and then designed and synthesized several 2-aminopyridine derivatives containing a truncated side chain to avoid the hydrophobic pocket that differentiates human and rat nNOS in an attempt to explore alternative binding poses along the substrate access channel of human nNOS. Introduction of an N-methylethane-1,2-diamine side chain and conformational constraints such as benzonitrile and pyridine as the middle aromatic linker were sufficient to increase human and rat nNOS binding affinity and inducible and endothelial NOS selectivity. We found that 14b is a potent inhibitor; the binding modes with human and rat nNOS are unexpected, inducing side chain rotamer changes in Gln478 (rat) at the top of the active site. Compound 19c exhibits Ki values of 24 and 55 nM for rat and human nNOS, respectively, with 153-fold iNOS and 1040-fold eNOS selectivity. 19c has 18% oral bioavailability.
Co-reporter:Yinan Zhang; Kevin Tianmeng Zhao; Susan G. Fox; Jinho Kim; Donald R. Kirsch; Robert J. Ferrante; Richard I. Morimoto
Journal of Medicinal Chemistry 2015 Volume 58(Issue 15) pp:5942-5949
Publication Date(Web):July 17, 2015
DOI:10.1021/acs.jmedchem.5b00561
Pyrazolone derivatives have previously been found to be inhibitors of Cu/Zn superoxide dismutase 1 (SOD1)-dependent protein aggregation, which extended survival of an amyotrophic lateral sclerosis (ALS) mouse model. On the basis of ADME analysis, we describe herein a new series of tertiary amine-containing pyrazolones and their structure–activity relationships. Further conversion to the conjugate salts greatly improved their solubility. Phosphate compound 17 exhibited numerous benefits both to cellular activity and to CNS-related drug-like properties in vitro and in vivo, including microsomal stability, tolerated toxicity, and blood–brain barrier permeation. These results indicate that tertiary amine pyrazolones comprise a valuable class of ALS drug candidates.
Co-reporter:Maris A. Cinelli; Huiying Li; Anthony V. Pensa; Soosung Kang; Linda J. Roman; Pavel Martásek; Thomas L. Poulos
Journal of Medicinal Chemistry 2015 Volume 58(Issue 21) pp:8694-8712
Publication Date(Web):October 15, 2015
DOI:10.1021/acs.jmedchem.5b01330
Excess nitric oxide (NO) produced by neuronal nitric oxide synthase (nNOS) is implicated in neurodegenerative disorders. As a result, inhibition of nNOS and reduction of NO levels is desirable therapeutically, but many nNOS inhibitors are poorly bioavailable. Promising members of our previously reported 2-aminoquinoline class of nNOS inhibitors, although orally bioavailable and brain-penetrant, suffer from unfavorable off-target binding to other CNS receptors, and they resemble known promiscuous binders. Rearranged phenyl ether- and aniline-linked 2-aminoquinoline derivatives were therefore designed to (a) disrupt the promiscuous binding pharmacophore and diminish off-target interactions and (b) preserve potency, isoform selectivity, and cell permeability. A series of these compounds was synthesized and tested against purified nNOS, endothelial NOS (eNOS), and inducible NOS (iNOS) enzymes. One compound, 20, displayed high potency, selectivity, and good human nNOS inhibition, and retained some permeability in a Caco-2 assay. Most promisingly, CNS receptor counterscreening revealed that this rearranged scaffold significantly reduces off-target binding.
Co-reporter:Jeffrey K. Holden, Soosung Kang, Federico C. Beasley, Maris A. Cinelli, Huiying Li, Saurabh G. Roy, Dillon Dejam, Aimee L. Edinger, Victor Nizet, Richard B. Silverman, Thomas L. Poulos
Chemistry & Biology 2015 Volume 22(Issue 6) pp:785-792
Publication Date(Web):18 June 2015
DOI:10.1016/j.chembiol.2015.05.013
•Inhibitors selective toward bacterial nitric oxide synthase have been identified•These inhibitors are antimicrobial against MRSA•Crystallography reveals the structural basis for selectivity•NOS inhibitor library rapidly screened to identify potent inhibitorsBacterial infections associated with methicillin-resistant Staphylococcus aureus (MRSA) are a major economic burden to hospitals, and confer high rates of morbidity and mortality among those infected. Exploitation of novel therapeutic targets is thus necessary to combat this dangerous pathogen. Here, we report on the identification and characterization, including crystal structures, of two nitric oxide synthase (NOS) inhibitors that function as antimicrobials against MRSA. These data provide the first evidence that bacterial NOS (bNOS) inhibitors can work synergistically with oxidative stress to enhance MRSA killing. Crystal structures show that each inhibitor contacts an active site Ile residue in bNOS that is Val in the mammalian NOS isoforms. Mutagenesis studies show that the additional nonpolar contacts provided by the Ile in bNOS contribute to tighter binding toward the bacterial enzyme.Figure optionsDownload full-size imageDownload high-quality image (156 K)Download as PowerPoint slide
Co-reporter:Paramita Mukherjee; Huiying Li; Irina Sevrioukova; Georges Chreifi; Pavel Martásek; Linda J. Roman; Thomas L. Poulos
Journal of Medicinal Chemistry 2015 Volume 58(Issue 3) pp:1067-1088
Publication Date(Web):December 9, 2014
DOI:10.1021/jm501719e
Selective inhibition of neuronal nitric oxide synthase (nNOS) is an important therapeutic approach to target neurodegenerative disorders. However, the majority of the nNOS inhibitors developed are arginine mimetics and, therefore, suffer from poor bioavailability. We designed a novel strategy to combine a more pharmacokinetically favorable 2-imidazolylpyrimidine head with promising structural components from previous inhibitors. In conjunction with extensive structure–activity studies, several highly potent and selective inhibitors of nNOS were discovered. X-ray crystallographic analysis reveals that these type II inhibitors utilize the same hydrophobic pocket to gain strong inhibitory potency (13), as well as high isoform selectivity. Interestingly, select compounds from this series (9) showed good permeability and low efflux in a Caco-2 assay, suggesting potential oral bioavailability, and exhibited minimal off-target binding to 50 central nervous system receptors. Furthermore, even with heme-coordinating groups in the molecule, modifying other pharmacophoric fragments minimized undesirable inhibition of cytochrome P450s from human liver microsomes.
Co-reporter:Jeffrey K. Holden; Soosung Kang; Scott A. Hollingsworth; Huiying Li; Nathan Lim; Steven Chen; He Huang; Fengtian Xue; Wei Tang; Richard B. Silverman;Thomas L. Poulos
Journal of Medicinal Chemistry 2015 Volume 58(Issue 2) pp:994-1004
Publication Date(Web):December 18, 2014
DOI:10.1021/jm501723p
Inhibition of bacterial nitric oxide synthase (bNOS) has the potential to improve the efficacy of antimicrobials used to treat infections by Gram-positive pathogens Staphylococcus aureus and Bacillus anthracis. However, inhibitor specificity toward bNOS over the mammalian NOS (mNOS) isoforms remains a challenge because of the near identical NOS active sites. One key structural difference between the NOS isoforms is the amino acid composition of the pterin cofactor binding site that is adjacent to the NOS active site. Previously, we demonstrated that a NOS inhibitor targeting both the active and pterin sites was potent and functioned as an antimicrobial (Holden, , Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 18127). Here we present additional crystal structures, binding analyses, and bacterial killing studies of inhibitors that target both the active and pterin sites of a bNOS and function as antimicrobials. Together, these data provide a framework for continued development of bNOS inhibitors, as each molecule represents an excellent chemical scaffold for the design of isoform selective bNOS inhibitors.
Co-reporter:Hyunbeom Lee, Hoang V. Le, Rui Wu, Emma Doud, Ruslan Sanishvili, John F. Kellie, Phillip D. Compton, Boobalan Pachaiyappan, Dali Liu, Neil L. Kelleher, and Richard B. Silverman
ACS Chemical Biology 2015 Volume 10(Issue 9) pp:2087
Publication Date(Web):June 25, 2015
DOI:10.1021/acschembio.5b00212
When γ-aminobutyric acid (GABA), the major inhibitory neurotransmitter in the mammalian central nervous system, falls below a threshold level, seizures occur. One approach to raise GABA concentrations is to inhibit GABA aminotransferase (GABA-AT), a pyridoxal 5′-phosphate-dependent enzyme that degrades GABA. We have previously developed (1S,3S)-3-amino-4-difluoromethylene-1-cyclopentanoic acid (CPP-115), which is 186 times more efficient in inactivating GABA-AT than vigabatrin, the only FDA-approved inactivator of GABA-AT. We also developed (E)- and (Z)-(1S,3S)-3-amino-4-fluoromethylenyl-1-cyclopentanoic acid (1 and 2, respectively), monofluorinated analogs of CPP-115, which are comparable to vigabatrin in inactivating GABA-AT. Here, we report the mechanism of inactivation of GABA-AT by 1 and 2. Both produce a metabolite that induces disruption of the Glu270–Arg445 salt bridge to accommodate interaction between the metabolite formyl group and Arg445. This is the second time that Arg445 has interacted with a ligand and is involved in GABA-AT inactivation, thereby confirming the importance of Arg445 in future inactivator design.
Co-reporter:Jeffrey K. Holden, Dillon Dejam, Matthew C. Lewis, He Huang, Soosung Kang, Qing Jing, Fengtian Xue, Richard B. Silverman, and Thomas L. Poulos
Biochemistry 2015 Volume 54(Issue 26) pp:4075-4082
Publication Date(Web):June 11, 2015
DOI:10.1021/acs.biochem.5b00431
Nitric oxide generated by bacterial nitric oxide synthase (NOS) increases the susceptibility of Gram-positive pathogens Staphylococcus aureus and Bacillus anthracis to oxidative stress, including antibiotic-induced oxidative stress. Not surprisingly, NOS inhibitors also improve the effectiveness of antimicrobials. Development of potent and selective bacterial NOS inhibitors is complicated by the high active site sequence and structural conservation shared with the mammalian NOS isoforms. To exploit bacterial NOS for the development of new therapeutics, recognition of alternative NOS surfaces and pharmacophores suitable for drug binding is required. Here, we report on a wide number of inhibitor-bound bacterial NOS crystal structures to identify several compounds that interact with surfaces unique to the bacterial NOS. Although binding studies indicate that these inhibitors weakly interact with the NOS active site, many of the inhibitors reported here provide a revised structural framework for the development of new antimicrobials that target bacterial NOS. In addition, mutagenesis studies reveal several key residues that unlock access to bacterial NOS surfaces that could provide the selectivity required to develop potent bacterial NOS inhibitors.
Co-reporter:Ehud Zigmond, Ami Ben Ya’acov, Hyunbeom Lee, Yoav Lichtenstein, Zvi Shalev, Yoav Smith, Lidya Zolotarov, Ehud Ziv, Rony Kalman, Hoang V. Le, Hejun Lu, Richard B. Silverman, and Yaron Ilan
ACS Medicinal Chemistry Letters 2015 Volume 6(Issue 8) pp:840
Publication Date(Web):May 29, 2015
DOI:10.1021/acsmedchemlett.5b00153
Hepatocellular carcinoma is the second leading cause of cancer death worldwide. DNA microarray analysis identified the ornithine aminotransferase (OAT) gene as a prominent gene overexpressed in hepatocellular carcinoma (HCC) from Psammomys obesus. In vitro studies demonstrated inactivation of OAT by gabaculine (1), a neurotoxic natural product, which suppressed in vitro proliferation of two HCC cell lines. Alpha-fetoprotein (AFP) secretion, a biomarker for HCC, was suppressed by gabaculine in both cell lines, but not significantly. Because of the active site similarity between GABA aminotransferase (GABA-AT) and OAT, a library of 24 GABA-AT inhibitors was screened to identify a more selective inhibitor of OAT. (1S,3S)-3-Amino-4-(hexafluoropropan-2-ylidene)cyclopentane-1-carboxylic acid (2) was found to be an inactivator of OAT that only weakly inhibits GABA-AT, l-aspartate aminotransferase, and l-alanine aminotransferase. In vitro administration of 2 significantly suppressed AFP secretion in both Hep3B and HepG2 HCC cells; in vivo, 2 significantly suppressed AFP serum levels and tumor growth in HCC-harboring mice, even at 0.1 mg/kg. Overexpression of the OAT gene in HCC and the ability to block the growth of HCC by OAT inhibitors support the role of OAT as a potential therapeutic target to inhibit HCC growth. This is the first demonstration of suppression of HCC by an OAT inactivator.Keywords: alpha fetoprotein; antitumor agent; GABA aminotransferase; Hepatocellular carcinoma; ornithine aminotransferase; selective inhibitors
Co-reporter:Mohammad A. Khanfar, Luisa Quinti, Hua Wang, Johnathan Nobles, Aleksey G. Kazantsev, and Richard B. Silverman
ACS Medicinal Chemistry Letters 2015 Volume 6(Issue 5) pp:607
Publication Date(Web):March 26, 2015
DOI:10.1021/acsmedchemlett.5b00075
Inhibitors of sirtuin-2 (SIRT2) deacetylase have been shown to be protective in various models of Huntington’s disease (HD) by decreasing polyglutamine aggregation, a hallmark of HD pathology. The present study was directed at optimizing the potency of SIRT2 inhibitors containing the 3-(benzylsulfonamido)benzamide scaffold and improving their metabolic stability. Molecular modeling and docking studies revealed an unfavorable role of the sulfonamide moiety for SIRT2 binding. This prompted us to replace the sulfonamide with thioether, sulfoxide, or sulfone groups. The thioether analogues were the most potent SIRT2 inhibitors with a two- to three-fold increase in potency relative to their corresponding sulfonamide analogues. The newly synthesized compounds also demonstrated higher SIRT2 selectivity over SIRT1 and SIRT3. Two thioether-derived compounds (17 and 18) increased α-tubulin acetylation in a dose-dependent manner in at least one neuronal cell line, and 18 was found to inhibit polyglutamine aggregation in PC12 cells.Keywords: 3-(benzylthio)benzamide; docking; Huntington’s disease; polyglutamine aggregation; SIRT2
Co-reporter:Soosung Kang, Mizuki Watanabe, J.C. Jacobs, Masaya Yamaguchi, Samira Dahesh, Victor Nizet, Thomas S. Leyh, Richard B. Silverman
European Journal of Medicinal Chemistry 2015 90() pp: 448-461
Publication Date(Web):
DOI:10.1016/j.ejmech.2014.11.040
Co-reporter:Paramita Mukherjee, Maris A. Cinelli, Soosung Kang and Richard B. Silverman
Chemical Society Reviews 2014 vol. 43(Issue 19) pp:6814-6838
Publication Date(Web):19 Feb 2014
DOI:10.1039/C3CS60467E
Nitric oxide (NO) is an important signaling molecule in the human body, playing a crucial role in cell and neuronal communication, regulation of blood pressure, and in immune activation. However, overproduction of NO by the neuronal isoform of nitric oxide synthase (nNOS) is one of the fundamental causes underlying neurodegenerative disorders and neuropathic pain. Therefore, developing small molecules for selective inhibition of nNOS over related isoforms (eNOS and iNOS) is therapeutically desirable. The aims of this review focus on the regulation and dysregulation of NO signaling, the role of NO in neurodegeneration and pain, the structure and mechanism of nNOS, and the use of this information to design selective inhibitors of this enzyme. Structure-based drug design, the bioavailability and pharmacokinetics of these inhibitors, and extensive target validation through animal studies are addressed.
Co-reporter:He Huang ; Huiying Li ; Sun Yang ; Georges Chreifi ; Pavel Martásek ; Linda J. Roman ; Frank L. Meyskens ; Thomas L. Poulos
Journal of Medicinal Chemistry 2014 Volume 57(Issue 3) pp:686-700
Publication Date(Web):January 21, 2014
DOI:10.1021/jm401252e
Selective inhibitors of neuronal nitric oxide synthase (nNOS) are regarded as valuable and powerful agents with therapeutic potential for the treatment of chronic neurodegenerative pathologies and human melanoma. Here, we describe a novel hybrid strategy that combines the pharmacokinetically promising thiophene-2-carboximidamide fragment and structural features of our previously reported potent and selective aminopyridine inhibitors. Two inhibitors, 13 and 14, show low nanomolar inhibitory potency (Ki = 5 nM for nNOS) and good isoform selectivities (nNOS over eNOS [440- and 540-fold, respectively] and over iNOS [260- and 340-fold, respectively]). The crystal structures of these nNOS–inhibitor complexes reveal a new hot spot that explains the selectivity of 14 and why converting the secondary to tertiary amine leads to enhanced selectivity. More importantly, these compounds are the first highly potent and selective nNOS inhibitory agents that exhibit excellent in vitro efficacy in melanoma cell lines.
Co-reporter:Maris A. Cinelli ; Huiying Li ; Georges Chreifi ; Pavel Martásek ; Linda J. Roman ; Thomas L. Poulos
Journal of Medicinal Chemistry 2014 Volume 57(Issue 4) pp:1513-1530
Publication Date(Web):January 28, 2014
DOI:10.1021/jm401838x
Since high levels of nitric oxide (NO) are implicated in neurodegenerative disorders, inhibition of the neuronal isoform of nitric oxide synthase (nNOS) and reduction of NO levels are therapeutically desirable. Nonetheless, many nNOS inhibitors mimic l-arginine and are poorly bioavailable. 2-Aminoquinoline-based scaffolds were designed with the hope that they could (a) mimic aminopyridines as potent, isoform-selective arginine isosteres and (b) possess chemical properties more conducive to oral bioavailability and CNS penetration. A series of these compounds was synthesized and assayed against purified nNOS enzymes, endothelial NOS (eNOS), and inducible NOS (iNOS). Several compounds built on a 7-substituted 2-aminoquinoline core are potent and isoform-selective; X-ray crystallography indicates that aminoquinolines exert inhibitory effects by mimicking substrate interactions with the conserved active site glutamate residue. The most potent and selective compounds, 7 and 15, were tested in a Caco-2 assay and showed good permeability and low efflux, suggesting high potential for oral bioavailability.
Co-reporter:Soosung Kang ; Wei Tang ; Huiying Li ; Georges Chreifi ; Pavel Martásek ; Linda J. Roman ; Thomas L. Poulos
Journal of Medicinal Chemistry 2014 Volume 57(Issue 10) pp:4382-4396
Publication Date(Web):April 23, 2014
DOI:10.1021/jm5004182
Overproduction of NO by nNOS is implicated in the pathogenesis of diverse neuronal disorders. Since NO signaling is involved in diverse physiological functions, selective inhibition of nNOS over other isoforms is essential to minimize side effects. A series of α-amino functionalized aminopyridine derivatives (3–8) were designed to probe the structure–activity relationship between ligand, heme propionate, and H4B. Compound 8R was identified as the most potent and selective molecule of this study, exhibiting a Ki of 24 nM for nNOS, with 273-fold and 2822-fold selectivity against iNOS and eNOS, respectively. Although crystal structures of 8R complexed with nNOS and eNOS revealed a similar binding mode, the selectivity stems from the distinct electrostatic environments in two isoforms that result in much lower inhibitor binding free energy in nNOS than in eNOS. These findings provide a basis for further development of simple, but even more selective and potent, nNOS inhibitors.
Co-reporter:Mohammad A. Khanfar, Luisa Quinti, Hua Wang, Soo Hyuk Choi, Aleksey G. Kazantsev, Richard B. Silverman
European Journal of Medicinal Chemistry 2014 Volume 76() pp:414-426
Publication Date(Web):9 April 2014
DOI:10.1016/j.ejmech.2014.02.003
•Inhibitors of SIRT2 are protective in models of Huntington's disease (HD).•Inhibitors of SIRT2 decrease polyglutamine aggregation in HD.•Sulfobenzoic acid derivatives shown to be selective SIRT2 inhibitors.•Bioactive in α-tubulin K40 acetylation in two neuronal cell lines.•Bioactive in polyglutamine aggregation assay.Inhibitors of sirtuin-2 deacetylase (SIRT2) have been shown to be protective in various models of Huntington's disease (HD) by decreasing polyglutamine aggregation, a hallmark of HD pathology. The present study was directed at optimizing the potency of SIRT2 inhibitors containing the neuroprotective sulfobenzoic acid scaffold and improving their pharmacology. To achieve that goal, 176 analogues were designed, synthesized, and tested in deacetylation assays against the activities of major human sirtuins SIRT1-3. This screen yielded 15 compounds with enhanced potency for SIRT2 inhibition and 11 compounds having SIRT2 inhibition equal to reference compound AK-1. The newly synthesized compounds also demonstrated higher SIRT2 selectivity over SIRT1 and SIRT3. These candidates were subjected to a dose–response bioactivity assay, measuring an increase in α-tubulin K40 acetylation in two neuronal cell lines, which yielded five compounds bioactive in both cell lines and eight compounds bioactive in at least one of the cell lines tested. These bioactive compounds were subsequently tested in a tertiary polyglutamine aggregation assay, which identified five inhibitors. ADME properties of the bioactive SIRT2 inhibitors were assessed, which revealed a significant improvement of the pharmacological properties of the new entities, reaching closer to the goal of a clinically-viable candidate.
Co-reporter:Paul C. Trippier, Kevin Tianmeng Zhao, Susan G. Fox, Isaac T. Schiefer, Radhia Benmohamed, Jason Moran, Donald R. Kirsch, Richard I. Morimoto, and Richard B. Silverman
ACS Chemical Neuroscience 2014 Volume 5(Issue 9) pp:823
Publication Date(Web):July 7, 2014
DOI:10.1021/cn500147v
Amyotrophic lateral sclerosis (ALS) is a progressive and ultimately fatal neurodegenerative disease. Pyrazolone containing small molecules have shown significant disease attenuating efficacy in cellular and murine models of ALS. Pyrazolone based affinity probes were synthesized to identify high affinity binding partners and ascertain a potential biological mode of action. Probes were confirmed to be neuroprotective in PC12-SOD1G93A cells. PC12-SOD1G93A cell lysates were used for protein pull-down, affinity purification, and subsequent proteomic analysis using LC-MS/MS. Proteomics identified the 26S proteasome regulatory subunit 4 (PSMC1), 26S proteasome regulatory subunit 6B (PSMC4), and T-complex protein 1 (TCP-1) as putative protein targets. Coincubation with appropriate competitors confirmed the authenticity of the proteomics results. Activation of the proteasome by pyrazolones was demonstrated in the absence of exogenous proteasome inhibitor and by restoration of cellular protein degradation of a fluorogenic proteasome substrate in PC12-SOD1G93A cells. Importantly, supplementary studies indicated that these molecules do not induce a heat shock response. We propose that pyrazolones represent a rare class of molecules that enhance proteasomal activation in the absence of a heat shock response and may have therapeutic potential in ALS.Keywords: Amyotrophic lateral sclerosis; Neurodegeneration; Drug Discovery; Proteasome activator; Pyrazolone; Target identification
Co-reporter:Qing Jing, Huiying Li, Linda J. Roman, Pavel Martásek, Thomas L. Poulos, and Richard B. Silverman
ACS Medicinal Chemistry Letters 2014 Volume 5(Issue 1) pp:56-60
Publication Date(Web):November 5, 2013
DOI:10.1021/ml400381s
The three important mammalian isozymes of nitric oxide synthase (NOS) are neuronal NOS (nNOS), endothelial NOS (eNOS), and inducible NOS (iNOS). Inhibitors of nNOS show promise as treatments for neurodegenerative diseases. Eight easily synthesized compounds containing either one (20a,b) or two (9a–d; 15a,b) 2-amino-4-methylpyridine groups with a chiral pyrrolidine linker were designed as selective nNOS inhibitors. Inhibitor 9c is the best of these compounds, having a potency of 9.7 nM and dual selectivity of 693 and 295 against eNOS and iNOS, respectively. Crystal structures of nNOS complexed with either 9a or 9c show a double-headed binding mode, where each 2-aminopyridine headgroup interacts with either a nNOS active site Glu residue or a heme propionate. In addition, the pyrrolidine nitrogen of 9c contributes additional hydrogen bonds to the heme propionate, resulting in a unique binding orientation. In contrast, the lack of hydrogen bonds from the pyrrolidine of 9a to the heme propionate allows the inhibitor to adopt two different binding orientations. Both 9a and 9c bind to eNOS in a single-headed mode, which is the structural basis for the isozyme selectivity.Keywords: enzyme inhibition; neurodegenerative diseases; Nitric oxide; nitric oxide synthase;
Co-reporter:Roman Davydov, Kristin Jansen Labby, Sarah E. Chobot, Dmitriy A. Lukoyanov, Brian R. Crane, Richard B. Silverman, and Brian M. Hoffman
Biochemistry 2014 Volume 53(Issue 41) pp:
Publication Date(Web):September 24, 2014
DOI:10.1021/bi500485z
Nitric oxide synthase (NOS) catalyzes the conversion of l-arginine to l-citrulline and NO in a two-step process involving the intermediate Nω-hydroxy-l-arginine (NHA). It was shown that Cpd I is the oxygenating species for l-arginine; the hydroperoxo ferric intermediate is the reactive intermediate with NHA. Methylation of the Nω-OH and Nω-H of NHA significantly inhibits the conversion of NHA into NO and l-citrulline by mammalian NOS. Kinetic studies now show that Nω-methylation of NHA has a qualitatively similar effect on H2O2-dependent catalysis by bacterial gsNOS. To elucidate the effect of methylating Nω-hydroxy l-arginine on the properties and reactivity of the one-electron-reduced oxy-heme center of NOS, we have applied cryoreduction/annealing/EPR/ENDOR techniques. Measurements of solvent kinetic isotope effects during 160 K cryoannealing cryoreduced oxy-gsNOS/NHA confirm the hydroperoxo ferric intermediate as the catalytically active species of step two. Product analysis for cryoreduced samples with methylated NHA’s, NHMA, NMOA, and NMMA, annealed to 273 K, show a correlation of yields of l-citrulline with the intensity of the g 2.26 EPR signal of the peroxo ferric species trapped at 77 K, which converts to the reactive hydroperoxo ferric state. There is also a correlation between the yield of l-citrulline in these experiments and kobs for the H2O2-dependent conversion of the substrates by gsNOS. Correspondingly, no detectable amount of cyanoornithine, formed when Cpd I is the reactive species, was found in the samples. Methylation of the NHA guanidinium Nω-OH and Nω-H inhibits the second NO-producing reaction by favoring protonation of the ferric-peroxo to form unreactive conformers of the ferric-hydroperoxo state. It is suggested that this is caused by modification of the distal-pocket hydrogen-bonding network of oxy gsNOS and introduction of an ordered water molecule that facilitates delivery of the proton(s) to the one-electron-reduced oxy-heme moiety. These results illustrate how variations in the properties of the substrate can modulate the reactivity of a monooxygenase.
Co-reporter:Huiying Li, Joumana Jamal, Silvia Delker, Carla Plaza, Haitao Ji, Qing Jing, He Huang, Soosung Kang, Richard B. Silverman, and Thomas L. Poulos
Biochemistry 2014 Volume 53(Issue 32) pp:5272-5279
Publication Date(Web):August 4, 2014
DOI:10.1021/bi500561h
Many pyrrolidine-based inhibitors highly selective for neuronal nitric oxide synthase (nNOS) over endothelial NOS (eNOS) exhibit dramatically different binding modes. In some cases, the inhibitor binds in a 180° flipped orientation in nNOS relative to eNOS. From the several crystal structures we have determined, we know that isoform selectivity correlates with the rotamer position of a conserved tyrosine residue that H-bonds with a heme propionate. In nNOS, this Tyr more readily adopts the out-rotamer conformation, while in eNOS, the Tyr tends to remain fixed in the original in-rotamer conformation. In the out-rotamer conformation, inhibitors are able to form better H-bonds with the protein and heme, thus increasing inhibitor potency. A segment of polypeptide that runs along the surface near the conserved Tyr has long been thought to be the reason for the difference in Tyr mobility. Although this segment is usually disordered in both eNOS and nNOS, sequence comparisons and modeling from a few structures show that this segment is structured quite differently in eNOS and nNOS. In this study, we have probed the importance of this surface segment near the Tyr by making a few mutants in the region followed by crystal structure determinations. In addition, because the segment near the conserved Tyr is highly ordered in iNOS, we also determined the structure of an iNOS–inhibitor complex. This new structure provides further insight into the critical role that mobility plays in isoform selectivity.
Co-reporter:Qing Jing, Huiying Li, Linda J. Roman, Pavel Martásek, Thomas L. Poulos, Richard B. Silverman
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 18) pp:4504-4510
Publication Date(Web):15 September 2014
DOI:10.1016/j.bmcl.2014.07.079
To develop potent and selective nNOS inhibitors, a new series of double-headed molecules with chiral linkers that derive from natural amino acid derivatives have been designed and synthesized. The new structures integrate a thiophenecarboximidamide head with two types of chiral linkers, presenting easy synthesis and good inhibitory properties. Inhibitor (S)-9b exhibits a potency of 14.7 nM against nNOS and is 1134 and 322-fold more selective for nNOS over eNOS and iNOS, respectively. Crystal structures show that the additional binding between the aminomethyl moiety of 9b and propionate A on the heme and tetrahydrobiopterin (H4B) in nNOS, but not eNOS, contributes to its high selectivity. This work demonstrates the advantage of integrating known structures into structure optimization, and it should be possible to more readily develop compounds that incorporate bioavailability with these advanced features. Moreover, this integrative strategy is a general approach in new drug discovery.
Co-reporter:Guoyao Xia, Radhia Benmohamed, Richard I. Morimoto, Donald R. Kirsch, Richard B. Silverman
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 21) pp:5098-5101
Publication Date(Web):1 November 2014
DOI:10.1016/j.bmcl.2014.08.066
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease characterized by the progressive loss of motor neurons, leading to muscle weakness, paralysis, and death, most often from respiratory failure. Over 200 pyrimidine-2,4,6-trione (PYT) small molecules, which prevent aggregation and reduce the associated toxicity of mutant superoxide dismutase 1 (SOD1) found in patients with familial ALS, have been synthesized and tested. One of the compounds (1,3-bis(2-phenylethyl)pyrimidine-2,4,6(1H,3H,5H)-trione, (1) was previously found to have an excellent combination of potency efficacy, and some desirable pharmacokinetic properties. To improve the solubility and metabolic stability properties of this compound, deuterium and fluorine were introduced into 1. New analogs with better solubility, plasma stability, and human microsome stability were identified.
Co-reporter:Paul C. Trippier, Radhia Benmohamed, Donald R. Kirsch, Richard B. Silverman
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 2) pp:691
Publication Date(Web):15 January 2014
DOI:10.1016/j.bmcl.2013.12.027
Co-reporter:Paul C. Trippier ; Kristin Jansen Labby ; Dustin D. Hawker ; Jan J. Mataka
Journal of Medicinal Chemistry 2013 Volume 56(Issue 8) pp:3121-3147
Publication Date(Web):March 4, 2013
DOI:10.1021/jm3015926
The development of new therapeutics for the treatment of neurodegenerative pathophysiologies currently stands at a crossroads. This presents an opportunity to transition future drug discovery efforts to target disease modification, an area in which much still remains unknown. In this Perspective we examine recent progress in the areas of neurodegenerative drug discovery, focusing on some of the most common targets and mechanisms: N-methyl-d-aspartic acid (NMDA) receptors, voltage gated calcium channels (VGCCs), neuronal nitric oxide synthase (nNOS), oxidative stress from reactive oxygen species, and protein aggregation. These represent the key players identified in neurodegeneration and are part of a complex, intertwined signaling cascade. The synergistic delivery of two or more compounds directed against these targets, along with the design of small molecules with multiple modes of action, should be explored in pursuit of more effective clinical treatments for neurodegenerative diseases.
Co-reporter:Soosung Kang ; Garry Cooper ; Sara Fernandez Dunne ; Chi-Hao Luan ; D. James Surmeier
Journal of Medicinal Chemistry 2013 Volume 56(Issue 11) pp:4786-4797
Publication Date(Web):May 7, 2013
DOI:10.1021/jm4005048
CaV1.3 L-type calcium channels (LTCCs) have been a potential target for Parkinson’s disease since calcium ion influx through the channel was implicated in the generation of mitochondrial oxidative stress, causing cell death in the dopaminergic neurons. Selective inhibition of CaV1.3 over other LTCC isoforms, especially CaV1.2, is critical to minimize potential side effects. We recently identified pyrimidinetriones (PYTs) as a CaV1.3-selective scaffold; here we report the structure–activity relationship of PYTs with both CaV1.3 and CaV1.2 LTCCs. By variation of the substituents on the cyclopentyl and arylalkyl groups of PYT, SAR studies allowed characterization of the CaV1.3 and CaV1.2 LTCCs binding sites. The SAR also identified four important moieties that either retain selectivity or enhance binding affinity. Our study represents a significant enhancement of the SAR of PYTs at CaV1.3 and CaV1.2 LTCCs and highlights several advances in the lead optimization and diversification of this family of compounds for drug development.
Co-reporter:He Huang ; Huiying Li ; Pavel Martásek ; Linda J. Roman ; Thomas L. Poulos
Journal of Medicinal Chemistry 2013 Volume 56(Issue 7) pp:3024-3032
Publication Date(Web):March 1, 2013
DOI:10.1021/jm4000984
Nitric oxide synthases (NOSs) comprise three closely related isoforms that catalyze the oxidation of l-arginine to l-citrulline and the important second messenger nitric oxide (NO). Pharmacological selective inhibition of neuronal NOS (nNOS) has the potential to be therapeutically beneficial in various neurodegenerative diseases. Here, we present a structure-guided, selective nNOS inhibitor design based on the crystal structure of lead compound 1 in nNOS. The best inhibitor, 7, exhibited low nanomolar inhibitory potency and good isoform selectivities (nNOS over eNOS and iNOS are 472-fold and 239-fold, respectively). Consistent with the good selectivity, 7 binds to nNOS and eNOS with different binding modes. The distinctly different binding modes of 7, driven by the critical residue Asp597 in nNOS, offers compelling insight to explain its isozyme selectivity, which should guide future drug design programs.
Co-reporter:Yinan Zhang ; Radhia Benmohamed ; He Huang ; Tian Chen ; Cindy Voisine ; Richard I. Morimoto ; Donald R. Kirsch
Journal of Medicinal Chemistry 2013 Volume 56(Issue 6) pp:2665-2675
Publication Date(Web):February 27, 2013
DOI:10.1021/jm400079a
The arylsulfanylpyrazolone and aryloxanylpyrazolone scaffolds previously were reported to inhibit Cu/Zn superoxide dismutase 1 dependent protein aggregation and to extend survival in the ALS mouse model. However, further evaluation of these compounds indicated weak pharmacokinetic properties and a relatively low maximum tolerated dose. On the basis of an ADME analysis, a new series of compounds, the arylazanylpyrazolones, has been synthesized, and structure–activity relationships were determined. The SAR results showed that the pyrazolone ring is critical to cellular protection. The NMR, IR, and computational analyses suggest that phenol-type tautomers of the pyrazolone ring are the active pharmacophore with the arylazanylpyrazolone analogues. A comparison of experimental and calculated IR spectra is shown to be a valuable method to identify the predominant tautomer.
Co-reporter:Kristin Jansen Labby, Huiying Li, Linda J. Roman, Pavel Martásek, Thomas L. Poulos, and Richard B. Silverman
Biochemistry 2013 Volume 52(Issue 18) pp:
Publication Date(Web):April 15, 2013
DOI:10.1021/bi301571v
Nitric oxide synthase (NOS) catalyzes the conversion of l-arginine to l-citrulline through the intermediate Nω-hydroxy-l-arginine (NHA), producing nitric oxide, an important mammalian signaling molecule. Several disease states are associated with improper regulation of nitric oxide production, making NOS a therapeutic target. The first step of the NOS reaction has been well-characterized and is presumed to proceed through a compound I heme species, analogous to the cytochrome P450 mechanism. The second step, however, is enzymatically unprecedented and is thought to occur via a ferric peroxo heme species. To gain insight into the details of this unique second step, we report here the synthesis of NHA analogues bearing guanidinium methyl or ethyl substitutions and their investigation as either inhibitors of or alternate substrates for NOS. Radiolabeling studies reveal that Nω-methoxy-l-arginine, an alternative NOS substrate, produces citrulline, nitric oxide, and methanol. On the basis of these results, we propose a mechanism for the second step of NOS catalysis in which a methylated nitric oxide species is released and is further metabolized by NOS. Crystal structures of our NHA analogues bound to nNOS have been determined, revealing the presence of an active site water molecule only in the presence of singly methylated analogues. Bulkier analogues displace this active site water molecule; a different mechanism is proposed in the absence of the water molecule. Our results provide new insights into the steric and stereochemical tolerance of the NOS active site and substrate capabilities of NOS.
Co-reporter:Qing Jing, Huiying Li, Jianguo Fang, Linda J. Roman, Pavel Martásek, Thomas L. Poulos, Richard B. Silverman
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 17) pp:5323-5331
Publication Date(Web):1 September 2013
DOI:10.1016/j.bmc.2013.06.014
In certain neurodegenerative diseases damaging levels of nitric oxide (NO) are produced by neuronal nitric oxide synthase (nNOS). It, therefore, is important to develop inhibitors selective for nNOS that do not interfere with other NOS isoforms, especially endothelial NOS (eNOS), which is critical for proper functioning of the cardiovascular system. While we have been successful in developing potent and isoform-selective inhibitors, such as lead compounds 1 and 2, the ease of synthesis and bioavailability have been problematic. Here we describe a new series of compounds including crystal structures of NOS-inhibitor complexes that integrate the advantages of easy synthesis and good biological properties compared to the lead compounds. These results provide the basis for additional structure–activity relationship (SAR) studies to guide further improvement of isozyme selective inhibitors.
Co-reporter:Huiying Li, Fengtian Xue, James M. Kraus II, Haitao Ji, Kristin Jansen Labby, Jan Mataka, Silvia L. Delker, Pavel Martásek, Linda J. Roman, Thomas L. Poulos, Richard B. Silverman
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 5) pp:1333-1343
Publication Date(Web):1 March 2013
DOI:10.1016/j.bmc.2012.12.019
Inhibitors of neuronal nitric oxide synthase have been proposed as therapeutics for the treatment of different types of neurological disorders. On the basis of a cis-3,4-pyrrolidine scaffold, a series of trans-cyclopropyl- and methyl-containing nNOS inhibitors have been synthesized. The insertion of a rigid electron-withdrawing cyclopropyl ring decreases the basicity of the adjacent amino group, which resulted in decreased inhibitory activity of these inhibitors compared to the parent compound. Nonetheless, three of them exhibited double-digit nanomolar inhibition with high nNOS selectivity on the basis of in vitro enzyme assays. Crystal structures of nNOS and eNOS with these inhibitors bound provide a basis for detailed structure–activity relationship (SAR) studies. The conclusions from these studies will be used as a guide in the future development of selective NOS inhibitors.
Co-reporter:Yinan Zhang, Richard B. Silverman
Tetrahedron Letters 2013 Volume 54(Issue 6) pp:573-575
Publication Date(Web):6 February 2013
DOI:10.1016/j.tetlet.2012.11.085
A novel method to construct the 1-aryl-3-piperidone scaffold is described here. Starting from 3,5-dichloroaniline, a seven-step synthesis, without the use of protecting groups, generates the desired 3-piperidone ring in an overall yield of 30% through a key Morita–Baylis–Hillman reaction and ring-closing metathesis, providing easy access to diverse and useful heterocycles.
Co-reporter:Qing Jing, Huiying Li, Georges Chreifi, Linda J. Roman, Pavel Martásek, Thomas L. Poulos, Richard B. Silverman
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 20) pp:5674-5679
Publication Date(Web):15 October 2013
DOI:10.1016/j.bmcl.2013.08.034
To develop potent and selective nNOS inhibitors, new double-headed molecules with chiral linkers that derive from natural amino acids or their derivatives have been designed. The new structures contain two ether bonds, which greatly simplifies the synthesis and accelerates structure optimization. Inhibitor (R)-6b exhibits a potency of 32 nM against nNOS and is 475 and 244 more selective for nNOS over eNOS and iNOS, respectively. Crystal structures show that the additional binding between the aminomethyl moiety of 6b and the two heme propionates in nNOS, but not eNOS, is the structural basis for its high selectivity. This work demonstrates the importance of stereochemistry in this class of molecules, which significantly influences the potency and selectivity of the inhibitors. The structure–activity information gathered here provides a guide for future structure optimization.
Co-reporter:Amit Walia, Soosung Kang, and Richard B Silverman
The Journal of Organic Chemistry 2013 Volume 78(Issue 21) pp:10931-10937
Publication Date(Web):September 27, 2013
DOI:10.1021/jo401778e
Primary amines can be readily doubly protected as N-substituted 2,5-dimethylpyrroles. Although this protecting group is stable toward strong bases and nucleophiles, long reaction times are required for both the protection and deprotection steps, generally resulting in low deprotection yields. By employing microwave irradiation, protection and deprotection reaction times are dramatically reduced. Furthermore, deprotection with dilute hydrochloric acid in ethanol increases reaction yields. Diverse deprotection conditions have been developed in conjunction with microwave irradiation, so that protection as an N-substituted 2,5-dimethylpyrrole can be orthogonal to other standard amine protecting groups, such as tert-butyloxycarbonyl (Boc), carbobenzyloxy (Cbz), and 9-fluorenylmethyloxycarbonyl (Fmoc).
Co-reporter:Jose I. Juncosa, Hyunbeom Lee, Richard B. Silverman
Analytical Biochemistry 2013 440(2) pp: 145-149
Publication Date(Web):15 September 2013
DOI:10.1016/j.ab.2013.05.025
We have developed two new continuous coupled assays for ornithine-δ-aminotransferase (OAT) that are more sensitive than previous methods, measure activity in real time, and can be carried out in multiwell plates for convenience and high throughput. The first assay is based on the reduction of Δ1-pyrroline-5-carboxylate (P5C), generated from ornithine by OAT, using human pyrroline 5-carboxylate reductase 1 (PYCR1), which results in the concomitant oxidation of NADH (nicotinamide adenine dinucleotide, reduced form) to NAD+ (nicotinamide adenine dinucleotide, oxidized form). This procedure was found to be three times more sensitive than previous methods and is suitable for the study of small molecules as inhibitors or inactivators of OAT or as a method to determine OAT activity in unknown samples. The second method involves the detection of l-glutamate, produced during the regeneration of the cofactor pyridoxal 5’-phosphate (PLP) of OAT by an unamplified modification of the commercially available Amplex Red l-glutamate detection kit (Life Technologies). This assay is recommended for the determination of the substrate activity of small molecules against OAT; measuring the transformation of l-ornithine at high concentrations by this assay is complicated by the fact that it also acts as a substrate for the l-glutamate oxidase (GluOx) reporter enzyme.
Co-reporter:Qing Jing;Soosung Kang;Jeffrey K. Holden;Thomas L. Poulos;Huiying Li;Jerry Richo
PNAS 2013 Volume 110 (Issue 45 ) pp:18127-18131
Publication Date(Web):2013-11-05
DOI:10.1073/pnas.1314080110
Nitric oxide (NO) produced by bacterial NOS functions as a cytoprotective agent against oxidative stress in Staphylococcus aureus, Bacillus anthracis, and Bacillus subtilis. The screening of several NOS-selective inhibitors uncovered two inhibitors with potential antimicrobial properties. These
two compounds impede the growth of B. subtilis under oxidative stress, and crystal structures show that each compound exhibits a unique binding mode. Both compounds serve
as excellent leads for the future development of antimicrobials against bacterial NOS-containing bacteria.
Co-reporter:He Huang, Haitao Ji, Huiying Li, Qing Jing, Kristin Jansen Labby, Pavel Martásek, Linda J. Roman, Thomas L. Poulos, and Richard B. Silverman
Journal of the American Chemical Society 2012 Volume 134(Issue 28) pp:11559-11572
Publication Date(Web):June 25, 2012
DOI:10.1021/ja302269r
The reduction of pathophysiologic levels of nitric oxide through inhibition of neuronal nitric oxide synthase (nNOS) has the potential to be therapeutically beneficial in various neurodegenerative diseases. We have developed a series of pyrrolidine-based nNOS inhibitors that exhibit excellent potencies and isoform selectivities (J. Am. Chem. Soc.2010, 132, 5437). However, there are still important challenges, such as how to decrease the multiple positive charges derived from basic amino groups, which contribute to poor bioavailability, without losing potency and/or selectivity. Here we present an interdisciplinary study combining molecular docking, crystallography, molecular dynamics simulations, synthesis, and enzymology to explore potential pharmacophoric features of nNOS inhibitors and to design potent and selective monocationic nNOS inhibitors. The simulation results indicate that different hydrogen bond patterns, electrostatic interactions, hydrophobic interactions, and a water molecule bridge are key factors for stabilizing ligands and controlling ligand orientation. We find that a heteroatom in the aromatic head or linker chain of the ligand provides additional stability and blocks the substrate binding pocket. Finally, the computational insights are experimentally validated with double-headed pyridine analogues. The compounds reported here are among the most potent and selective monocationic pyrrolidine-based nNOS inhibitors reported to date, and 10 shows improved membrane permeability.
Co-reporter:Richard B. Silverman
Journal of Medicinal Chemistry 2012 Volume 55(Issue 2) pp:567-575
Publication Date(Web):December 14, 2011
DOI:10.1021/jm201650r
Co-reporter:Tian Chen ; Radhia Benmohamed ; Jinho Kim ; Karen Smith ; Daniel Amante ; Richard I. Morimoto ; Donald R. Kirsch ; Robert J. Ferrante ∞
Journal of Medicinal Chemistry 2012 Volume 55(Issue 1) pp:515-527
Publication Date(Web):December 22, 2011
DOI:10.1021/jm2014277
Amyotrophic lateral sclerosis (ALS) is an orphan neurodegenerative disease currently without a cure. The arylsulfanyl pyrazolone (ASP) scaffold was one of the active scaffolds identified in a cell-based high throughput screening assay targeting mutant Cu/Zn superoxide dismutase 1 (SOD1) induced toxicity and aggregation as a marker for ALS. The initial ASP hit compounds were potent and had favorable ADME properties but had poor microsomal and plasma stability. Here, we identify the microsomal metabolite and describe synthesized analogues of these ASP compounds to address the rapid metabolism. Both in vitro potency and pharmacological properties of the ASP scaffold have been dramatically improved via chemical modification to the corresponding sulfone and ether derivatives. One of the ether analogues (13), with superior potency and in vitro pharmacokinetic properties, was tested in vivo for its pharmacokinetic profile, brain penetration, and efficacy in an ALS mouse model. The analogue showed sustained blood and brain levels in vivo and significant activity in the mouse model of ALS, thus validating the new aryloxanyl pyrazolone scaffold as an important novel therapeutic lead for the treatment of this neurodegenerative disorder.
Co-reporter:Yue Pan ; Madina R. Gerasimov ; Trine Kvist ; Petrine Wellendorph ; Karsten K. Madsen ; Elena Pera ; Hyunbeom Lee ; Arne Schousboe ; Mary Chebib ; Hans Bräuner-Osborne ; Cheryl M. Craft ; Jonathan D. Brodie ∞; Wynne K. Schiffer ; Stephen L. Dewey ; Steven R. Miller ×
Journal of Medicinal Chemistry 2012 Volume 55(Issue 1) pp:357-366
Publication Date(Web):November 30, 2011
DOI:10.1021/jm201231w
Vigabatrin, a GABA aminotransferase (GABA-AT) inactivator, is used to treat infantile spasms and refractory complex partial seizures and is in clinical trials to treat addiction. We evaluated a novel GABA-AT inactivator (1S, 3S)-3-amino-4-difluoromethylenyl-1-cyclopentanoic acid (CPP-115, compound 1) and observed that it does not exhibit other GABAergic or off-target activities and is rapidly and completely orally absorbed and eliminated. By use of in vivo microdialysis techniques in freely moving rats and microPET imaging techniques, 1 produced similar inhibition of cocaine-induced increases in extracellular dopamine and in synaptic dopamine in the nucleus accumbens at 1/300 to 1/600 the dose of vigabatrin. It also blocks expression of cocaine-induced conditioned place preference at a dose 1/300 that of vigabatrin. Electroretinographic (ERG) responses in rats treated with 1, at doses 20–40 times higher than those needed to treat addiction in rats, exhibited reductions in ERG responses, which were less than the reductions observed in rats treated with vigabatrin at the same dose needed to treat addiction in rats. In conclusion, 1 can be administered at significantly lower doses than vigabatrin, which suggests a potential new treatment for addiction with a significantly reduced risk of visual field defects.
Co-reporter:Lada Klaić, Richard I. Morimoto, and Richard B. Silverman
ACS Chemical Biology 2012 Volume 7(Issue 5) pp:928
Publication Date(Web):March 1, 2012
DOI:10.1021/cb200539u
The natural product celastrol (1) possesses numerous beneficial therapeutic properties and affects numerous cellular pathways. The mechanism of action and cellular target(s) of celastrol, however, remain unresolved. While a number of studies have proposed that the activity of celastrol is mediated through reaction with cysteine residues, these observations have been based on studies with specific proteins or by in vitro analysis of a small fraction of the proteome. In this study, we have investigated the spatial and structural requirements of celastrol for the design of suitable affinity probes to identify cellular binding partners of celastrol. Although celastrol has several potential sites for modification, some of these were not synthetically amenable or yielded unstable analogues. Conversion of the carboxylic acid functionality to amides and long-chain analogues, however, yielded bioactive compounds that induced the heat shock response (HSR) and antioxidant response and inhibited Hsp90 activity. This led to the synthesis of biotinylated celastrols (23 and 24) that were used as affinity reagents in extracts of human Panc-1 cells to identify Annexin II, eEF1A, and β-tubulin as potential targets of celastrol.
Co-reporter:Kristin Jansen Labby, Fengtian Xue, James M. Kraus, Haitao Ji, Jan Mataka, Huiying Li, Pavel Martásek, Linda J. Roman, Thomas L. Poulos, Richard B. Silverman
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 7) pp:2435-2443
Publication Date(Web):1 April 2012
DOI:10.1016/j.bmc.2012.01.037
Selective neuronal nitric oxide synthase (nNOS) inhibitors have therapeutic applications in the treatment of numerous neurodegenerative diseases. Here we report the synthesis and evaluation of a series of inhibitors designed to have increased cell membrane permeability via intramolecular hydrogen bonding. Their potencies were examined in both purified enzyme and cell-based assays; a comparison of these results demonstrates that two of the new inhibitors display significantly increased membrane permeability over previous analogs. NMR spectroscopy provides evidence of intramolecular hydrogen bonding under physiological conditions in two of the inhibitors. Crystal structures of the inhibitors in the nNOS active site confirm the predicted non-intramolecular hydrogen bonded binding mode. Intramolecular hydrogen bonding may be an effective approach for increasing cell membrane permeability without affecting target protein binding.
Co-reporter:Yinan Zhang, Radhia Benmohamed, Wei Zhang, Jinho Kim, Christina K. Edgerly, Yaoqiu Zhu, Richard I. Morimoto, Robert J. Ferrante, Donald R. Kirsch, and Richard B. Silverman
ACS Medicinal Chemistry Letters 2012 Volume 3(Issue 7) pp:584
Publication Date(Web):May 22, 2012
DOI:10.1021/ml3000963
Cyclohexane 1,3-diones were identified as a class of molecules exhibiting a protective effect against mutant SOD1 induced toxicity in PC-12 cells, but an optimized analogue had little or no effect on life extension in the G93A SOD1 mouse model for amyotrophic lateral sclerosis (ALS). Additional testing showed that these compounds were inactive in neurons, and further analogue synthesis was carried out to identify compounds with neuronal activity. Starting from two racemic derivatives that were active in cortical neurons, two potent analogues (1b and 2b) were resolved, which were protective against mutant SOD1 induced toxicity in PC-12 cells. Both compounds were found to be active in cortical neurons and presented good ADME profiles in vitro. On the basis of these results, an ALS mouse trial with 1b was carried out, which showed slightly greater life extension than the FDA-approved ALS drug riluzole, thereby validating cyclohexane 1,3-diones as a novel therapeutic class for the treatment of ALS.Keywords: amyotrophic lateral sclerosis (ALS); Cyclohexane 1,3-diones; mutant SOD1; PC-12 cells, cortical neurons; protein aggregation; superoxide dismutase 1 (SOD1);
Co-reporter:Wei Zhang, Radhia Benmohamed, Anthony C. Arvanites, Richard I. Morimoto, Robert J. Ferrante, Donald R. Kirsch, Richard B. Silverman
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 2) pp:1029-1045
Publication Date(Web):15 January 2012
DOI:10.1016/j.bmc.2011.11.039
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease characterized by the progressive loss of motor neurons. Currently, there is only one FDA-approved treatment for ALS (riluzole), and that drug only extends life, on average, by 2–3 months. Mutations in Cu/Zn superoxide dismutase (SOD1) are found in familial forms of the disease and have played an important role in the study of ALS pathophysiology. On the basis of their activity in a PC12-G93A-YFP high-throughput screening assay, several bioactive compounds have been identified and classified as cyclohexane-1,3-dione (CHD) derivatives. A concise and efficient synthetic route has been developed to provide diverse CHD analogs. The structural modification of the CHD scaffold led to the discovery of a more potent analog (26) with an EC50 of 700 nM having good pharmacokinetic properties, such as high solubility, low human and mouse metabolic potential, and relatively good plasma stability. It was also found to efficiently penetrate the blood–brain barrier. However, compound 26 did not exhibit any significant life span extension in the ALS mouse model. It was found that, although 26 was active in PC12 cells, it had poor activity in other cell types, including primary cortical neurons, indicating that it can penetrate into the brain, but is not active in neuronal cells, potentially due to poor selective cell penetration. Further structural modification of the CHD scaffold was aimed at improving global cell activity as well as maintaining potency. Two new analogs (71 and 73) were synthesized, which had significantly enhanced cortical neuronal cell permeability, as well as similar potency to that of 26 in the PC12-G93A assay. These CHD analogs are being investigated further as novel therapeutic candidates for ALS.
Co-reporter:Dustin D. Hawker, Richard B. Silverman
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 19) pp:5763-5773
Publication Date(Web):1 October 2012
DOI:10.1016/j.bmc.2012.08.009
Two principal neurotransmitters are involved in the regulation of mammalian neuronal activity, namely, γ-aminobutyric acid (GABA), an inhibitory neurotransmitter, and l-glutamic acid, an excitatory neurotransmitter. Low GABA levels in the brain have been implicated in epilepsy and several other neurological diseases. Because of GABA’s poor ability to cross the blood–brain barrier (BBB), a successful strategy to raise brain GABA concentrations is the use of a compound that does cross the BBB and inhibits or inactivates GABA aminotransferase (GABA-AT), the enzyme responsible for GABA catabolism. Vigabatrin, a mechanism-based inactivator of GABA-AT, is currently a successful therapeutic for epilepsy, but has harmful side effects, leaving a need for improved GABA-AT inactivators. Here, we report the synthesis and evaluation of a series of heteroaromatic GABA analogues as substrates of GABA-AT, which will be used as the basis for the design of novel enzyme inactivators.
Co-reporter:Paul C. Trippier, Radhia Benmohamed, Donald R. Kirsch, Richard B. Silverman
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 21) pp:6647-6650
Publication Date(Web):1 November 2012
DOI:10.1016/j.bmcl.2012.08.114
Amyotrophic lateral sclerosis (ALS) is a debilitating and fatal neurodegenerative disease. Although the cause remains unknown, misfolded protein aggregates are seen in neurons of sporadic ALS patients, and familial ALS mutations, including mutations in superoxide dismutase 1 (SOD1), produce proteins with an increased propensity to misfold and aggregate. A structure activity relationship of a lead scaffold exhibiting neuroprotective activity in a G93A-SOD1 mouse model for ALS has been further investigated in a model PC12 cellular assay. Synthesis of biotinylated probes at the N1 nitrogen of the pyrazolone ring gave compounds (5d–e) that retained activity within 10-fold of the proton-bearing lead compound (5a) and were equipotent with a sterically less cumbersome N1-methyl substituted analogue (5b). However, when methyl substitution was introduced at N1 and N2 of the pyrazolone ring, the compound was inactive (5c). These data led us to investigate further the pharmacophoric nature of the pyrazolone unit. A range of N1 substitutions were tolerated, leading to the identification of an N1-benzyl substituted pyrazolone (5m), equipotent with 5a. Substitution at N2 or excision of N2, however, removed all activity. Therefore, the hydrogen bond donating ability of the N2–H of the pyrazolone ring appears to be a critical part of the structure, which will influence further analogue synthesis.
Co-reporter:James M. Kraus, Hunter C. Gits, Richard B. Silverman
Tetrahedron Letters 2012 Volume 53(Issue 11) pp:1319-1322
Publication Date(Web):14 March 2012
DOI:10.1016/j.tetlet.2011.12.120
Co-reporter:Yinan Zhang and Richard B. Silverman
The Journal of Organic Chemistry 2012 Volume 77(Issue 7) pp:3462-3467
Publication Date(Web):March 5, 2012
DOI:10.1021/jo300239e
The direct amination of α-haloacetoacetates with anilines is described. Compared to existing methods, this simple protocol provides an attractive strategy to prepare diverse γ-anilino-β-ketoesters in one step. Good to excellent yields of the amination products were obtained under robust conditions, providing versatile and useful scaffolds.
Co-reporter:Lada Klaić ; Paul C. Trippier ; Rama K. Mishra ; Richard I. Morimoto
Journal of the American Chemical Society 2011 Volume 133(Issue 49) pp:19634-19637
Publication Date(Web):November 16, 2011
DOI:10.1021/ja208359a
Celastrol, an important natural product and Hsp90 inhibitor with a wide range of biological and medical activities and broad use as a biological probe, acts by an as yet undetermined mode of action. It is known to undergo Michael additions with biological sulfur nucleophiles. Here it is demonstrated that nucleophiles add to the pharmacophore of celastrol in a remarkable stereospecific manner. Extensive characterization of the addition products has been obtained using NMR spectrometry, nuclear Overhauser effects, and density functional theory to determine facial selectivity and gain insight into the orbital interactions of the reactive centers. This stereospecificity of celastrol may be important to its protein target selectivity.
Co-reporter:Guoyao Xia ; Radhia Benmohamed ; Jinho Kim ; Anthony C. Arvanites ; Richard I. Morimoto ; Robert J. Ferrante ; Donald R. Kirsch
Journal of Medicinal Chemistry 2011 Volume 54(Issue 7) pp:2409-2421
Publication Date(Web):March 4, 2011
DOI:10.1021/jm101549k
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease characterized by the progressive loss of motor neurons, leading to muscle weakness, paralysis, and death, most often from respiratory failure. The only FDA-approved drug for the treatment of ALS, riluzole, only extends the median survival in patients by 2−3 months. There is an urgent need for novel therapeutic strategies for this devastating disease. Using a high-throughput screening assay targeting an ALS cultured cell model (PC12-G93A-YFP cell line), we previously identified three chemotypes that were neuroprotective. We present a further detailed analysis of one promising scaffold from that group, pyrimidine-2,4,6-triones (PYTs), characterizing a number of PYT analogues using SAR and ADME. The PYT compounds show good potency, superior ADME data, low toxicity, brain penetration, and excellent oral bioavailability. Compounds from this series show 100% efficacy in the protection assay with a good correlation in activity between the protection and protein aggregation assays. The modifications of the PYT scaffold presented here suggest that this chemical structure may be a novel drug candidate scaffold for use in clinical trials in ALS.
Co-reporter:Fengtian Xue ; Jianguo Fang ; Silvia L. Delker ; Huiying Li ; Pavel Martásek ¥; Linda J. Roman ; Thomas L. Poulos ▲
Journal of Medicinal Chemistry 2011 Volume 54(Issue 7) pp:2039-2048
Publication Date(Web):March 16, 2011
DOI:10.1021/jm101071n
We report novel neuronal nitric oxide synthase (nNOS) inhibitors based on a symmetric double-headed aminopyridine scaffold. The inhibitors were designed from crystal structures of leads 1 and 2 (Delker, S. L.; Ji, H.; Li, H.; Jamal, J.; Fang, J.; Xue, F.; Silverman, R. B.; Poulos, T. L.Unexpected binding modes of nitric oxide synthase inhibitors effective in the prevention of cerebral palsy. J. Am. Chem. Soc. 2010, 132, 5437−5442) and synthesized using a highly efficient route. The best inhibitor, 3j, showed low nanomolar inhibitory potency and modest isoform selectivity. It also exhibited enhanced membrane permeability. Inhibitor 3j binds to both the substrate site and the pterin site in nNOS but only to the substrate site in eNOS. These compounds provide a basis for further development of novel, potent, isoform selective, and bioavailable inhibitors for nNOS.
Co-reporter:Fengtian Xue ; James M. Kraus ; Kristin Jansen Labby ; Haitao Ji ; Jan Mataka ; Guoyao Xia ; Huiying Li ; Silvia L. Delker ; Linda J. Roman ; Pavel Martásek ; Thomas L. Poulos
Journal of Medicinal Chemistry 2011 Volume 54(Issue 18) pp:6399-6403
Publication Date(Web):August 2, 2011
DOI:10.1021/jm200411j
We report an efficient synthetic route to chiral pyrrolidine inhibitors of neuronal nitric oxide synthase (nNOS) and crystal structures of the inhibitors bound to nNOS and to endothelial NOS. The new route enables versatile structure–activity relationship studies on the pyrrolidine-based scaffold, which can be beneficial for further development of nNOS inhibitors. The X-ray crystal structures of five new fluorine-containing inhibitors bound to nNOS provide insights into the effect of the fluorine atoms on binding.
Co-reporter:Tian Chen, Radhia Benmohamed, Anthony C. Arvanites, Hantamalala Ralay Ranaivo, Richard I. Morimoto, Robert J. Ferrante, D. Martin Watterson, Donald R. Kirsch, Richard B. Silverman
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 1) pp:613-622
Publication Date(Web):1 January 2011
DOI:10.1016/j.bmc.2010.10.052
Amyotrophic lateral sclerosis (ALS) is an orphan neurodegenerative disease currently without a cure. Mutations in copper/zinc superoxide dismutase 1 (SOD1) have been implicated in the pathophysiology of this disease. Using a high-throughput screening assay expressing mutant G93A SOD1, two bioactive chemical hit compounds (1 and 2), identified as arylsulfanyl pyrazolones, were identified. The structural optimization of this scaffold led to the generation of a more potent analogue (19) with an EC50 of 170 nM. To determine the suitability of this class of compounds for further optimization, 1 was subjected to a battery of pharmacokinetic assays; most of the properties of 1 were good for a screening hit, except it had a relatively rapid clearance and short microsomal half-life stability. Compound 2 was found to be blood–brain barrier penetrating with a brain/plasma ratio = 0.19. The optimization of this class of compounds could produce novel therapeutic candidates for ALS patients.
Co-reporter:Wenxin Gu, Richard B. Silverman
Tetrahedron Letters 2011 Volume 52(Issue 42) pp:5438-5440
Publication Date(Web):19 October 2011
DOI:10.1016/j.tetlet.2011.07.132
(S)-2-Boc-Amino-8-(R)-(tert-butyldimethylsilanyloxy)decanoic acid, the Boc-protected precursor of an unusual amino acid residue for the synthesis of microsporin B, was synthesized. The key steps include a Suzuki coupling followed by asymmetric homogeneous hydrogenation.
Co-reporter:Wenxin Gu and Richard B. Silverman
The Journal of Organic Chemistry 2011 Volume 76(Issue 20) pp:8287-8293
Publication Date(Web):September 14, 2011
DOI:10.1021/jo201453x
Omuralide, a transformation product of the microbial metabolite lactacystin, was the first molecule discovered as a specific inhibitor of the proteasome and is unique in that it specifically inhibits the proteolytic activity of the 20S subunit of the proteasome without inhibiting any other protease activities of the cell. The total syntheses of omuralide and (+)-lactacystin are reported. An important key intermediate is synthesized at an early stage, which allows analogues of these two natural products to be made readily.
Co-reporter:Silvia L Delker ; Haitao Ji ; Huiying Li ; Joumana Jamal ; Jianguo Fang ; Fengtian Xue ; Richard B. Silverman ;Thomas L. Poulos
Journal of the American Chemical Society 2010 Volume 132(Issue 15) pp:5437-5442
Publication Date(Web):March 25, 2010
DOI:10.1021/ja910228a
Selective inhibition of the neuronal isoform of nitric oxide synthase NOS (nNOS) has been shown to prevent brain injury and is important for the treatment of various neurodegenerative disorders. However, given the high active site conservation among all three NOS isoforms, the design of selective inhibitors is an extremely challenging problem. Here we present the structural basis for why novel and potent nNOS inhibitors exhibit the highest level of selectivity over eNOS reported so far (∼3,800-fold). By using a combination of crystallography, computational methods, and site-directed mutagenesis, we found that inhibitor chirality and an unanticipated structural change of the target enzyme control both the orientation and selectivity of these novel nNOS inhibitors. A new hot spot generated as a result of enzyme elasticity provides important information for the future fragment-based design of selective NOS inhibitors.
Co-reporter:Fengtian Xue ; Huiying Li ; Silvia L. Delker ; Jianguo Fang ; Pavel Martásek ; Linda J. Roman ; Thomas L. Poulos
Journal of the American Chemical Society 2010 Volume 132(Issue 40) pp:14229-14238
Publication Date(Web):September 15, 2010
DOI:10.1021/ja106175q
In our efforts to discover neuronal isoform selective nitric oxide synthase (NOS) inhibitors, we have developed a series of compounds containing a pyrrolidine ring with two stereogenic centers. The enantiomerically pure compounds, (S,S) versus (R,R), exhibited two different binding orientations, with (R,R) inhibitors showing much better potency and selectivity. To improve the bioavailability of these inhibitors, we have introduced a CF2 moiety geminal to an amino group in the long tail of one of these inhibitors, which reduced its basicity, resulting in compounds with monocationic character under physiological pH conditions. Biological evaluations have led to a nNOS inhibitor with a Ki of 36 nM and high selectivity for nNOS over eNOS (3800-fold) and iNOS (1400-fold). MM-PBSA calculations indicated that the low pKa NH is, at least, partially protonated when bound to the active site. A comparison of rat oral bioavailability of the difluorinated compound to the parent molecule shows 22% for the difluorinated compound versus essentially no oral bioavailability for the parent compound. This indicates that the goal of this research to make compounds with only one protonated nitrogen atom at physiological pH to allow for membrane permeability, but which can become protonated when bound to NOS, has been accomplished.
Co-reporter:Haitao Ji ; Silvia L. Delker ; Huiying Li ; Pavel Martásek ; Linda J. Roman ; Thomas L. Poulos
Journal of Medicinal Chemistry 2010 Volume 53(Issue 21) pp:7804-7824
Publication Date(Web):October 19, 2010
DOI:10.1021/jm100947x
Neuronal nitric oxide synthase (nNOS) represents an important therapeutic target for the prevention of brain injury and the treatment of various neurodegenerative disorders. A series of trans-substituted amino pyrrolidinomethyl 2-aminopyridine derivatives (8−34) was designed and synthesized. A structure−activity relationship analysis led to the discovery of low nanomolar nNOS inhibitors ((±)-32 and (±)-34) with more than 1000-fold selectivity for nNOS over eNOS. Four enantiomerically pure isomers of 3′-[2′′-(3′′′-fluorophenethylamino)ethoxy]pyrrolidin-4′-yl}methyl}-4-methylpyridin-2-amine (4) also were synthesized. It was found that (3′R,4′R)-4 can induce enzyme elasticity to generate a new “hot spot” for ligand binding. The inhibitor adopts a unique binding mode, the same as that observed for (3′R,4′R)-3′-[2′′-(3′′′-fluorophenethylamino)ethylamino]pyrrolidin-4′-yl}methyl}-4-methylpyridin-2-amine ((3′R,4′R)-3) ( J. Am. Chem. Soc. 2010, 132 (15), 5437−5442). On the basis of structure−activity relationships of 8−34 and different binding conformations of the cis and trans isomers of 3 and 4, critical structural requirements of the NOS active site for ligand binding are revealed.
Co-reporter:Silvia L. Delker, Fengtian Xue, Huiying Li, Joumana Jamal, Richard B. Silverman, and Thomas L. Poulos
Biochemistry 2010 Volume 49(Issue 51) pp:
Publication Date(Web):October 30, 2010
DOI:10.1021/bi1013479
In previous studies [Delker, S. L., et al. (2010), J. Am. Chem. Soc. 132, 5437−5442], we determined the crystal structures of neuronal nitric oxide synthase (nNOS) in complex with nNOS-selective chiral pyrrolidine inhibitors, designed to have an aminopyridine group bound over the heme where it can electrostatically interact with the conserved active site Glu residue. However, in addition to the expected binding mode with the (S,S)-cis inhibitors, an unexpected “flipped” orientation was observed for the (R,R)-cis enantiomers. In the flipped mode, the aminopyridine extends out of the active site where it interacts with one heme propionate. This prompted us to design and synthesize symmetric “double-headed” inhibitors with an aminopyridine at each end of a bridging ring structure [Xue, F., Delker, S. L., Li, H., Fang, J., Jamal, J., Martásek, P., Roman, L. J., Poulos, T. L., and Silverman, R. B. Symmetric double-headed aminopyridines, a novel strategy for potent and membrane-permeable inhibitors of neuronal nitric oxide synthase. J. Med. Chem. (submitted for publication)]. One aminopyridine should interact with the active site Glu and the other with the heme propionate. Crystal structures of these double-headed aminopyridine inhibitors in complexes with nNOS show unexpected and significant protein and heme conformational changes induced by inhibitor binding that result in removal of the tetrahydrobiopterin (H4B) cofactor and creation of a new Zn2+ site. These changes are due to binding of a second inhibitor molecule that results in the displacement of H4B and the placement of the inhibitor pyridine group in position to serve as a Zn2+ ligand together with Asp, His, and a chloride ion. Binding of the second inhibitor molecule and generation of the Zn2+ site do not occur in eNOS. Structural requirements for creation of the new Zn2+ site in nNOS were analyzed in detail. These observations open the way for the potential design of novel inhibitors selective for nNOS.
Co-reporter:Takashi Kudoh, Chan Sun Park, Scott T. Lefurgy, Meihao Sun, Theodore Michels, Thomas S. Leyh, Richard B. Silverman
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 3) pp:1124-1134
Publication Date(Web):1 February 2010
DOI:10.1016/j.bmc.2009.12.050
Survival of the human pathogen Streptococcus pneumoniae requires a functional mevalonate pathway, which produces isopentenyl diphosphate, the essential building block of isoprenoids. Flux through this pathway appears to be regulated at the mevalonate kinase (MK) step, which is strongly feedback-inhibited by diphosphomevalonate (DPM), the penultimate compound in the pathway. The human mevalonate pathway is not regulated by DPM, making the bacterial pathway an attractive antibiotic target. Since DPM has poor drug characteristics, being highly charged, we propose to use unphosphorylated, cell-permeable prodrugs based on mevalonate that will be phosphorylated in turn by MK and phosphomevalonate kinase (PMK) to generate the active compound in situ. To test the limits of this approach, we synthesized a series of C3-substituted mevalonate analogues to probe the steric and electronic requirements of the MK and PMK active sites. MK and PMK accepted substrates with up to two additional carbons, showing a preference for small substituents. This result establishes the feasibility of using a prodrug strategy for DPM-based antibiotics in S. pneumoniae and identified several analogues to be tested as inhibitors of MK. Among the substrates accepted by both enzymes were cyclopropyl, vinyl, and ethynyl mevalonate analogues that, when diphosphorylated, might be mechanism-based inactivators of the next enzyme in the pathway, diphosphomevalonate decarboxylase.
Co-reporter:Che-Chien Chang, Song Cao, Soosung Kang, Li Kai, Xinyong Tian, Prativa Pandey, Sara Fernandez Dunne, Chi-Hao Luan, D. James Surmeier, Richard B. Silverman
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 9) pp:3147-3158
Publication Date(Web):1 May 2010
DOI:10.1016/j.bmc.2010.03.038
L-type Ca2+ channels in mammalian brain neurons have either a CaV1.2 or CaV1.3 pore-forming subunit. Recently, it was shown that CaV1.3 Ca2+ channels underlie autonomous pacemaking in adult dopaminergic neurons in the substantia nigra pars compacta, and this reliance renders them sensitive to toxins used to create animal models of Parkinson’s disease. Antagonism of these channels with the dihydropyridine antihypertensive drug isradipine diminishes the reliance on Ca2+ and the sensitivity of these neurons to toxins, pointing to a potential neuroprotective strategy. However, for neuroprotection without an antihypertensive side effect, selective CaV1.3 channel antagonists are required. In an attempt to identify potent and selective antagonists of CaV1.3 channels, 124 dihydropyridines (4-substituted-1,4-dihydropyridine-3,5-dicarboxylic diesters) were synthesized. The antagonism of heterologously expressed CaV1.2 and CaV1.3 channels was then tested using electrophysiological approaches and the FLIPR Calcium 4 assay. Despite the large diversity in substitution on the dihydropyridine scaffold, the most CaV1.3 selectivity was only about twofold. These results support a highly similar dihydropyridine binding site at both CaV1.2 and CaV1.3 channels and suggests that other classes of compounds need to be identified for CaV1.3 selectivity.Hundred and twenty four analogues made to identify a selective CaV1.3 Ca+2 channel antagonist.
Co-reporter:Fengtian Xue, Huiying Li, Jianguo Fang, Linda J. Roman, Pavel Martásek, Thomas L. Poulos, Richard B. Silverman
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 21) pp:6258-6261
Publication Date(Web):1 November 2010
DOI:10.1016/j.bmcl.2010.08.096
Selective inhibition of the neuronal isoform of nitric oxide synthase (nNOS) over endothelial nitric oxide synthase (eNOS) and inducible nitric oxide synthase (iNOS) has become a promising strategy for the discovery of new therapeutic agents for neurodegenerative diseases. However, because of the high sequence homology of different isozymes in the substrate binding pocket, developing inhibitors with both potency and excellent isoform selectivity remains a challenging problem. Herein, we report the evaluation of a recently discovered peripheral hydrophobic pocket (Tyr706, Leu337, and Met336) that opens up upon inhibitor binding and its potential in designing potent and selective nNOS inhibitors using three compounds, 2a, 2b, and 3. Crystal structure results show that inhibitors 2a and 3 adopted the same binding mode as lead compound 1. We also found that hydrophobic interactions between the 4-methyl group of the aminopyridine ring of these compounds with the side chain of Met336, as well as the π–π stacking interaction between the pyridinyl motif and the side chain of Tyr706 are important for the high potency and selectivity of these nNOS inhibitors.
Co-reporter:Fengtian Xue, Richard B. Silverman
Tetrahedron Letters 2010 Volume 51(Issue 18) pp:2536-2538
Publication Date(Web):5 May 2010
DOI:10.1016/j.tetlet.2010.03.009
We report a fast N→O tert-butyloxycarbonyl (Boc) migration of the imide (3R,4R)-tert-butyl 3-((6-(bis(tert-butoxycarbonyl)amino)-4-methylpyridin-2-yl)methyl)-4-hydroxypyrrolidine-1-carboxylate (2) via a base-generated alkoxide. The mechanism of the migration is intramolecular, involving an unusual nine-membered cyclic transition state.
Co-reporter:Jeffrey D. Martell ; Huiying Li ; Tzanko Doukov ; Pavel Martásek ; Linda J. Roman ; Michael Soltis ; Thomas L. Poulos
Journal of the American Chemical Society 2009 Volume 132(Issue 2) pp:798-806
Publication Date(Web):December 16, 2009
DOI:10.1021/ja908544f
The heme−thioether ligand interaction often occurs between heme iron and native methionine ligands, but thioether-based heme-coordinating (type II) inhibitors are uncommon due to the difficulty in stabilizing the Fe−S bond. Here, a thioether-based inhibitor (3) of neuronal nitric oxide synthase (nNOS) was designed, and its binding was characterized by spectrophotometry and crystallography. A crystal structure of inhibitor 3 coordinated to heme iron was obtained, representing, to our knowledge, the first crystal structure of a thioether inhibitor complexed to any heme enzyme. A series of related potential inhibitors (4−8) also were evaluated. Compounds 4−8 were all found to be type I (non-heme-coordinating) inhibitors of ferric nNOS, but 4 and 6−8 were found to switch to type II upon heme reduction to the ferrous state, reflecting the higher affinity of thioethers for ferrous heme than for ferric heme. Contrary to what has been widely thought, thioether−heme ligation was found not to increase inhibitor potency, illustrating the intrinsic weakness of the thioether−ferric heme linkage. Subtle changes in the alkyl groups attached to the thioether sulfur caused drastic changes in the binding conformation, indicating that hydrophobic contacts play a crucial role in stabilizing the thioether−heme coordination.
Co-reporter:Jianguo Fang ; Haitao Ji ; Graham R. Lawton ; Fengtian Xue ; Linda J. Roman
Journal of Medicinal Chemistry 2009 Volume 52(Issue 14) pp:4533-4537
Publication Date(Web):June 18, 2009
DOI:10.1021/jm900380j
A common dichotomy exists in inhibitor design: should the compounds be designed to block the enzymes of animals in the preclinical studies or to inhibit the human enzyme? We report that a single mutation of Leu-337 in rat neuronal nitric oxide synthase (nNOS) to His makes the enzyme resemble human nNOS more than rat nNOS. We expect that the approach used in this study can unite the dichotomy and speed up the process of inhibitor design and development.
Co-reporter:Haitao Ji ; Huiying Li ; Pavel Martásek ; Linda J. Roman ; Thomas L. Poulos
Journal of Medicinal Chemistry 2009 Volume 52(Issue 3) pp:779-797
Publication Date(Web):January 6, 2009
DOI:10.1021/jm801220a
Selective inhibition of neuronal nitric oxide synthase (nNOS) has been shown to prevent brain injury and is important for the treatment of various neurodegenerative disorders. This study shows that not only greater inhibitory potency and isozyme selectivity but more druglike properties can be achieved by fragment hopping. On the basis of the structure of lead molecule 6, fragment hopping effectively extracted the minimal pharmacophoric elements in the active site of nNOS for ligand hydrophobic and steric interactions and generated appropriate lipophilic fragments for lead optimization. More potent and selective inhibitors with better druglike properties were obtained within the design of 20 derivatives (compounds 7−26). Our structure-based inhibitor design for nNOS and SAR analysis reveal the robustness and efficiency of fragment hopping in lead discovery and structural optimization, which implicates a broad application of this approach to many other therapeutic targets for which known druglike small-molecule modulators are still limited.
Co-reporter:Graham R. Lawton, Hantamalala Ralay Ranaivo, Laura K. Chico, Haitao Ji, Fengtian Xue, Pavel Martásek, Linda J. Roman, D. Martin Watterson, Richard B. Silverman
Bioorganic & Medicinal Chemistry 2009 Volume 17(Issue 6) pp:2371-2380
Publication Date(Web):15 March 2009
DOI:10.1016/j.bmc.2009.02.017
Overproduction of nitric oxide by neuronal nitric oxide synthase (nNOS) has been linked to several neurodegenerative diseases. We have recently designed potent and isoform selective inhibitors of nNOS, but the lead compound contains several basic functional groups. A large number of charges and hydrogen bond donors can impede the ability of molecules to cross the blood brain barrier and thereby limit the effectiveness of potential neurological therapeutics. Replacement of secondary amines in our lead compound with neutral ether and amide groups was made to increase bioavailability and to determine if the potency and selectivity of the inhibitor would be impacted. An ether analogue has been identified that retains a similar potency and selectivity to that of the lead compound, and shows increased ability to penetrate the blood brain barrier.
Co-reporter:Richard B. Silverman, Graham R. Lawton, Hantamalala Ralay Ranaivo, Laura K. Chico, Jiwon Seo, D. Martin Watterson
Bioorganic & Medicinal Chemistry 2009 Volume 17(Issue 21) pp:7593-7605
Publication Date(Web):1 November 2009
DOI:10.1016/j.bmc.2009.08.065
Several prodrug approaches were taken to mask amino groups in two potent and selective neuronal nitric oxide synthase (nNOS) inhibitors containing either a primary or secondary amino group to lower the charge and improve blood–brain barrier (BBB) penetration. The primary amine was masked as an azide and the secondary amine as an amide or carbamate. The azide was not reduced to the amine under a variety of in vitro and ex vivo conditions. Despite the decrease in charge of the amino group as an amide and as carbamates, BBB penetration did not increase. It appears that the uses of azides as prodrugs for primary amines or amides and carbamates as prodrugs for secondary amines are not universally effective for CNS applications.
Co-reporter:Jianguo Fang, Richard B. Silverman
Analytical Biochemistry 2009 Volume 390(Issue 1) pp:74-78
Publication Date(Web):1 July 2009
DOI:10.1016/j.ab.2009.04.004
Nitric oxide synthase (NOS) inhibitors are potential drug candidates because it has been well demonstrated that excessive production of nitric oxide critically contributes to a range of diseases. Most inhibitors have been screened in vitro using recombinant enzymes, leading to the discovery of a variety of potent compounds. To make inhibition studies more physiologically relevant and bridge the gap between the in vitro assay and in vivo studies, we report here a cellular model for screening NOS inhibitors. Stable transformants were generated by overexpressing rat neuronal NOS in HEK 293T cells. The enzyme was activated by introducing calcium ions into cells, and its activity was assayed by determining the amount of nitrite that was formed in culture medium using the Griess reagent. We tested a few NOS inhibitors with this assay and found that the method is sensitive, versatile, and easy to use. The cell-based assay provides more information than in vitro assays regarding the bioavailability of NOS inhibitors, and it is suitable for high-throughput screening.
Co-reporter:RichardB. Silverman
Angewandte Chemie 2008 Volume 120( Issue 19) pp:3552-3556
Publication Date(Web):
DOI:10.1002/ange.200704280
Co-reporter:RichardB. Silverman
Angewandte Chemie International Edition 2008 Volume 47( Issue 19) pp:3500-3504
Publication Date(Web):
DOI:10.1002/anie.200704280
Co-reporter:Erik P. Erdal, Pavel Martásek, Linda J. Roman, Richard B. Silverman
Bioorganic & Medicinal Chemistry 2007 Volume 15(Issue 18) pp:6096-6108
Publication Date(Web):15 September 2007
DOI:10.1016/j.bmc.2007.06.038
Nitric oxide (NO) is an important second messenger molecule for blood pressure homeostasis, as a neurotransmitter, and in the immune defense system. Excessive NO can lead to neurodegeneration and connective tissue damage. Three different isozymes of the enzyme nitric oxide synthase regulate NO production in endothelial (eNOS), neuronal (nNOS), and macrophage (iNOS) cells. Whereas creating a lower level of NO in some cells could be beneficial, it also could be detrimental to the protective effects that NO has on other cells. Therefore, it is essential that therapeutic NOS inhibitors be made that are subtype selective. Previously, we reported a series of nitroarginine-containing dipeptide amides as potent and selective nNOS inhibitors. Here we synthesize peptidomimetic hydroxyethylene isosteres of these dipeptide amides for potential increased bioavailability. None of the compounds is as potent or selective as the dipeptide amides, but they exhibit good inhibition and selectivity. When the terminal amino group was converted to a hydroxyl group, potency and selectivity greatly diminished, supporting the importance of the terminal amino group for binding.
Co-reporter:Elizabeth A. Litzinger, Pavel Martásek, Linda J. Roman, Richard B. Silverman
Bioorganic & Medicinal Chemistry 2006 Volume 14(Issue 9) pp:3185-3198
Publication Date(Web):1 May 2006
DOI:10.1016/j.bmc.2005.12.043
Based on computer modeling of the active site of nitric oxide synthases (NOS), a series of 10 amidine compounds (9–18) was designed including potential inhibitors that involve the coordination of side-chain functional groups with the iron of the heme cofactor. The most potent and selective compound was the methylthio amidine analogue 9, which was more potent than l-nitroarginine with 185-fold selectivity for inhibition of neuronal NOS over endothelial NOS. It also exhibited time-dependent inhibition, but did not involve the mechanism previously proposed for other amidine inhibitors of NOS. None of the compounds, however, exhibited heme-binding characteristics according to absorption spectroscopy.
Co-reporter:Wenxin Gu, Inna Nusinzon, Ronald D. Smith Jr., Curt M. Horvath, Richard B. Silverman
Bioorganic & Medicinal Chemistry 2006 Volume 14(Issue 10) pp:3320-3329
Publication Date(Web):15 May 2006
DOI:10.1016/j.bmc.2005.12.047
Suberoylanilide hydroxamic acid (SAHA), an inhibitor of histone deacetylase, is used in clinical trials for a variety of advanced cancers. Twelve new analogs of SAHA were synthesized and tested as in vitro inhibitors of isolated histone deacetylases (HDACS) and in vivo inhibitors of interferon regulated transcriptional responses (a marker for HDAC activity). The analogs containing an α-mercaptoketone or an α-thioacetoxyketone were more potent than SAHA in both assays.
Co-reporter:Bessie N.A. Mbadugha, Jiwon Seo, Haitao Ji, Pavel Martásek, Linda J. Roman, Thomas M. Shea, Huiying Li, Thomas L. Poulos, Richard B. Silverman
Bioorganic & Medicinal Chemistry 2006 Volume 14(Issue 11) pp:3681-3690
Publication Date(Web):1 June 2006
DOI:10.1016/j.bmc.2006.01.044
The X-ray structure of previously studied dipeptidomimetic inhibitors bound in the active site of neuronal nitric oxide synthase (nNOS) presented a possibility for optimizing the strength of enzyme–inhibitor interactions as well as for enhancing bioavailability. These desirable properties may be attainable by replacement of the terminal amino group of the parent compounds (1–6) with a hydroxyl group (11–13, and 18–20). The hypothesized effect would be twofold: first, a change from a positively charged amino group to a neutral hydroxyl group might afford more drug-like character and blood–brain barrier permeability to the inhibitors; second, as suggested by docking studies, the incorporated hydroxyl group might displace an active site water molecule with which the terminal amino group of the original compounds indirectly hydrogen bonds. In vitro activity assays of the hydroxyl-terminated analogs (11–13 and 18–20) showed greater than an order of magnitude increase in Ki values (decreased potency) relative to the amino-terminated compounds. These experimental data support the importance to enzyme binding of a potential electrostatic interaction relative to a hydrogen bonding interaction.
Co-reporter:Deanna J. Mitchell, Dejan Nikolic, Richard B. van Breemen, Richard B. Silverman
Bioorganic & Medicinal Chemistry Letters 2001 Volume 11(Issue 13) pp:1757-1760
Publication Date(Web):9 July 2001
DOI:10.1016/S0960-894X(01)00302-X
Incubation of 1-phenylcyclopropylamine with bovine liver MAO (MAO B), followed by complete enzymatic digestion to single amino acid residues and subsequent analysis by on-line liquid chromatography–electrospray ionization mass spectrometry, was used to investigate the resulting flavin adduct structure.Incubation of 1-phenylcyclopropylamine with bovine liver MAO (MAO B), followed by complete enzymatic digestion to single amino acid residues and subsequent analysis by on-line liquid chromatography–electrospray ionization mass spectrometry, was used to investigate the structure of the resulting flavin adduct.
Co-reporter:Paramita Mukherjee, Maris A. Cinelli, Soosung Kang and Richard B. Silverman
Chemical Society Reviews 2014 - vol. 43(Issue 19) pp:NaN6838-6838
Publication Date(Web):2014/02/19
DOI:10.1039/C3CS60467E
Nitric oxide (NO) is an important signaling molecule in the human body, playing a crucial role in cell and neuronal communication, regulation of blood pressure, and in immune activation. However, overproduction of NO by the neuronal isoform of nitric oxide synthase (nNOS) is one of the fundamental causes underlying neurodegenerative disorders and neuropathic pain. Therefore, developing small molecules for selective inhibition of nNOS over related isoforms (eNOS and iNOS) is therapeutically desirable. The aims of this review focus on the regulation and dysregulation of NO signaling, the role of NO in neurodegeneration and pain, the structure and mechanism of nNOS, and the use of this information to design selective inhibitors of this enzyme. Structure-based drug design, the bioavailability and pharmacokinetics of these inhibitors, and extensive target validation through animal studies are addressed.

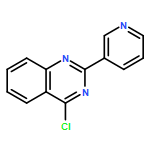









![Bicyclo[2.2.1]heptan-2-amine](http://img.cochemist.com/ccimg/900/822-98-0.png)
![Bicyclo[2.2.1]heptan-2-amine](http://img.cochemist.com/ccimg/900/822-98-0_b.png)
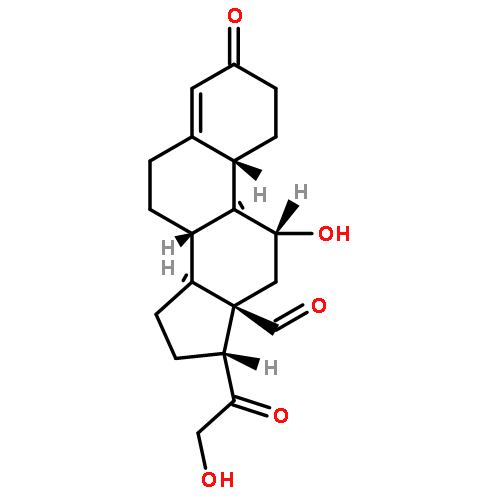












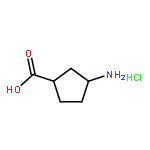
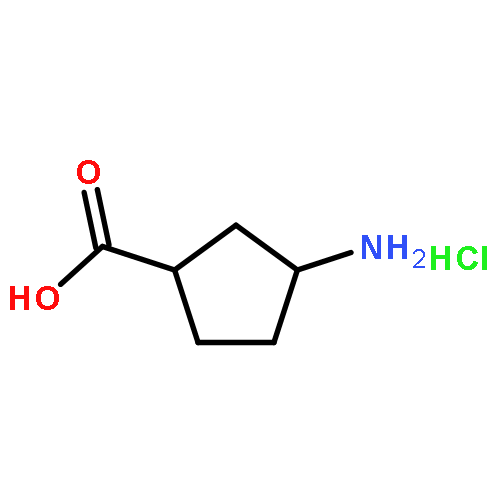








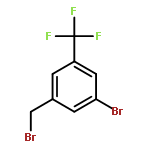
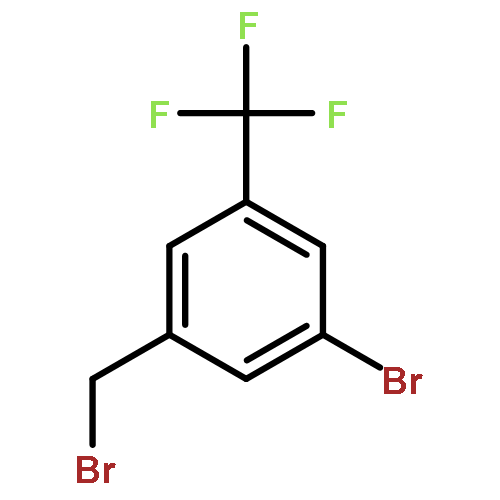
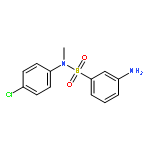
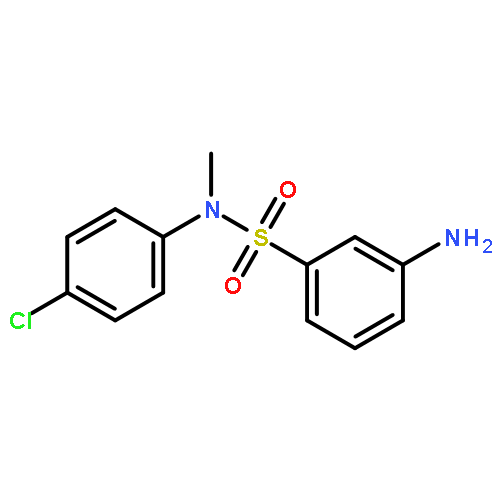
![L-Ornithine, N5-[(hydroxyamino)iminomethyl]-N5-methyl-](http://img.cochemist.com/ccimg/864000/863987-43-3.png)
![L-Ornithine, N5-[(hydroxyamino)iminomethyl]-N5-methyl-](http://img.cochemist.com/ccimg/864000/863987-43-3_b.png)
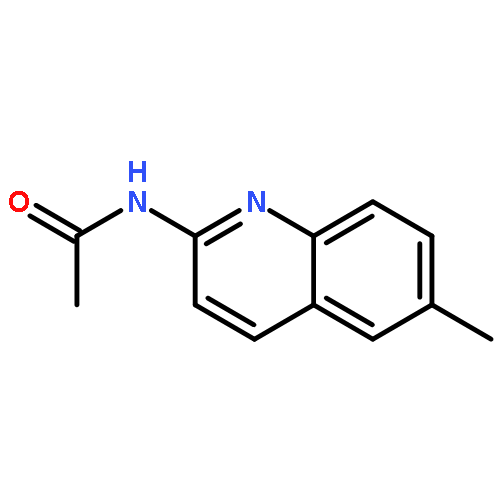
![Acetamide, N-[6-(bromomethyl)-2-quinolinyl]-](http://img.cochemist.com/ccimg/863600/863549-36-4.png)
![Acetamide, N-[6-(bromomethyl)-2-quinolinyl]-](http://img.cochemist.com/ccimg/863600/863549-36-4_b.png)
![Ethanol, 2-[(3-fluorophenyl)methoxy]-](http://img.cochemist.com/ccimg/851900/851859-50-2.png)
![Ethanol, 2-[(3-fluorophenyl)methoxy]-](http://img.cochemist.com/ccimg/851900/851859-50-2_b.png)
![[1-(tert-Butoxycarbonyl)-1,2,3,6-tetrahydropyridine-4-yl]boronic acid](http://img.cochemist.com/ccimg/844600/844501-00-4.png)
![[1-(tert-Butoxycarbonyl)-1,2,3,6-tetrahydropyridine-4-yl]boronic acid](http://img.cochemist.com/ccimg/844600/844501-00-4_b.png)



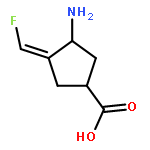

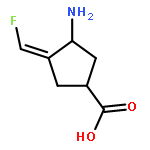
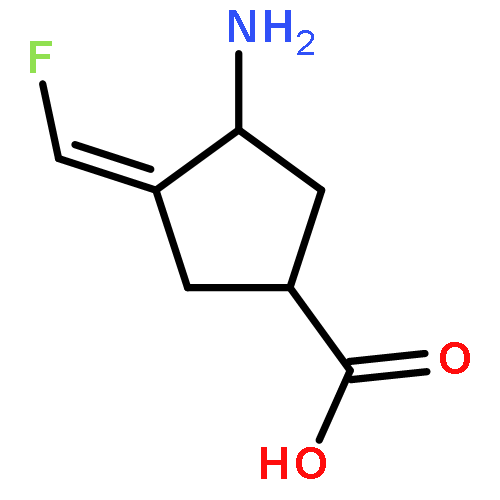
![4,7,10,13-Tetraoxa-16-azaheneicosanoic acid, 21-[(3aS,4S,6aR)-hexahydro-2-oxo-1H-thieno[3,4-d]imidazol-4-yl]-17-oxo-, hydrazide](/data/chemimg/3743000/756525-97-0.png)
![4,7,10,13-Tetraoxa-16-azaheneicosanoic acid, 21-[(3aS,4S,6aR)-hexahydro-2-oxo-1H-thieno[3,4-d]imidazol-4-yl]-17-oxo-, hydrazide](/data/chemimg/3743000/756525-97-0_b.png)
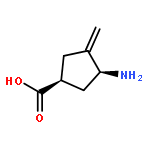

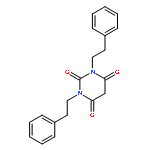
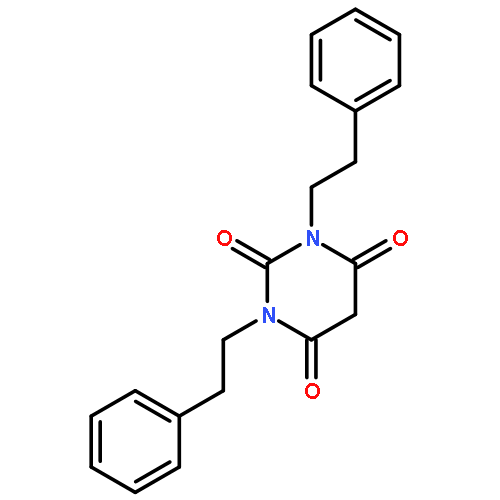
![3-[(4-Methoxy-phenyl)-methyl-sulfamoyl]-benzoic acid](http://img.cochemist.com/ccimg/379300/379254-75-8.png)
![3-[(4-Methoxy-phenyl)-methyl-sulfamoyl]-benzoic acid](http://img.cochemist.com/ccimg/379300/379254-75-8_b.png)
![Benzoic acid, 3-[[(4-chlorophenyl)methylamino]sulfonyl]-](http://img.cochemist.com/ccimg/361500/361473-14-5.png)
![Benzoic acid, 3-[[(4-chlorophenyl)methylamino]sulfonyl]-](http://img.cochemist.com/ccimg/361500/361473-14-5_b.png)

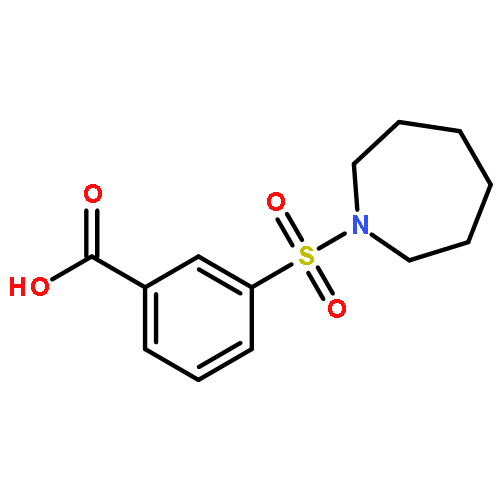
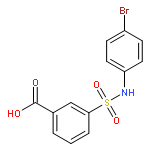
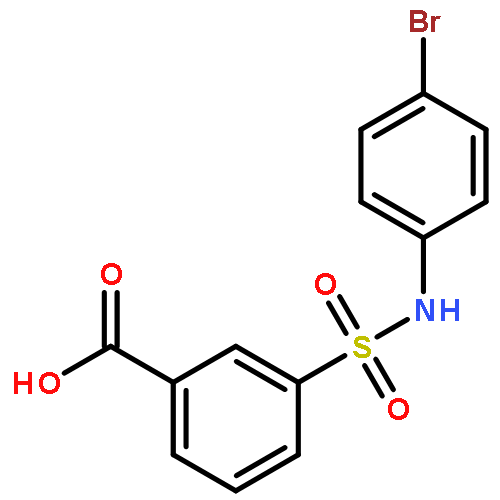
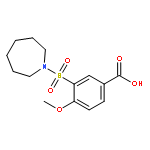
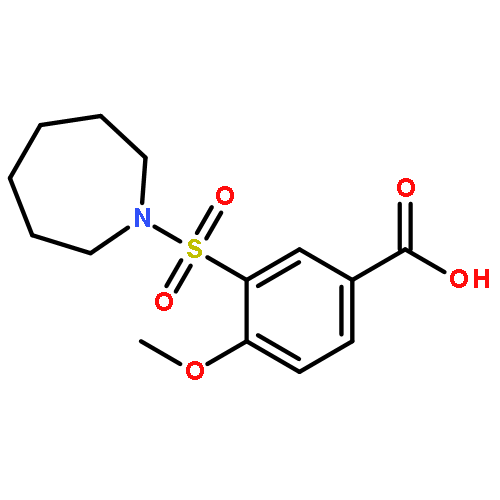


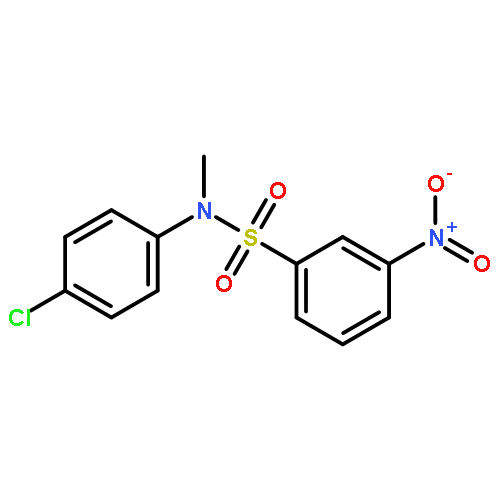
![METHYL 4-[4-(CHLOROMETHYL)-5-METHYL-1,3-OXAZOL-2-YL]BENZOATE](/data/chemimg/413600/213190-12-6.png)
![METHYL 4-[4-(CHLOROMETHYL)-5-METHYL-1,3-OXAZOL-2-YL]BENZOATE](/data/chemimg/413600/213190-12-6_b.png)
![9H-Xanthene-9-propanoicacid, a-amino-a-[(1S,2S)-2-carboxycyclopropyl]-,(aS)-](http://img.cochemist.com/ccimg/202000/201943-63-7.png)
![9H-Xanthene-9-propanoicacid, a-amino-a-[(1S,2S)-2-carboxycyclopropyl]-,(aS)-](http://img.cochemist.com/ccimg/202000/201943-63-7_b.png)
![1-[2-(3-fluorophenyl)ethyl]piperazine](http://img.cochemist.com/ccimg/188500/188400-93-3.png)
![1-[2-(3-fluorophenyl)ethyl]piperazine](http://img.cochemist.com/ccimg/188500/188400-93-3_b.png)
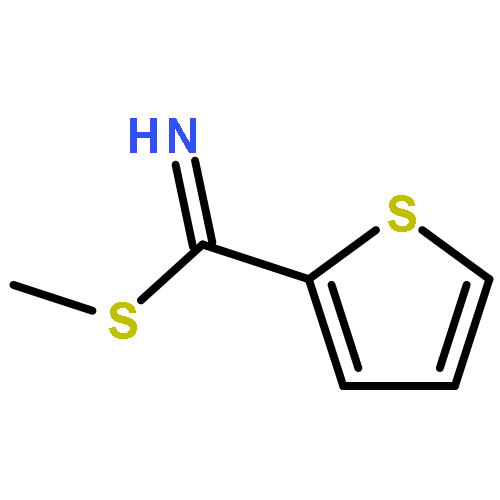


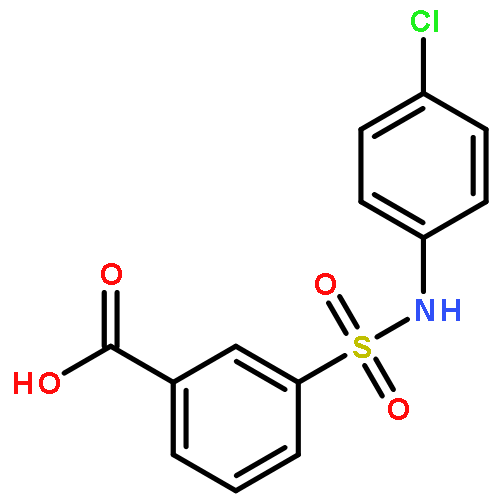


![1-(2,6-DIMETHYL-1-PIPERIDINYL)-2-({1-[2-OXO-2-(1-PYRROLIDINYL)ETHYL]-1H-BENZIMIDAZOL-2-YL}SULFANYL)ETHANONE](http://img.cochemist.com/ccimg/134900/134833-83-3.png)
![1-(2,6-DIMETHYL-1-PIPERIDINYL)-2-({1-[2-OXO-2-(1-PYRROLIDINYL)ETHYL]-1H-BENZIMIDAZOL-2-YL}SULFANYL)ETHANONE](http://img.cochemist.com/ccimg/134900/134833-83-3_b.png)
![(1R,4S)-2-Azabicyclo[2.2.1]heptan-3-one](http://img.cochemist.com/ccimg/134100/134003-02-4.png)
![(1R,4S)-2-Azabicyclo[2.2.1]heptan-3-one](http://img.cochemist.com/ccimg/134100/134003-02-4_b.png)


![2-Butanone, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/120600/120591-36-8.png)
![2-Butanone, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/120600/120591-36-8_b.png)


![Benzenamine, 3,3'-[methylenebis(oxy)]bis-](http://img.cochemist.com/ccimg/119400/119380-88-0.png)
![Benzenamine, 3,3'-[methylenebis(oxy)]bis-](http://img.cochemist.com/ccimg/119400/119380-88-0_b.png)


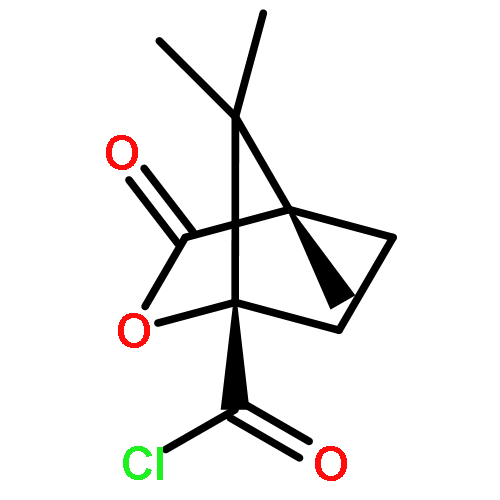
![Benzene, 1-[(2-bromoethoxy)methyl]-3-fluoro-](http://img.cochemist.com/ccimg/103100/103061-59-2.png)
![Benzene, 1-[(2-bromoethoxy)methyl]-3-fluoro-](http://img.cochemist.com/ccimg/103100/103061-59-2_b.png)


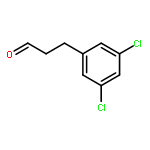
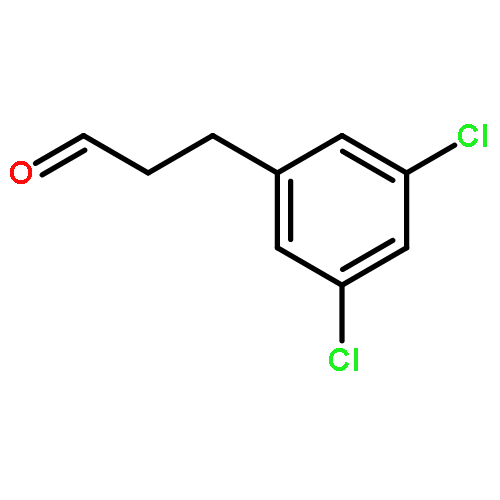
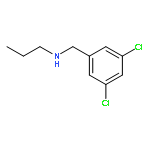

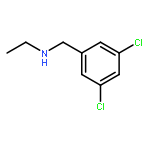
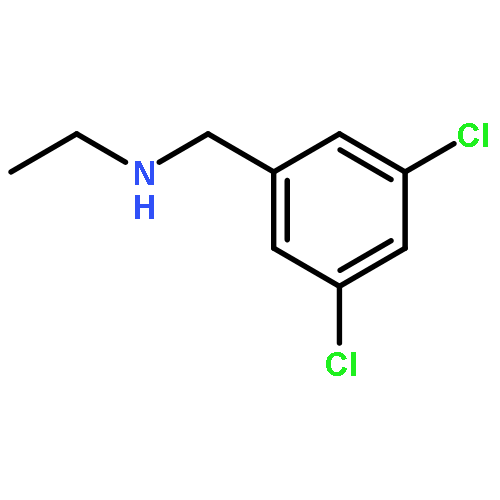





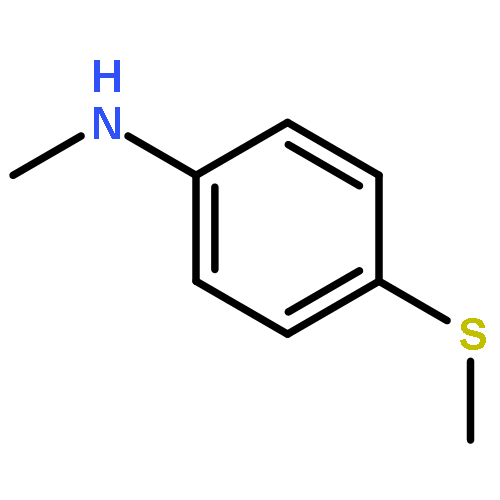
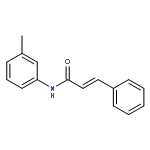
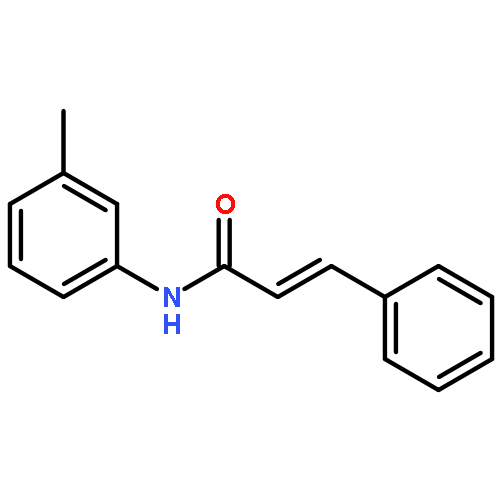
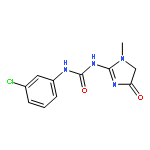
![L-ORNITHINE, N2-[(1,1-DIMETHYLETHOXY)CARBONYL]-, 1,1-DIMETHYLETHYL ESTER](http://img.cochemist.com/ccimg/53100/53054-03-8.png)
![L-ORNITHINE, N2-[(1,1-DIMETHYLETHOXY)CARBONYL]-, 1,1-DIMETHYLETHYL ESTER](http://img.cochemist.com/ccimg/53100/53054-03-8_b.png)



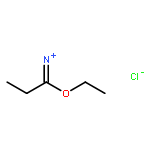

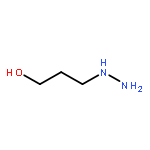
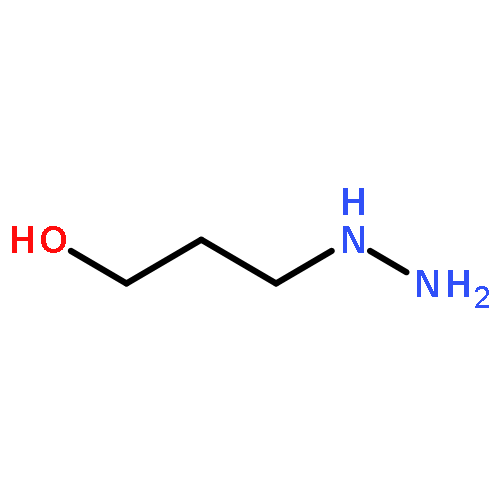



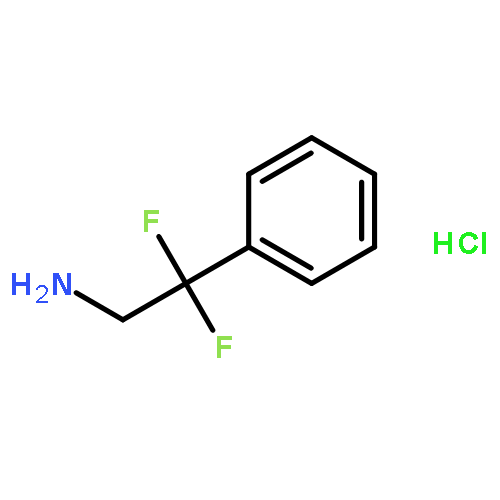
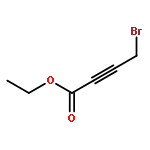
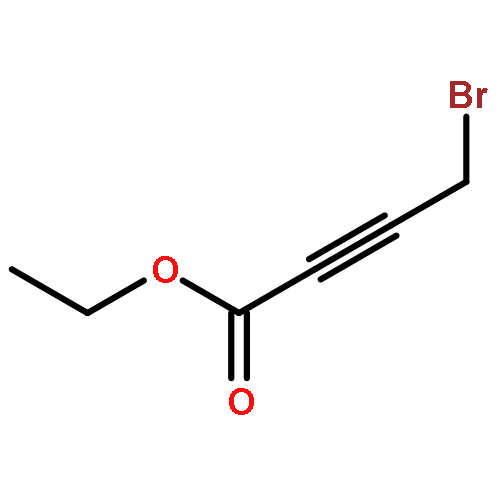
![N-[(2,4-DICHLOROPHENYL)METHYL]PROPAN-1-AMINE](http://img.cochemist.com/ccimg/39200/39180-81-9.png)
![N-[(2,4-DICHLOROPHENYL)METHYL]PROPAN-1-AMINE](http://img.cochemist.com/ccimg/39200/39180-81-9_b.png)
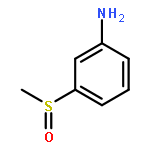
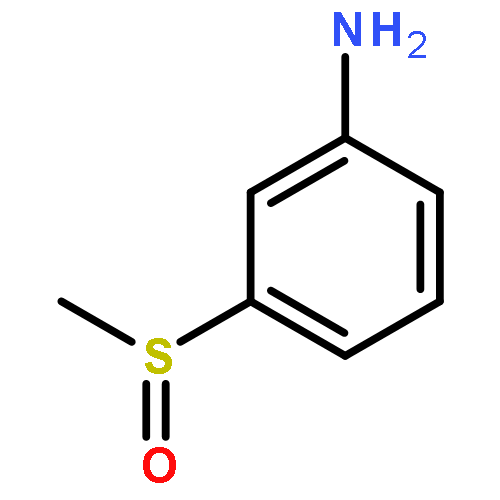
![6-methyl-2-[(E)-phenyldiazenyl]pyridin-3-ol](http://img.cochemist.com/ccimg/32000/31993-01-8.png)
![6-methyl-2-[(E)-phenyldiazenyl]pyridin-3-ol](http://img.cochemist.com/ccimg/32000/31993-01-8_b.png)




![1,1'-[ethane-1,2-diylbis(oxy)]bis(3-nitrobenzene)](http://img.cochemist.com/ccimg/26000/25986-11-2.png)
![1,1'-[ethane-1,2-diylbis(oxy)]bis(3-nitrobenzene)](http://img.cochemist.com/ccimg/26000/25986-11-2_b.png)
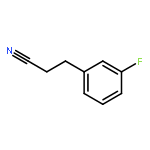

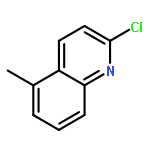
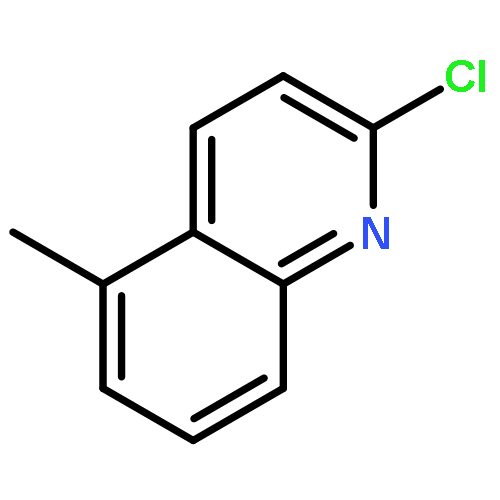


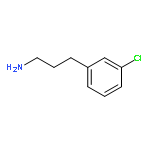

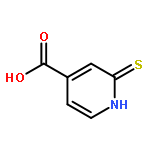
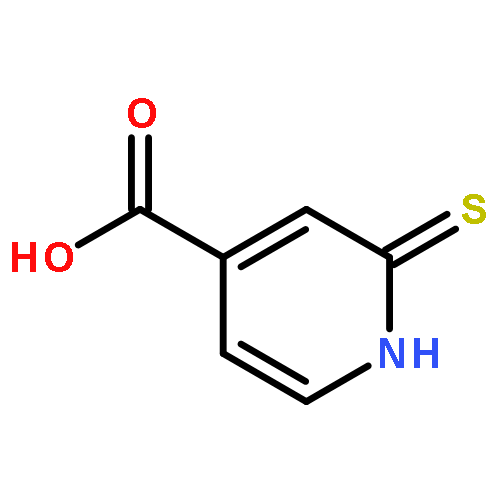
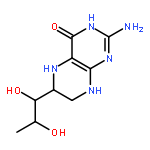

![Benzene,1,1'-[1,3-propanediylbis(oxy)]bis[3-nitro- (9CI)](http://img.cochemist.com/ccimg/14500/14467-65-3.png)
![Benzene,1,1'-[1,3-propanediylbis(oxy)]bis[3-nitro- (9CI)](http://img.cochemist.com/ccimg/14500/14467-65-3_b.png)
![benzene, 1,1'-[methylenebis(oxy)]bis[3-nitro-](http://img.cochemist.com/ccimg/13900/13831-55-5.png)
![benzene, 1,1'-[methylenebis(oxy)]bis[3-nitro-](http://img.cochemist.com/ccimg/13900/13831-55-5_b.png)
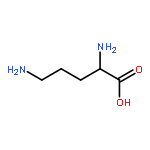
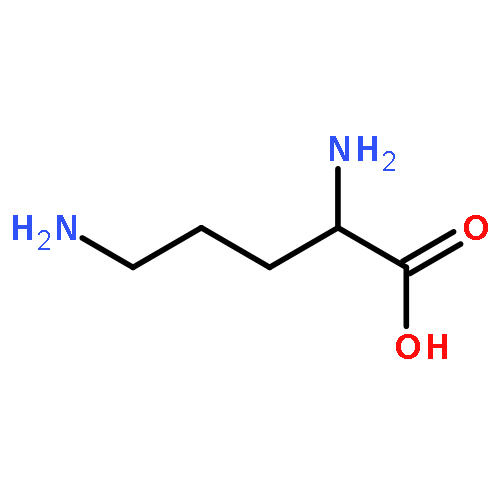


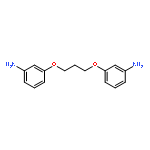
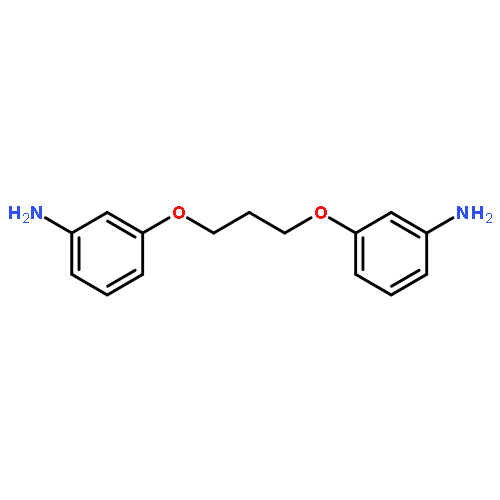
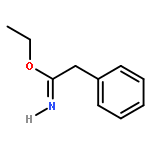
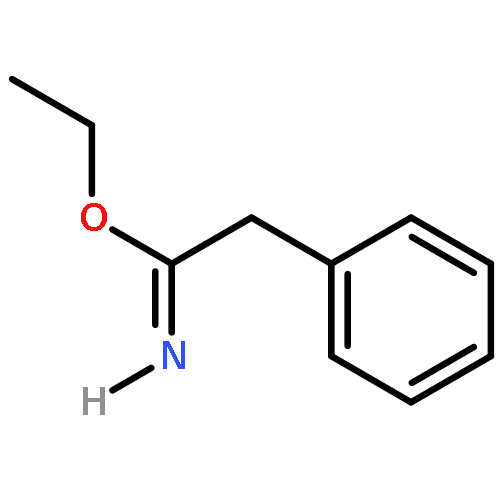


![Bicyclo[3.1.0]hexan-2-one](http://img.cochemist.com/ccimg/4200/4160-49-0.png)
![Bicyclo[3.1.0]hexan-2-one](http://img.cochemist.com/ccimg/4200/4160-49-0_b.png)
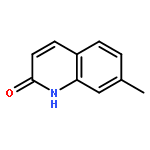
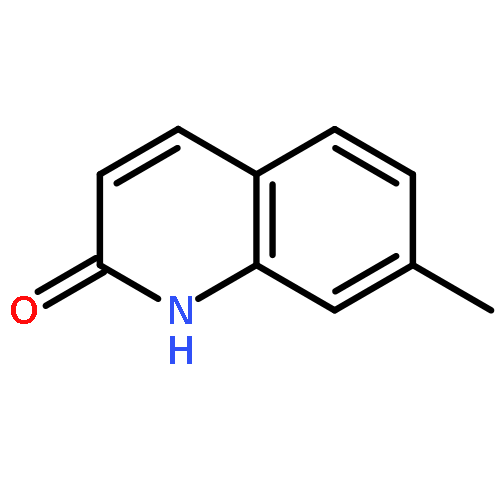
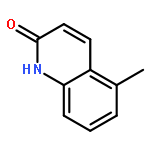
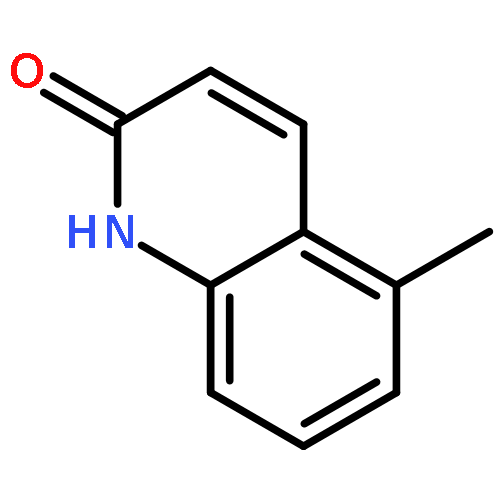



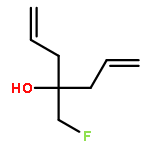
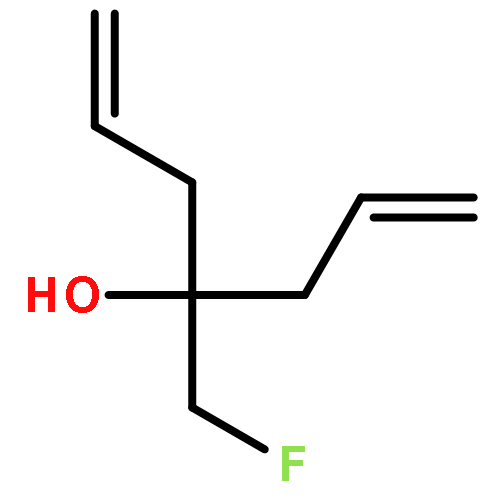
![[1,1'-Biphenyl]-3,3'-diamine](http://img.cochemist.com/ccimg/2100/2050-89-7.png)
![[1,1'-Biphenyl]-3,3'-diamine](http://img.cochemist.com/ccimg/2100/2050-89-7_b.png)
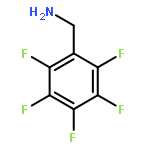
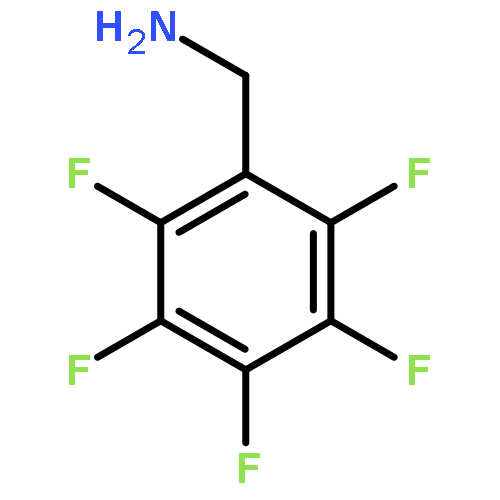
![Phosphonium,[(3-nitrophenyl)methyl]triphenyl-, bromide (1:1)](http://img.cochemist.com/ccimg/1600/1530-41-2.png)
![Phosphonium,[(3-nitrophenyl)methyl]triphenyl-, bromide (1:1)](http://img.cochemist.com/ccimg/1600/1530-41-2_b.png)


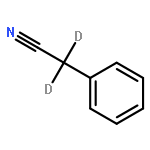
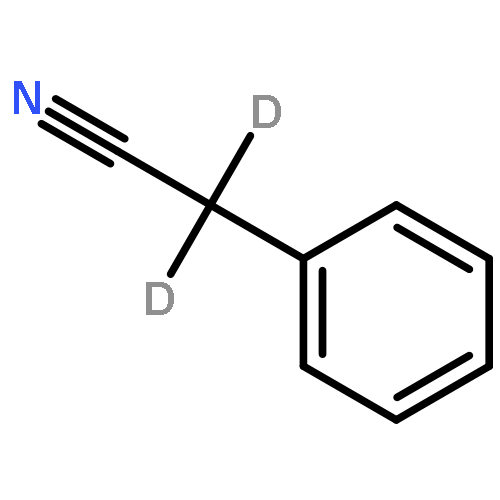
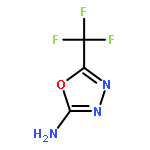
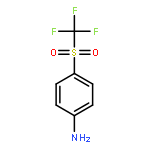
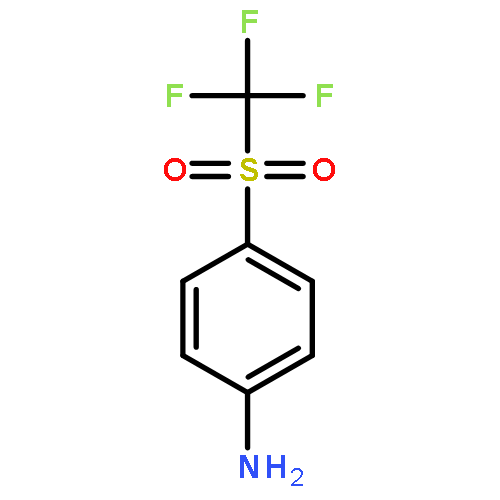
















![3-[1-(methylamino)ethyl]phenol](http://img.cochemist.com/ccimg/923100/923035-06-7.png)
![3-[1-(methylamino)ethyl]phenol](http://img.cochemist.com/ccimg/923100/923035-06-7_b.png)
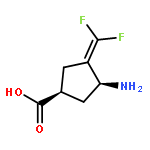
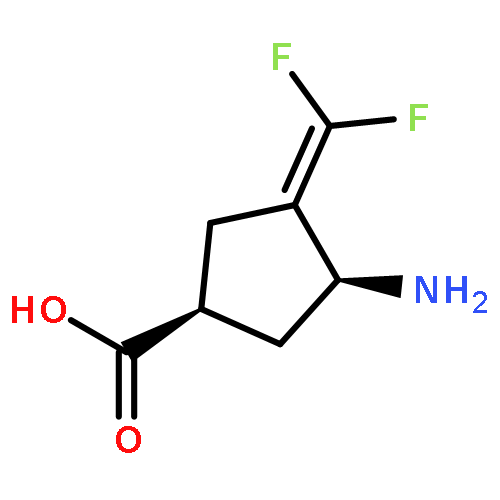

![Carbamic acid, methyl[(3-nitrophenyl)methyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/180100/180002-02-2.png)
![Carbamic acid, methyl[(3-nitrophenyl)methyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/180100/180002-02-2_b.png)
![Carbamic acid, [(3-aminophenyl)methyl]methyl-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/167800/167756-90-3.png)
![Carbamic acid, [(3-aminophenyl)methyl]methyl-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/167800/167756-90-3_b.png)


![L-Leucinamide,N-[(phenylmethoxy)carbonyl]-L-leucyl-N-[(1S)-1-formyl-3-methylbutyl]-](http://img.cochemist.com/ccimg/133500/133407-82-6.png)
![L-Leucinamide,N-[(phenylmethoxy)carbonyl]-L-leucyl-N-[(1S)-1-formyl-3-methylbutyl]-](http://img.cochemist.com/ccimg/133500/133407-82-6_b.png)


![[4-(2-METHYLPHENYL)-1,2-OXAZOL-5-YL]METHANOL](http://img.cochemist.com/ccimg/78600/78507-19-4.png)
![[4-(2-METHYLPHENYL)-1,2-OXAZOL-5-YL]METHANOL](http://img.cochemist.com/ccimg/78600/78507-19-4_b.png)


![Phenol, 3-[(dimethylamino)methyl]-](http://img.cochemist.com/ccimg/60800/60760-04-5.png)
![Phenol, 3-[(dimethylamino)methyl]-](http://img.cochemist.com/ccimg/60800/60760-04-5_b.png)
![L-Ornithine,N5-[(hydroxyamino)iminomethyl]-](http://img.cochemist.com/ccimg/53100/53054-07-2.png)
![L-Ornithine,N5-[(hydroxyamino)iminomethyl]-](http://img.cochemist.com/ccimg/53100/53054-07-2_b.png)
![PHENOL, 3-[2-(DIMETHYLAMINO)ETHOXY]-](http://img.cochemist.com/ccimg/50600/50544-44-0.png)
![PHENOL, 3-[2-(DIMETHYLAMINO)ETHOXY]-](http://img.cochemist.com/ccimg/50600/50544-44-0_b.png)





![Carbamic acid,N-[(3-hydroxyphenyl)methyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/28400/28387-66-8.png)
![Carbamic acid,N-[(3-hydroxyphenyl)methyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/28400/28387-66-8_b.png)



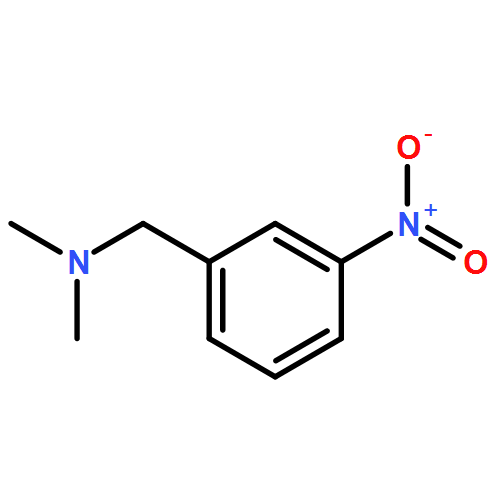

![Phenol,4-[(dimethylamino)methyl]-](http://img.cochemist.com/ccimg/200/103-87-7.png)
![Phenol,4-[(dimethylamino)methyl]-](http://img.cochemist.com/ccimg/200/103-87-7_b.png)






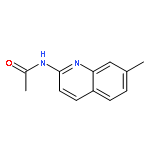
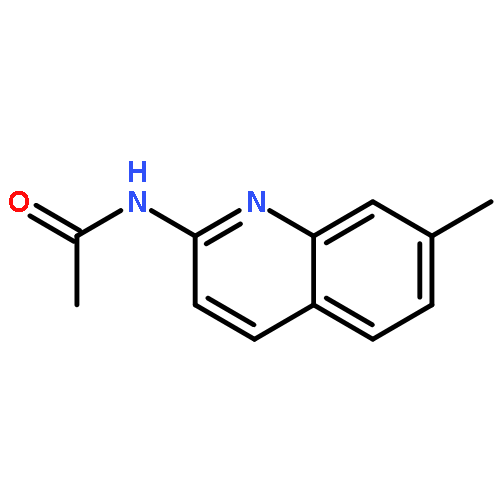
![Acetamide, N-[7-(bromomethyl)-2-quinolinyl]-](http://img.cochemist.com/ccimg/863600/863549-38-6.png)
![Acetamide, N-[7-(bromomethyl)-2-quinolinyl]-](http://img.cochemist.com/ccimg/863600/863549-38-6_b.png)


![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8.png)
![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8_b.png)







