Co-reporter:Wei Liu;Houying Li;Haijuan Qin;Wei Zhao
Chemical Research in Chinese Universities 2017 Volume 33( Issue 2) pp:213-220
Publication Date(Web):2017 April
DOI:10.1007/s40242-017-6371-z
An unexpected decarboxamidation of α-arylsulfonyl Weinreb amides as a side reaction under the standard acylating conditions was found in Weinreb amides chemistry. The control experiments for mechanism study disclosed that α-sulfo group was necessary, and α-quaternary carbon was the key factor for the reaction. Meanwhile, an efficient method was established for the preparation of secondary alkyl arylsulfones by this unexpected C―C bond cleavage reaction using excess Grignard reagent.
Co-reporter:Xiaobin Tang;Zhenghui Li;Yonghong Li
Chemical Research in Chinese Universities 2015 Volume 31( Issue 1) pp:71-77
Publication Date(Web):2015 February
DOI:10.1007/s40242-015-4309-x
A series of novel saccharin derivatives containing 1,2,3-triazole moiety was synthesized in satisfactory yields. The structures of all the compounds were elucidated and confirmed by Fourier transform infrared(FTIR) spectroscopy, 1H and 13C nuclear magnetic resonance(1H NMR, 13C NMR) spectroscopy, and high resolution mass spectrometry(HRMS). The bioassays indicated that most of the title compounds displayed some extent herbicidal activities at 100 μg/mL.
Co-reporter:Zhenfei Guo;Zhihui Yan;Xiaowei Zhou;Quan Wang;Meiqi Lu
Medicinal Chemistry Research 2015 Volume 24( Issue 5) pp:1814-1829
Publication Date(Web):2015 May
DOI:10.1007/s00044-014-1259-7
Several series of novel 1,4-disubstituted 1,2,3-triazoles were prepared using the Huisgen cycloaddition reaction and evaluated as inhibitors against caspase-3/-7. The 1,2-benzisothiazol-3-one-derived 1,4-disubstituted 1,2,3-triazoles containing an urea group had dramatically increased inhibitory activity in vitro compared to the others, and the most potent caspase-3 inhibitor was found to be N-((1-(4-fluorophenyl)-1H-1,2,3-triazol-4-yl)methyl)-3-oxobenzoisothiazole-2(3H)-carboxamide 14c with IC50-values of 11.0 ± 1.2 nM. Meanwhile, the compound 14c showed significant protection against apoptosis in human Jurkat T cells, as determined by annexin V-FITC/7-AAD. Moreover, in order to better rationalize the action and the binding mode of these compounds, docking studies were carried out.
Co-reporter:Wei Liu, He-Min Zhu, Guo-Jun Niu, En-Zhi Shi, Jie Chen, Bo Sun, Wei-Qiang Chen, Hong-Gang Zhou, Cheng Yang
Bioorganic & Medicinal Chemistry 2014 Volume 22(Issue 1) pp:292-302
Publication Date(Web):1 January 2014
DOI:10.1016/j.bmc.2013.11.028
The Severe Acute Respiratory Syndrome (SARS) is a serious life-threatening and strikingly mortal respiratory illness caused by SARS-CoV. SARS-CoV which contains a chymotrypsin-like main protease analogous to that of the main picornavirus protease, 3CLpro. 3CLpro plays a pivotal role in the viral replication cycle and is a potential target for SARS inhibitor development. A series of isatin derivatives as possible SARS-CoV 3CLpro inhibitors was designed, synthesized, and evaluated by in vitro protease assay using fluorogenic substrate peptide, in which several showed potent inhibition against the 3CLpro. Structure–activity relationship was analyzed, and possible binding interaction modes were proposed by molecular docking studies. Among all compounds, 8k1 showed most potent inhibitory activity against 3CLpro (IC50 = 1.04 μM). These results indicated that these inhibitors could be potentially developed into anti-SARS drugs.
Co-reporter:Lixin Wu, Meiqi Lu, Zhihui Yan, Xiaobin Tang, Bo Sun, Wei Liu, Honggang Zhou, Cheng Yang
Bioorganic & Medicinal Chemistry 2014 Volume 22(Issue 8) pp:2416-2426
Publication Date(Web):15 April 2014
DOI:10.1016/j.bmc.2014.03.002
A novel series of 1,2-benzisothiazol-3-one derivatives was synthesized and their biological activities were evaluated for inhibiting caspase-3 and -7 activities, in which some of them showed low nanomolar potency against caspase-3 in vitro and significant protection against apoptosis in a camptothecin-induced Jurkat T cells system. Among the tested compounds, compound 5i exhibited the most potent caspase-3 inhibitory activity (IC50 = 1.15 nM). The molecular docking predicted the interactions and binding modes of the synthesized inhibitor in the caspase-3 active site.
Co-reporter:Zhenghui Li, Yang Pan, Weilong Zhong, Yunpeng Zhu, Yongle Zhao, Lixin Li, Wei Liu, Honggang Zhou, Cheng Yang
Bioorganic & Medicinal Chemistry 2014 22(24) pp: 6735-6745
Publication Date(Web):
DOI:10.1016/j.bmc.2014.11.005
Co-reporter:Dazhi Liu, Zhen Tian, Zhihui Yan, Lixin Wu, Yan Ma, Quan Wang, Wei Liu, Honggang Zhou, Cheng Yang
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 11) pp:2960-2967
Publication Date(Web):1 June 2013
DOI:10.1016/j.bmc.2013.03.075
A number of 1,2-benzisothiazol-3-one derivatives were prepared through structural modification of the original compound from high-throughput screening. Some analogues (e.g., 6b, 6r, 6s and 6w) were identified as novel and potent caspase inhibitors with IC50 of nanomolar. Structure–activity relationship (SAR) studies for caspase-3 inhibition were evaluated in vitro. Molecular modeling studies provided further insight into the interaction of this class of compounds with activated caspase-3. The present small molecule caspase-3 inhibitor with novel structures different from structures of known caspase inhibitors revealed a new direction for therapeutic strategies directed against diseases involving abnormally up-regulated apoptosis.
Co-reporter:Yuanpei Sun;Ning Zhang;Jian Wang;Yu Guo;Bo Sun;Honggang Zhou;Cheng Yang
Chinese Journal of Chemistry 2013 Volume 31( Issue 9) pp:1199-1206
Publication Date(Web):
DOI:10.1002/cjoc.201300392
Abstract
SARS CoV 3CLpro is known to be a promising target for development of therapeutic agents against the severe acute respiratory syndrome (SARS). A quinolinone compound 1 was selected via virtual screening, and it was synthetized and tested for enzymatic inhibition in vitro. Compound 1 showed potent inhibitory activity (IC50=0.44 µmol/L) toward SARS CoV 3CLpro. Further work on a series of quinolinone derivatives resulted in the discovery of the most potent compound 23, inhibiting SARS CoV 3CLpro with an IC50 of 36.86 nmol/L. The structure-activity relationships were also discussed.
Co-reporter:Xiao-xiao Sun;Tao Sun;Tai-yi Wang;Yan Zhang
Chemical Research in Chinese Universities 2013 Volume 29( Issue 6) pp:1098-1103
Publication Date(Web):2013 December
DOI:10.1007/s40242-013-3216-2
Aurora A is a cell cycle kinase linked to cancer. For the purpose of finding biologically active of novel compounds and providing new ideas for drug-design, we performed virtual screening in commercially available databases and got pyrazoleanthrone with promising inhibitory activity against Aurora A. Optimization of solvent accessible C7 position of pyrazoleanthrone made us get thirteen target compounds. These pyrazoleanthrone derivatives were evaluated by Aurora A inhibition assays in vitro. The results show that some target compounds could inhibit Aurora A kinase. Meanwhile, these title compounds were tested in vitro against hepatocellular carcinoma(HepG2) cells by the 3′-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide(MTT) method, showing that most of them had inhibitory potency. The inhibition rate of compound 6h was about 80% against HepG2 cells, and the IC50 value was 17.4 μmol/L, which would be considered for further study.
Co-reporter:Wei Liu;Yanting Tang;Yu Guo;Bo Sun;Hemin Zhu;Yanyan Xiao;Dan Dong;Cheng Yang
Applied Organometallic Chemistry 2012 Volume 26( Issue 4) pp:189-193
Publication Date(Web):
DOI:10.1002/aoc.2837
ABSTRACT
Fourteen new ferrocene derivatives containing urea linker were synthesized from the reaction of ferrocenecarbonyl azide with aromatic amines. The structures of the synthetic compounds were confirmed by 1H NMR, 13C NMR, IR, mass spectrometry and elemental analysis. These organometallic compounds were evaluated by in vitro protease assay using fluorogenic substrate peptide, and several showed potent inhibition against HIV-1 protease. Copyright © 2012 John Wiley & Sons, Ltd.








![1,5,6,7-tetrahydro-Cyclopent[f]indole-2,3-dione](http://img.cochemist.com/ccimg/383500/383425-01-2.png)
![1,5,6,7-tetrahydro-Cyclopent[f]indole-2,3-dione](http://img.cochemist.com/ccimg/383500/383425-01-2_b.png)




![Piperazine,1-methyl-4-[(4-nitrophenyl)sulfonyl]-](http://img.cochemist.com/ccimg/223800/223785-97-5.png)
![Piperazine,1-methyl-4-[(4-nitrophenyl)sulfonyl]-](http://img.cochemist.com/ccimg/223800/223785-97-5_b.png)


![Morpholine, 4-[(3,3-dichloro-2,3-dihydro-2-oxo-1H-indol-5-yl)sulfonyl]-](http://img.cochemist.com/ccimg/220500/220428-21-7.png)
![Morpholine, 4-[(3,3-dichloro-2,3-dihydro-2-oxo-1H-indol-5-yl)sulfonyl]-](http://img.cochemist.com/ccimg/220500/220428-21-7_b.png)






![L-Valinamide,N-acetyl-L-a-aspartyl-L-a-glutamyl-N-[(1S)-2-carboxy-1-formylethyl]-](http://img.cochemist.com/ccimg/169400/169332-60-9.png)
![L-Valinamide,N-acetyl-L-a-aspartyl-L-a-glutamyl-N-[(1S)-2-carboxy-1-formylethyl]-](http://img.cochemist.com/ccimg/169400/169332-60-9_b.png)





![1,2-Benzisothiazol-3(2H)-one, 2-[2-(4-methoxyphenyl)-2-oxoethyl]-,1,1-dioxide](http://img.cochemist.com/ccimg/75000/74921-64-5.png)
![1,2-Benzisothiazol-3(2H)-one, 2-[2-(4-methoxyphenyl)-2-oxoethyl]-,1,1-dioxide](http://img.cochemist.com/ccimg/75000/74921-64-5_b.png)











![BENZ[CD]INDOL-2(1H)-ONE, 1-BUTYL-](http://img.cochemist.com/ccimg/39300/39273-39-7.png)
![BENZ[CD]INDOL-2(1H)-ONE, 1-BUTYL-](http://img.cochemist.com/ccimg/39300/39273-39-7_b.png)


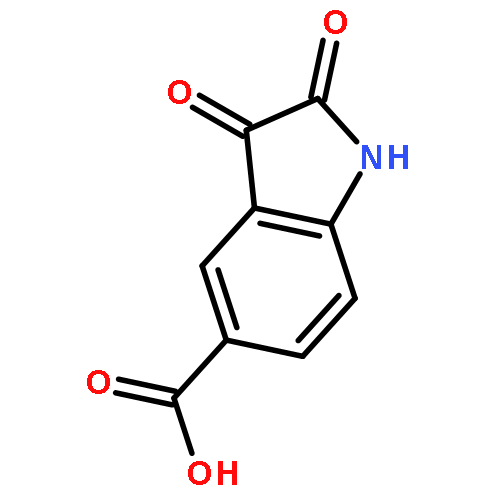



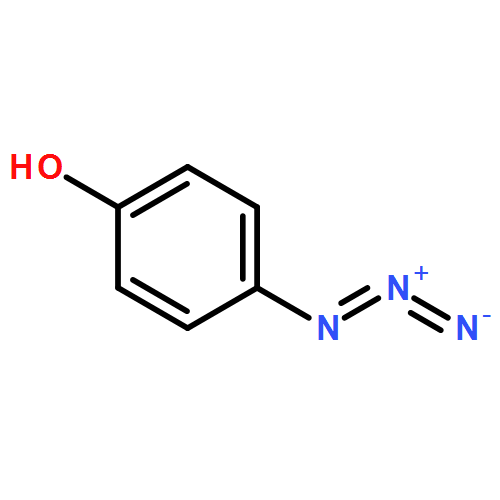






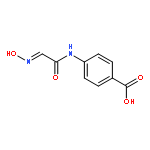





![2-(Hydroxymethyl)benzo[d]isothiazol-3(2H)-one 1,1-dioxide](http://img.cochemist.com/ccimg/14000/13947-20-1.png)
![2-(Hydroxymethyl)benzo[d]isothiazol-3(2H)-one 1,1-dioxide](http://img.cochemist.com/ccimg/14000/13947-20-1_b.png)
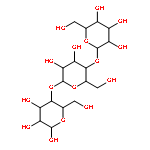











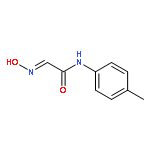
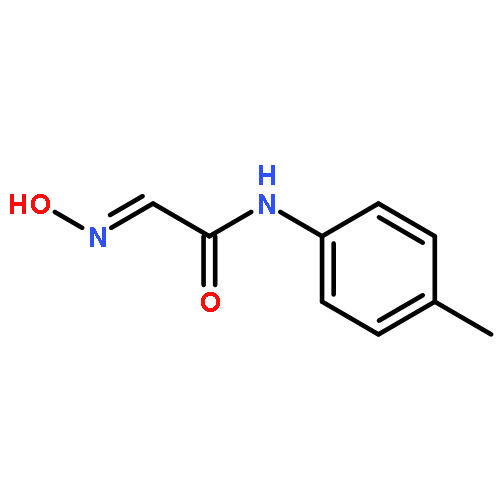
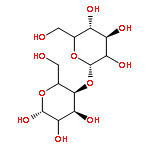
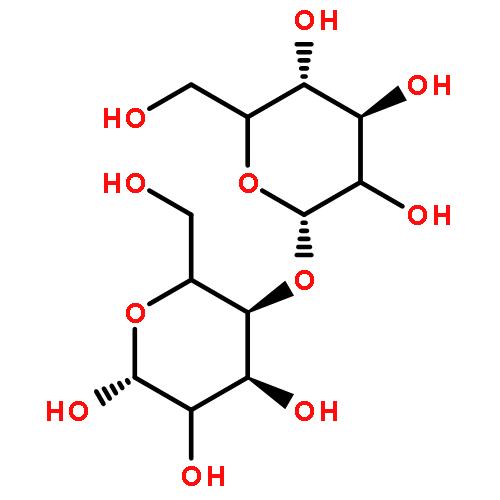
![4H-PYRAZOLO[1,5-C][1,3]THIAZOLE-2-CARBALDEHYDE](http://img.cochemist.com/ccimg/3300/3296-03-5.png)
![4H-PYRAZOLO[1,5-C][1,3]THIAZOLE-2-CARBALDEHYDE](http://img.cochemist.com/ccimg/3300/3296-03-5_b.png)



