Co-reporter:Nathan A. Bruender, Anthony P. Young, and Vahe Bandarian
Biochemistry 2015 Volume 54(Issue 18) pp:2903-2910
Publication Date(Web):May 1, 2015
DOI:10.1021/acs.biochem.5b00210
The radical S-adenosyl-l-methionine (SAM) superfamily is a large and growing group of enzymes that conduct complex radical-mediated transformations. A one-electron reduction of SAM via the +1 state of the cubane [4Fe-4S] cluster generates a 5′-deoxyadenosyl radical, which initiates turnover. The [4Fe-4S] cluster must be reduced from its resting +2 state to the catalytically active +1 oxidation state by an electron. In practice, dithionite or the Escherichia coli flavodoxin (EcFldA)/ferredoxin (flavodoxin):NADP+ oxidoreductase (Fpr)/NADPH system is used. Herein, we present a systematic investigation of the reductive activation of the radical SAM enzyme CDG synthase (BsQueE) from Bacillus subtilis comparing biological and chemical reductants. These data show that either of the flavodoxin homologues encoded by the B. subtilis genome, BsYkuN or BsYkuP, as well as a series of small molecule redox mediators, supports BsQueE activity. With dithionite as a reductant, the activity of BsQueE is ∼75-fold greater in the presence of BsYkuN and BsYkuP compared to that in the presence of dithionite alone. By contrast, EcFldA supports turnover to ∼10-fold greater levels than dithionite alone under the same conditions. Comparing the ratio of the rate of turnover to the apparent binding constant for the flavodoxin homologues reveals 10- and 240-fold preferences for BsYkuN over BsYkuP and EcFldA, respectively. The differential activation of the enzyme cannot be explained by the abortive cleavage of SAM. We conclude from these observations that the differential activation of BsQueE by Fld homologues may reside in the details of the interaction between the flavodoxin and the radical SAM enzyme.
Co-reporter:Anthony P. Young and Vahe Bandarian
Biochemistry 2015 Volume 54(Issue 23) pp:3569-3572
Publication Date(Web):June 8, 2015
DOI:10.1021/acs.biochem.5b00476
TYW1 catalyzes the formation of 4-demethylwyosine via the condensation of N-methylguanosine (m1G) with carbons 2 and 3 of pyruvate. In this study, labeled transfer ribonucleic acid (tRNA) and pyruvate were utilized to determine the site of hydrogen atom abstraction and regiochemistry of the pyruvate addition. tRNA containing a 2H-labeled m1G methyl group was used to identify the methyl group of m1G as the site of hydrogen atom abstraction by 5′-deoxyadenosyl radical. [2-13C1-3,3,3-2H3]Pyruvate was used to demonstrate retention of all the pyruvate protons, indicating that C2 of pyruvate forms the bridging carbon of the imidazoline ring and C3 the methyl.
Co-reporter:Micah T. Nelp ; Vahe Barian
Angewandte Chemie International Edition 2015 Volume 54( Issue 36) pp:10627-10629
Publication Date(Web):
DOI:10.1002/anie.201504505
Abstract
The biosynthesis of nitriles is known to occur through specialized pathways involving multiple enzymes; however, in bacterial and archeal biosynthesis of 7-deazapurines, a single enzyme, ToyM, catalyzes the conversion of the carboxylic acid containing 7-carboxy-7-deazaguanine (CDG) into its corresponding nitrile, 7-cyano-7-deazaguanine (preQ0). The mechanism of this unusual direct transformation was shown to proceed via the adenylation of CDG, which activates it to form the newly discovered amide intermediate 7-amido-7-deazaguanine (ADG). This is subsequently dehydrated to form the nitrile in a process that consumes a second equivalent of ATP. The authentic amide intermediate is shown to be chemically and kinetically competent. The ability of ToyM to activate two different substrates, an acid and an amide, accounts for this unprecedented one-enzyme catalysis of nitrile synthesis, and the differential rates of these two half reactions suggest that this catalytic ability is derived from an amide synthetase that gained a new function.
Co-reporter:Micah T. Nelp ; Vahe Barian
Angewandte Chemie 2015 Volume 127( Issue 36) pp:10773-10775
Publication Date(Web):
DOI:10.1002/ange.201504505
Abstract
The biosynthesis of nitriles is known to occur through specialized pathways involving multiple enzymes; however, in bacterial and archeal biosynthesis of 7-deazapurines, a single enzyme, ToyM, catalyzes the conversion of the carboxylic acid containing 7-carboxy-7-deazaguanine (CDG) into its corresponding nitrile, 7-cyano-7-deazaguanine (preQ0). The mechanism of this unusual direct transformation was shown to proceed via the adenylation of CDG, which activates it to form the newly discovered amide intermediate 7-amido-7-deazaguanine (ADG). This is subsequently dehydrated to form the nitrile in a process that consumes a second equivalent of ATP. The authentic amide intermediate is shown to be chemically and kinetically competent. The ability of ToyM to activate two different substrates, an acid and an amide, accounts for this unprecedented one-enzyme catalysis of nitrile synthesis, and the differential rates of these two half reactions suggest that this catalytic ability is derived from an amide synthetase that gained a new function.
Co-reporter:Micah T. Nelp, Andrei V. Astashkin, Linda A. Breci, Reid M. McCarty, and Vahe Bandarian
Biochemistry 2014 Volume 53(Issue 24) pp:
Publication Date(Web):June 10, 2014
DOI:10.1021/bi500260j
Nitrile hydratases (NHases) possess a mononuclear iron or cobalt cofactor whose coordination environment includes rare post-translationally oxidized cysteine sulfenic and sulfinic acid ligands. This cofactor is located in the α-subunit at the interfacial active site of the heterodimeric enzyme. Unlike canonical NHases, toyocamycin nitrile hydratase (TNHase) from Streptomyces rimosus is a unique three-subunit member of this family involved in the biosynthesis of pyrrolopyrimidine antibiotics. The subunits of TNHase are homologous to the α- and β-subunits of prototypical NHases. Herein we report the expression, purification, and characterization of the α-subunit of TNHase. The UV–visible, EPR, and mass spectra of the α-subunit TNHase provide evidence that this subunit alone is capable of synthesizing the active site complex with full post-translational modifications. Remarkably, the isolated post-translationally modified α-subunit is also catalytically active with the natural substrate, toyocamycin, as well as the niacin precursor 3-cyanopyridine. Comparisons of the steady state kinetic parameters of the single subunit variant to the heterotrimeric protein clearly show that the additional subunits impart substrate specificity and catalytic efficiency. We conclude that the α-subunit is the minimal sequence needed for nitrile hydration providing a simplified scaffold to study the mechanism and post-translational modification of this important class of catalysts.
Co-reporter:Reid M. McCarty, Carsten Krebs, and Vahe Bandarian
Biochemistry 2013 Volume 52(Issue 1) pp:
Publication Date(Web):November 29, 2012
DOI:10.1021/bi301156w
7-Carboxy-7-deazaguanine (CDG) synthase (QueE) catalyzes the complex heterocyclic radical-mediated conversion of 6-carboxy-5,6,7,8-tetrahydropterin (CPH4) to CDG in the third step of the biosynthetic pathway to all 7-deazapurines. Here we present a detailed characterization of QueE from Bacillus subtilis to delineate the mechanism of conversion of CPH4 to CDG. QueE is a member of the radical S-adenosyl-l-methionine (SAM) superfamily, all of which use a bound [4Fe-4S]+ cluster to catalyze the reductive cleavage of the SAM cofactor to generate methionine and a 5′-deoxyadenosyl radical (5′-dAdo•), which initiates enzymatic transformations requiring hydrogen atom abstraction. The ultraviolet–visible, electron paramagnetic resonance, and Mössbauer spectroscopic features of the homodimeric QueE point to the presence of a single [4Fe-4S] cluster per monomer. Steady-state kinetic experiments indicate a Km of 20 ± 7 μM for CPH4 and a kcat of 5.4 ± 1.2 min–1 for the overall transformation. The kinetically determined Kapp for SAM is 45 ± 1 μM. QueE is also magnesium-dependent and exhibits a Kapp for the divalent metal ion of 0.21 ± 0.03 mM. The SAM cofactor supports multiple turnovers, indicating that it is regenerated at the end of each catalytic cycle. The mechanism of rearrangement of QueE was probed with CPH4 isotopologs containing deuterium at C-6 or the two prochiral positions at C-7. These studies implicate 5′-dAdo• as the initiator of the ring contraction reaction catalyzed by QueE by abstraction of the H atom from C-6 of CPH4.
Co-reporter:Reid M. McCarty, Vahe Bandarian
Bioorganic Chemistry 2012 Volume 43() pp:15-25
Publication Date(Web):August 2012
DOI:10.1016/j.bioorg.2012.01.001
Pyrrolopyrimidine containing compounds, also known as 7-deazapurines, are a collection of purine-based metabolites that have been isolated from a variety of biological sources and have diverse functions which range from secondary metabolism to RNA modification. To date, nearly 35 compounds with the common 7-deazapurine core structure have been described. This article will illustrate the structural diversity of these compounds and review the current state of knowledge on the biosynthetic pathways that give rise to them.Graphical abstractHighlights► GTP is a precursor to all known 7-deazapurines. ► 7-Deazapurines are widely distributed in all kingdoms of life. ► Biosynthetic pathways for 7-deazapurines include several novel transformations.
Co-reporter:Anthony P. Young and Vahe Bandarian
Biochemistry 2011 Volume 50(Issue 49) pp:
Publication Date(Web):October 25, 2011
DOI:10.1021/bi2015053
TYW1 catalyzes the condensation of N-methylguanosine with two carbon atoms from an unknown second substrate to form 4-demethylwyosine, which is a common intermediate in the biosynthesis of all of the hypermodified RNA bases related to wybutosine found in eukaryal and archaeal tRNAPhe. Of the potential substrates examined, only incubation with pyruvate resulted in formation of 4-demethylwyosine. Moreover, incubation with C1, C2, C3, or C1,2,3-13C-labeled pyruvate showed that C2 and C3 are incorporated while C1 is not. The mechanistic implications of these results are discussed in the context of the structure of TYW1.
Co-reporter:Zachary D. Miles;Reid M. McCarty;Gabriella Molnar
PNAS 2011 Volume 108 (Issue 18 ) pp:7368-7372
Publication Date(Web):2011-05-03
DOI:10.1073/pnas.1018636108
Transfer RNA is one of the most richly modified biological molecules. Biosynthetic pathways that introduce these modifications
are underexplored, largely because their absence does not lead to obvious phenotypes under normal growth conditions. Queuosine
(Q) is a hypermodified base found in the wobble positions of tRNA Asp, Asn, His, and Tyr from bacteria to mankind. Using liquid
chromatography MS methods, we have screened 1,755 single gene knockouts of Escherichia coli and have identified the key final step in the biosynthesis of Q. The protein is homologous to B12-dependent iron-sulfur proteins involved in halorespiration. The recombinant Bacillus subtilis epoxyqueuosine (oQ) reductase catalyzes the conversion of oQ to Q in a synthetic substrate, as well as undermodified RNA
isolated from an oQ reductase knockout strain. The activity requires inclusion of a reductant and a redox mediator. Finally,
exogenously supplied cobalamin stimulates the activity. This work provides the framework for studies of the biosynthesis of
other modified RNA components, where lack of accessible phenotype or obvious gene clustering has impeded discovery. Moreover,
discovery of the elusive oQ reductase protein completes the biosynthetic pathway of Q.
Co-reporter:Reid M. McCarty, Árpád Somogyi, Guangxin Lin, Neil E. Jacobsen and Vahe Bandarian
Biochemistry 2009 Volume 48(Issue 18) pp:
Publication Date(Web):April 8, 2009
DOI:10.1021/bi900400e
Deazapurine-containing secondary metabolites comprise a broad range of structurally diverse nucleoside analogues found throughout biology, including various antibiotics produced by species of Streptomyces bacteria and the hypermodified tRNA bases queuosine and archaeosine. Despite early interest in deazapurines as antibiotic, antiviral, and antineoplastic agents, the biosynthetic route toward deazapurine production has remained largely elusive for more than 40 years. Here we present the first in vitro preparation of the deazapurine base preQ0, by the successive action of four enzymes. The pathway includes the conversion of the recently identified biosynthetic intermediate, 6-carboxy-5,6,7,8-tetrahydropterin, to a novel intermediate, 7-carboxy-7-deazaguanine (CDG), by an unusual transformation catalyzed by Bacillus subtilis QueE, a member of the radical SAM enzyme superfamily. The carboxylate moiety on CDG is converted subsequently to a nitrile to yield preQ0 by either B. subtilis QueC or Streptomyces rimosus ToyM in an ATP-dependent reaction, in which ammonia serves as the nitrogen source. The results presented here are consistent with early radiotracer studies on deazapurine biosynthesis and provide a unified pathway for the production of deazapurines in nature.
Co-reporter:Reid M. McCarty, Árpád Somogyi and Vahe Bandarian
Biochemistry 2009 Volume 48(Issue 11) pp:
Publication Date(Web):February 20, 2009
DOI:10.1021/bi9001437
To elucidate the early steps required during biosynthesis of a broad class of 7-deazapurine-containing natural products, we have studied the reaction catalyzed by Escherichia coli QueD, a 6-pyruvoyl-5,6,7,8-tetrahydropterin synthase (PTPS) homologue possibly involved in queuosine biosynthesis. While mammalian PTPS homologues convert 7,8-dihydroneopterin triphosphate (H2NTP) to 6-pyruvoyltetrahydropterin (PPH4) in biopterin biosynthesis, E. coli QueD catalyzes the conversion of H2NTP to 6-carboxy-5,6,7,8-tetrahydropterin (CPH4). E. coli QueD can also convert PPH4 and sepiapterin to CPH4, allowing a mechanism to be proposed.
Co-reporter:Reid M. McCarty, Vahe Bandarian
Chemistry & Biology 2008 Volume 15(Issue 8) pp:790-798
Publication Date(Web):25 August 2008
DOI:10.1016/j.chembiol.2008.07.012
Pyrrolopyrimidine nucleosides analogs, collectively referred to as deazapurines, are an important class of structurally diverse compounds found in a wide variety of biological niches. In this report, a cluster of genes from Streptomyces rimosus (ATCC 14673) involved in production of the deazapurine antibiotics sangivamycin and toyocamycin was identified. The cluster includes toyocamycin nitrile hydratase, an enzyme that catalyzes the conversion of toyocamycin to sangivamycin. In addition to this rare nitrile hydratase, the cluster encodes a GTP cyclohydrolase I, linking the biosynthesis of deazapurines to folate biosynthesis, and a set of purine salvage/biosynthesis genes, which presumably convert the guanine moiety from GTP to the adenine-like deazapurine base found in toyocamycin and sangivamycin. The gene cluster presented here could potentially serve as a model to allow identification of deazapurine biosynthetic pathways in other bacterial species.

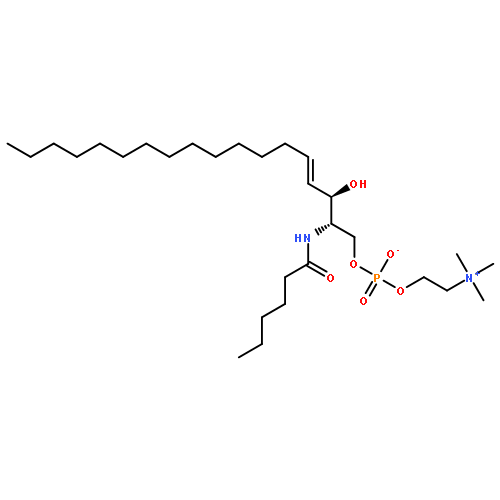
![[(E,2S,3R)-2-(dodecanoylamino)-3-hydroxyoctadec-4-enyl] 2-(trimethylazaniumyl)ethyl phosphate](http://img.cochemist.com/ccimg/475000/474923-21-2.png)
![[(E,2S,3R)-2-(dodecanoylamino)-3-hydroxyoctadec-4-enyl] 2-(trimethylazaniumyl)ethyl phosphate](http://img.cochemist.com/ccimg/475000/474923-21-2_b.png)
![4H-Pyrrolo[2,3-d]pyrimidin-4-one,2-amino-5-[[(3,4-dihydroxy-6-oxabicyclo[3.1.0]hex-2-yl)amino]methyl]-1,7-dihydro-7-b-D-ribofuranosyl-](http://img.cochemist.com/ccimg/107900/107865-20-3.png)
![4H-Pyrrolo[2,3-d]pyrimidin-4-one,2-amino-5-[[(3,4-dihydroxy-6-oxabicyclo[3.1.0]hex-2-yl)amino]methyl]-1,7-dihydro-7-b-D-ribofuranosyl-](http://img.cochemist.com/ccimg/107900/107865-20-3_b.png)
![triphosphoric acid, mono[(2R,3S)-3-(2-amino-1,4,7,8-tetrahydro-4-oxo-6-pteridinyl)-2,3-dihydroxypropyl] ester](http://img.cochemist.com/ccimg/20600/20574-65-6.png)
![triphosphoric acid, mono[(2R,3S)-3-(2-amino-1,4,7,8-tetrahydro-4-oxo-6-pteridinyl)-2,3-dihydroxypropyl] ester](http://img.cochemist.com/ccimg/20600/20574-65-6_b.png)
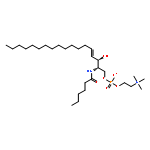
![2-Amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidine-5-carboxamide](http://img.cochemist.com/ccimg/124800/124738-80-3.png)
![2-Amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidine-5-carboxamide](http://img.cochemist.com/ccimg/124800/124738-80-3_b.png)
![Methyl 2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidine-5-carboxylate](http://img.cochemist.com/ccimg/124800/124738-76-7.png)
![Methyl 2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidine-5-carboxylate](http://img.cochemist.com/ccimg/124800/124738-76-7_b.png)
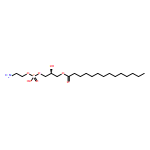
![2-amino-4,7-dihydro-4-oxo-3H-Pyrrolo[2,3-d]pyrimidine-5-carboxylic acid](http://img.cochemist.com/ccimg/92700/92636-62-9.png)
![2-amino-4,7-dihydro-4-oxo-3H-Pyrrolo[2,3-d]pyrimidine-5-carboxylic acid](http://img.cochemist.com/ccimg/92700/92636-62-9_b.png)
![2-amino-4,7-dihydro-4-oxo-3H-Pyrrolo[2,3-d]pyrimidine-5-carbonitrile](http://img.cochemist.com/ccimg/69300/69205-79-4.png)
![2-amino-4,7-dihydro-4-oxo-3H-Pyrrolo[2,3-d]pyrimidine-5-carbonitrile](http://img.cochemist.com/ccimg/69300/69205-79-4_b.png)





![7H-Pyrrolo[2,3-d]pyrimidine-5-carbonitrile,4-amino-7-b-D-ribofuranosyl-](http://img.cochemist.com/ccimg/700/606-58-6.png)
![7H-Pyrrolo[2,3-d]pyrimidine-5-carbonitrile,4-amino-7-b-D-ribofuranosyl-](http://img.cochemist.com/ccimg/700/606-58-6_b.png)





