Co-reporter:Stephanie Russell, Raphaël Rahmani, Amy J. Jones, Harriet L. Newson, Kevin Neilde, Ignacio Cotillo, Marzieh Rahmani Khajouei, Lori Ferrins, Sana Qureishi, Nghi Nguyen, Maria S. Martinez-Martinez, Donald F. Weaver, Marcel Kaiser, Jennifer Riley, John Thomas, Manu De Rycker, Kevin D. Read, Gavin R. Flematti, Eileen Ryan, Scott Tanghe, Ana Rodriguez, Susan A. Charman, Albane Kessler, Vicky M. Avery, Jonathan B. Baell, and Matthew J. Piggott
Journal of Medicinal Chemistry 2016 Volume 59(Issue 21) pp:9686-9720
Publication Date(Web):August 22, 2016
DOI:10.1021/acs.jmedchem.6b00442
The parasitic trypanosomes Trypanosoma brucei and T. cruzi are responsible for significant human suffering in the form of human African trypanosomiasis (HAT) and Chagas disease. Drugs currently available to treat these neglected diseases leave much to be desired. Herein we report optimization of a novel class of N-(2-(2-phenylthiazol-4-yl)ethyl)amides, carbamates, and ureas, which rapidly, selectively, and potently kill both species of trypanosome. The mode of action of these compounds is unknown but does not involve CYP51 inhibition. They do, however, exhibit clear structure–activity relationships, consistent across both trypanosome species. Favorable physicochemical parameters place the best compounds in CNS drug-like chemical space but, as a class, they exhibit poor metabolic stability. One of the best compounds (64a) cleared all signs of T. cruzi infection in mice when CYP metabolism was inhibited, with sterile cure achieved in one mouse. This family of compounds thus shows significant promise for trypanosomiasis drug discovery.
Co-reporter:Swapna Johnson, Raphaël Rahmani, Damien R. Drew, Melanie J. Williams, Mark Wilkinson, Yan Hong Tan, Johnny X. Huang, Christopher J. Tonkin, James G. Beeson, Jake Baum, Brian J. Smith, and Jonathan B. Baell
Journal of Medicinal Chemistry 2016 Volume 59(Issue 24) pp:10994-11005
Publication Date(Web):November 9, 2016
DOI:10.1021/acs.jmedchem.6b01109
Polymerization of the cytosolic protein actin is critical to cell movement and host cell invasion by the malaria parasite, Plasmodium falciparum. Any disruption to actin polymerization dynamics will render the parasite incapable of invading a host cell and thereby unable to cause infection. Here, we explore the potential of using truncated latrunculins as potential chemotherapeutics for the treatment of malaria. Exploration of the binding interactions of the natural actin inhibitor latrunculins with actin revealed how a truncated core of the inhibitor could retain its key interaction features with actin. This truncated core was synthesized and subjected to preliminary structure–activity relationship studies to generate a focused set of analogues. Biochemical analyses of these analogues demonstrate their 6-fold increased activity compared with that of latrunculin B against P. falciparum and a 16-fold improved selectivity ex vivo. These data establish the latrunculin core as a potential focus for future structure-based drug design of chemotherapeutics against malaria.
Co-reporter:Jonathan B. Baell
Journal of Natural Products 2016 Volume 79(Issue 3) pp:616-628
Publication Date(Web):February 22, 2016
DOI:10.1021/acs.jnatprod.5b00947
We have previously reported on classes of compounds that can interfere with bioassays via a number of different mechanisms and termed such compounds Pan Assay INterference compoundS, or PAINS. These compounds were defined on the basis of high-throughput data derived from vendor-supplied synthetics. The question therefore arises whether the concept of PAINS is relevant to compounds of natural origin. Here, it is shown that this is indeed the case, but that the context of the biological readout is an important factor that must be brought into consideration.
Co-reporter:Jitendra R. Harjani, Beow Keat Yap, Raymond S. Norton, Jonathan B. Baell
Tetrahedron 2016 Volume 72(Issue 23) pp:3256-3261
Publication Date(Web):9 June 2016
DOI:10.1016/j.tet.2016.04.051
A new method for the synthesis of cystathionine containing cyclic peptides has been developed. Conventionally such systems are typically made with relatively late-stage on-resin cyclisation involving reaction between a chlorohomoalanine and cysteine residue. We offer a different approach involving early incorporation of a cystathionine residue through on-resin reaction between a cysteine residue and an N-Alloc iodohomoalanine cumyl ester. Subsequent cyclisation then involves amide bond formation. The success of this method was demonstrated by applying it to the synthesis of a cystathionine analogue of the disulfide bridge cyclic peptide, N-acetylated cyclic peptide c[CVDINNNC]-NH2, a biologically active iNOS binding epitope mimetic that binds tightly to the protein, SPSB2. Our method expands the repertoire of synthetic options towards the construction of cystathionine containing cyclic peptides.
Co-reporter:Raphaël Rahmani; Kung Ban; Amy J. Jones; Lori Ferrins; Danny Ganame; Melissa L. Sykes; Vicky M. Avery; Karen L. White; Eileen Ryan; Marcel Kaiser; Susan A. Charman;Jonathan B. Baell
Journal of Medicinal Chemistry 2015 Volume 58(Issue 17) pp:6753-6765
Publication Date(Web):August 6, 2015
DOI:10.1021/acs.jmedchem.5b00438
From a whole-organism high throughput screen of approximately 87000 compounds against Trypanosoma brucei brucei, we recently identified eight new unique compounds for the treatment of human African trypanosomiasis. In an effort to understand the structure–activity relationships around these compounds, we report for the first time our results on a new class of trypanocides, the pyrazine carboxamides. Attracted by the low molecular weight (270 g·mol–1) of our starting hit (9) and its potency (0.49 μM), the SAR around the core was explored, leading to compounds having an EC50 as low as 25 nM against T. b. brucei and being more than 1500 times less toxic against mammalian L6 and HEK293 cell lines. The most potent compounds in the series were exquisitely selective for T. brucei over a panel of other protozoan parasites, showing an excellent correlation with the human infective parasite Trypanosoma brucei rhodesiense, the most potent compound (65) having an EC50 of 24 nM. The compounds are highly drug-like and are able to penetrate the CNS, their only limitation currently being their rate of microsomal metabolism. To that effect, efforts to identify potential metabolites of selected compounds are also reported.
Co-reporter:Jonathan B. Baell
ACS Medicinal Chemistry Letters 2015 Volume 6(Issue 3) pp:229
Publication Date(Web):February 9, 2015
DOI:10.1021/acsmedchemlett.5b00032
Whether identified through high throughput screening or in silico screening, and whether target-based or phenotypic, sets of hits will contain chemical con artists. Such pan-assay interference compounds (PAINS) and other subversive compounds continue to pollute the scientific literature. There are several angles of attack to aid identification of such nonprogressable molecules. One of these rules above all, and this is a demonstration of genuine structure–activity relationships. Recognition of this, which will require a greater effort in medicinal chemistry, will be of general benefit.
Co-reporter:Daniel L. Priebbenow, Robert. W. Gable, and Jonathan Baell
The Journal of Organic Chemistry 2015 Volume 80(Issue 9) pp:4412-4418
Publication Date(Web):April 13, 2015
DOI:10.1021/acs.joc.5b00250
A new method for the regioselective and stereoselective iodoacyloxylation of alkynes has been developed. This protocol utilizes a combination of an iodobenzene dicarboxylate and iodine to functionalize a series of activated and unactivated alkynes in an entirely selective and predictable fashion. The resultant iodo-enol esters were subsequently coupled with boronic acids to afford tetrasubstituted alkene derivatives, which could be further converted to the corresponding 1,1-disubstituted acetophenone.
Co-reporter:Lori Ferrins ; Michelle Gazdik ; Raphaël Rahmani ; Swapna Varghese ; Melissa L. Sykes ; Amy J. Jones ; Vicky M. Avery ; Karen L. White ; Eileen Ryan ; Susan A. Charman ; Marcel Kaiser ; Christel A. S. Bergström ◆;Jonathan B. Baell
Journal of Medicinal Chemistry 2014 Volume 57(Issue 15) pp:6393-6402
Publication Date(Web):June 30, 2014
DOI:10.1021/jm500191u
A whole-organism screen of approximately 87000 compounds against Trypanosoma brucei brucei identified a number of promising compounds for medicinal chemistry optimization. One of these classes of compounds we termed the pyridyl benzamides. While the initial hit had an IC50 of 12 μM, it was small enough to be attractive for further optimization, and we utilized three parallel approaches to develop the structure–activity relationships. We determined that the physicochemical properties for this class are generally favorable with particular positions identified that appear to block metabolism when substituted and others that modulate solubility. Our most active compound is 79, which has an IC50 of 0.045 μM against the human pathogenic strain Trypanosoma brucei rhodesiense and is more than 4000 times less active against the mammalian L6 cell line.
Co-reporter:Ryan M. Brady ; Amelia Vom ; Michael J. Roy ; Nathan Toovey ; Brian J. Smith ; Rebecca M. Moss ; Effie Hatzis ; David C. S. Huang ; John P. Parisot ; Hong Yang ; Ian P. Street ; Peter M. Colman ; Peter E. Czabotar ; Jonathan B. Baell ;Guillaume Lessene
Journal of Medicinal Chemistry 2014 Volume 57(Issue 4) pp:1323-1343
Publication Date(Web):January 23, 2014
DOI:10.1021/jm401948b
The prosurvival BCL-2 proteins are attractive yet challenging targets for medicinal chemists. Their involvement in the initiation and progression of many, if not all, tumors makes them prime targets for developing new anticancer therapies. We present our approach based on de novo structure-based drug design. Using known structural information from complexes engaging opposing members of the BCL-2 family of proteins, we designed peptidomimetic compounds using a benzoylurea scaffold to reproduce key interactions between these proteins. A library stemming from the initial de novo designed scaffold led to the discovery of ligands with low micromolar potency (KD = 4 μM) and selectivity for BCL-XL. These compounds bind in the canonical BH3 binding groove in a binding mode distinct from previously known BCL-2 inhibitors. The results of our study provide insight into the design of a new class of antagonists targeting a challenging class of protein–protein interactions.
Co-reporter:Adel A. Rashad, Amy J. Jones, Vicky M. Avery, Jonathan Baell, and Paul A. Keller
ACS Medicinal Chemistry Letters 2014 Volume 5(Issue 5) pp:496-500
Publication Date(Web):March 10, 2014
DOI:10.1021/ml400487t
The high throughput screening of a library of over 87,000 drug-like compounds against the African sleeping sickness parasite resulted in the discovery of hits with a wide range of molecular diversity. We report here the medicinal chemistry development of one such hit, a tetrahydroisoquinoline disulfonamide, with the synthesis and testing of 26 derivatives against the trypanosome subspecies. Activities in the 2–4 μM range were revealed with a selectivity index suitable for further development.Keywords: SAR; sulfonamides; T. brucei; tetrahydroquinoline;
Co-reporter:Jitendra R. Harjani, Andrew X. Tang, Raymond S. Norton, Jonathan B. Baell
Tetrahedron 2014 70(43) pp: 8047-8055
Publication Date(Web):
DOI:10.1016/j.tet.2014.08.036
Co-reporter:Silvia C. Teguh ; Nectarios Klonis ; Sandra Duffy ; Leonardo Lucantoni ; Vicky M. Avery ; Craig A. Hutton ; Jonathan B. Baell ;Leann Tilley
Journal of Medicinal Chemistry 2013 Volume 56(Issue 15) pp:6200-6215
Publication Date(Web):July 9, 2013
DOI:10.1021/jm400656s
A novel class of antimalarial compounds, based on an indol-3-yl linked to the 2-position of a 4-aminoquinoline moiety, shows promising activity against the malaria parasite, Plasmodium falciparum. Compounds with a quaternary nitrogen on the quinoline show improved activity against the chloroquine-resistant K1 strain. Nonquaternerized 4-aminoquinolines retain significant potency but are relatively less active against the K1 strain. Alkylation of the 4-amino group preferentially improves the activity against the chloroquine-sensitive 3D7 strain. The quinoline-indoles show only weak activity as inhibitors of β-hematin formation, and their activities are only weakly antagonized by a hemoglobinase inhibitor. The compounds appear to dissipate mitochondrial potential as an early event in their antimalarial action and therefore may exert their activity by compromising Plasmodium mitochondrial function. Interestingly, we observed a structural relationship between our compounds and the anticancer and anthelminthic compound, pyrvinium pamoate, which has also been proposed to exert its action via compromising mitochondrial function.
Co-reporter:Jonathan B. Baell
Journal of Chemical Information and Modeling 2013 Volume 53(Issue 1) pp:39-55
Publication Date(Web):November 30, 2012
DOI:10.1021/ci300461a
In establishing what we propose is the globe’s highest quality collection of available screening compounds, it is convincingly shown that the globe’s pool of such compounds is extremely shallow and can be represented by fewer than 350,000 compounds. To support our argument, we discuss and fully disclose our extensive battery of functional group filters. We discuss the use of PAINS filters and also show the effect of similarity exclusion on structure–activity relationships. We show why limited analogue representation requires screening at higher concentrations to capture hit classes for difficult targets that otherwise may be prosecuted unsuccessfully. We construct our arguments in a structurally focused manner to be most useful to medicinal chemists, the key players in drug discovery.
Co-reporter:Lori Ferrins, Raphaël Rahmani, Melissa L. Sykes, Amy J. Jones, Vicky M. Avery, Eliott Teston, Basmah Almohaywi, JieXiang Yin, Jason Smith, Chris Hyland, Karen L. White, Eileen Ryan, Michael Campbell, Susan A. Charman, Marcel Kaiser, Jonathan B. Baell
European Journal of Medicinal Chemistry 2013 Volume 66() pp:450-465
Publication Date(Web):August 2013
DOI:10.1016/j.ejmech.2013.05.007
•New class of trypanocidal agents.•3-(Oxazolo[4,5-b]pyridin-2-yl)anilides with clear SAR.•Potent inhibitors of Trypanosoma brucei, not toxic to mammalian cells.A whole organism high-throughput screen of approximately 87,000 compounds against Trypanosoma brucei brucei led to the recent discovery of several novel compound classes with low micromolar activity against this organism and without appreciable cytotoxicity to mammalian cells. Herein we report a structure–activity relationship (SAR) investigation around one of these hit classes, the 3-(oxazolo[4,5-b]pyridin-2-yl)anilides. Sharp SAR is revealed, with our most active compound (5) exhibiting an IC50 of 91 nM against the human pathogenic strain T.b. rhodesiense and being more than 700 times less toxic towards the L6 mammalian cell line. Physicochemical properties are attractive for many compounds in this series. For the most potent representatives, we show that solubility and metabolic stability are key parameters to target during future optimisation.
Co-reporter:Ryan M. Brady, Minmin Zhang, Robert Gable, Raymond S. Norton, Jonathan B. Baell
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 17) pp:4892-4895
Publication Date(Web):1 September 2013
DOI:10.1016/j.bmcl.2013.06.086
μ-Conotoxin KIIIA blocks voltage-gated sodium channels and displays potent analgesic activity in mice models for pain. Structure–activity studies with KIIIA have shown that residues important for sodium channel activity are presented on an α-helix. Herein, we report the de novo design and synthesis of a three-residue (Lys7, Trp8, His12) peptidomimetic based on a novel diketopiperazine (DKP) carboxamide scaffold.
Co-reporter:Jitendra R. Harjani; Beow Keat Yap; Eleanor W. W. Leung; Andrew Lucke; Sandra E. Nicholson; Martin J. Scanlon; David K. Chalmers; Philip E. Thompson; Raymond S. Norton;Jonathan B. Baell
Journal of Medicinal Chemistry () pp:
Publication Date(Web):May 23, 2016
DOI:10.1021/acs.jmedchem.6b00386
SPRY domain-containing suppressor of cytokine signaling box protein (SPSB) 2-deficient macrophages have been found to exhibit prolonged expression of inducible nitric oxide synthase (iNOS) and enhanced killing of persistent pathogens, suggesting that inhibitors of the SPSB2−iNOS interaction have potential as novel anti-infectives. In this study, we describe the design, synthesis, and characterization of cyclic peptidomimetic inhibitors of the SPSB2–iNOS interaction constrained by organic linkers to improve stability and druggability. SPR, ITC, and 19F NMR analyses revealed that the most potent cyclic peptidomimetic bound to the iNOS binding site of SPSB2 with low nanomolar affinity (KD 29 nM), a 10-fold improvement over that of the linear peptide DINNN (KD 318 nM), and showed strong inhibition of SPSB2–iNOS interaction in macrophage cell lysates. This study exemplifies a novel approach to cyclize a Type II β-turn linear peptide and provides a foundation for future development of this group of inhibitors as new anti-infectives.
Co-reporter:Daniel L. Priebbenow, Lisa Barbaro and Jonathan B. Baell
Organic & Biomolecular Chemistry 2016 - vol. 14(Issue 40) pp:NaN9628-9628
Publication Date(Web):2016/09/13
DOI:10.1039/C6OB01893A
Multi-drug resistant tuberculosis (MDR-TB) is of growing global concern and threatens to undermine increasing efforts to control the worldwide spread of tuberculosis (TB). Bedaquiline has recently emerged as a new drug developed to specifically treat MDR-TB. Despite being highly effective as a result of its unique mode of action, bedaquiline has been associated with significant toxicities and as such, safety concerns are limiting its clinical use. In order to access pharmaceutical agents that exhibit an improved safety profile for the treatment of MDR-TB, new synthetic pathways to facilitate the preparation of bedaquiline and analogues thereof have been discovered.









![1,2,4-TRIAZINE, 3-[(1,1-DIMETHYLETHYL)THIO]-](http://img.cochemist.com/ccimg/808800/808756-22-1.png)
![1,2,4-TRIAZINE, 3-[(1,1-DIMETHYLETHYL)THIO]-](http://img.cochemist.com/ccimg/808800/808756-22-1_b.png)
![6H-PURIN-6-ONE, 2-AMINO-8-[(2,3-DIHYDROXYPROPYL)THIO]-1,7-DIHYDRO-](http://img.cochemist.com/ccimg/758000/757992-04-4.png)
![6H-PURIN-6-ONE, 2-AMINO-8-[(2,3-DIHYDROXYPROPYL)THIO]-1,7-DIHYDRO-](http://img.cochemist.com/ccimg/758000/757992-04-4_b.png)






![Pyrrolidine, 1-[(3-amino-6-phenylpyrazinyl)carbonyl]-](http://img.cochemist.com/ccimg/625500/625459-35-0.png)
![Pyrrolidine, 1-[(3-amino-6-phenylpyrazinyl)carbonyl]-](http://img.cochemist.com/ccimg/625500/625459-35-0_b.png)










![6H-PURIN-6-ONE, 2-AMINO-8-[[(4-CHLOROPHENYL)METHYL]THIO]-1,7-DIHYDRO-](http://img.cochemist.com/ccimg/486500/486408-88-2.png)
![6H-PURIN-6-ONE, 2-AMINO-8-[[(4-CHLOROPHENYL)METHYL]THIO]-1,7-DIHYDRO-](http://img.cochemist.com/ccimg/486500/486408-88-2_b.png)





![6,7-DIHYDRO-[1,4]DIOXINO[2,3-F][1,3]BENZOTHIAZOL-2-AMINE](http://img.cochemist.com/ccimg/313300/313223-82-4.png)
![6,7-DIHYDRO-[1,4]DIOXINO[2,3-F][1,3]BENZOTHIAZOL-2-AMINE](http://img.cochemist.com/ccimg/313300/313223-82-4_b.png)
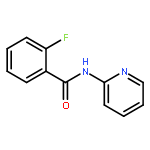
![[1,1'-Biphenyl]-2-carboxamide, N-2-pyridinyl-](http://img.cochemist.com/ccimg/258000/257941-41-6.png)
![[1,1'-Biphenyl]-2-carboxamide, N-2-pyridinyl-](http://img.cochemist.com/ccimg/258000/257941-41-6_b.png)


![7-Hydroxy-5-oxo-5H-thiazolo[3,2-a]pyrimidine-6-carboxylic acid methyl ester](http://img.cochemist.com/ccimg/224400/224313-88-6.png)
![7-Hydroxy-5-oxo-5H-thiazolo[3,2-a]pyrimidine-6-carboxylic acid methyl ester](http://img.cochemist.com/ccimg/224400/224313-88-6_b.png)


![1,2,4-Triazine, 3-[(1-methylethyl)thio]-](http://img.cochemist.com/ccimg/187100/187099-43-0.png)
![1,2,4-Triazine, 3-[(1-methylethyl)thio]-](http://img.cochemist.com/ccimg/187100/187099-43-0_b.png)










































![N6,N6-Dimethylbenzo[d]thiazole-2,6-diamine](http://img.cochemist.com/ccimg/64400/64334-41-4.png)
![N6,N6-Dimethylbenzo[d]thiazole-2,6-diamine](http://img.cochemist.com/ccimg/64400/64334-41-4_b.png)



















![1-{2-[2-(4-CHLOROPHENOXY)ETHOXY]-2-(2,4-DICHLOROPHENYL)ETHYL}-1H-<WBR />IMIDAZOLE NITRATE (1:1)](http://img.cochemist.com/ccimg/31000/30924-29-9.png)
![1-{2-[2-(4-CHLOROPHENOXY)ETHOXY]-2-(2,4-DICHLOROPHENYL)ETHYL}-1H-<WBR />IMIDAZOLE NITRATE (1:1)](http://img.cochemist.com/ccimg/31000/30924-29-9_b.png)







![4-Thiomorpholinamine,3-methyl-N-[(5-nitro-2-furanyl)methylene]-, 1,1-dioxide](http://img.cochemist.com/ccimg/23300/23256-30-6.png)
![4-Thiomorpholinamine,3-methyl-N-[(5-nitro-2-furanyl)methylene]-, 1,1-dioxide](http://img.cochemist.com/ccimg/23300/23256-30-6_b.png)
![2-Aminobenzo[d]thiazole-6-sulfonamide](http://img.cochemist.com/ccimg/18200/18101-58-1.png)
![2-Aminobenzo[d]thiazole-6-sulfonamide](http://img.cochemist.com/ccimg/18200/18101-58-1_b.png)
![5-Methyl-4,5,6,7-tetrahydrothiazolo[5,4-c]pyridin-2-amine](http://img.cochemist.com/ccimg/17900/17899-48-8.png)
![5-Methyl-4,5,6,7-tetrahydrothiazolo[5,4-c]pyridin-2-amine](http://img.cochemist.com/ccimg/17900/17899-48-8_b.png)
































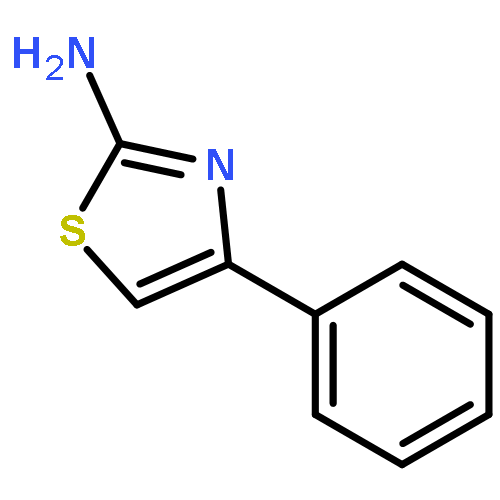
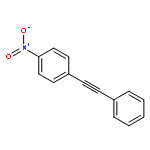
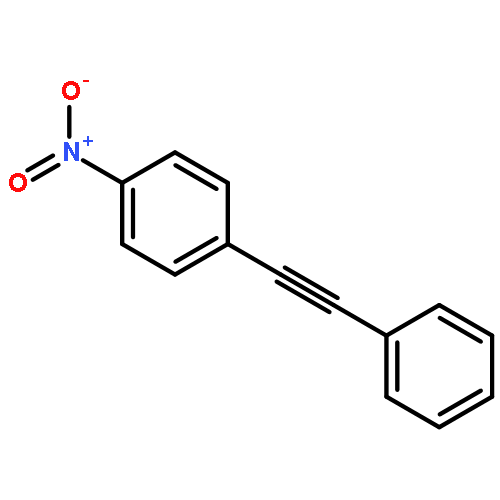


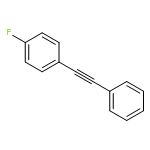


![1,3,5-Naphthalenetrisulfonicacid,8,8'-[carbonylbis[imino-3,1-phenylenecarbonylimino(4-methyl-3,1-phenylene)carbonylimino]]bis-](http://img.cochemist.com/ccimg/200/145-63-1.png)
![1,3,5-Naphthalenetrisulfonicacid,8,8'-[carbonylbis[imino-3,1-phenylenecarbonylimino(4-methyl-3,1-phenylene)carbonylimino]]bis-](http://img.cochemist.com/ccimg/200/145-63-1_b.png)






![6H-Purin-6-one, 2-amino-1,7-dihydro-8-[(2-oxo-2-phenylethyl)thio]-](http://img.cochemist.com/ccimg/51100/51041-04-4.png)
![6H-Purin-6-one, 2-amino-1,7-dihydro-8-[(2-oxo-2-phenylethyl)thio]-](http://img.cochemist.com/ccimg/51100/51041-04-4_b.png)

