Co-reporter:Dennis Pingen, Çiğdem Altıntaş, Max Rudolf Schaller and Dieter Vogt
Dalton Transactions 2016 vol. 45(Issue 29) pp:11765-11771
Publication Date(Web):10 Jun 2016
DOI:10.1039/C6DT01525E
A Ru-based half sandwich complex used in amine and alcohol racemization reactions was found to be active in the splitting of secondary amines to primary amines using NH3. Conversions up to 80% along with very high selectivities were achieved. However, after about 80% conversion the catalyst lost activity. Similar to Shvo's catalyst, the complex might deactivate under the influence of ammonia. It was revealed that not NH3 but mainly the primary amine is responsible for the deactivation.
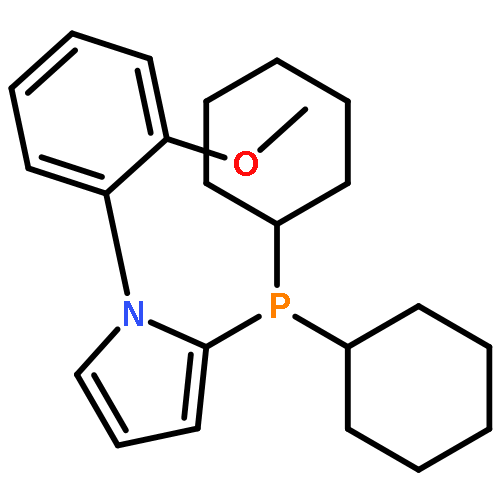
Co-reporter:Anna Falk;Alberto Cavalieri;Gary S. Nichol;Hans-Günther Schmalz
Advanced Synthesis & Catalysis 2015 Volume 357( Issue 14-15) pp:3317-3320
Publication Date(Web):
DOI:10.1002/adsc.201500644
Co-reporter:Evert H. Boymans, P. T. Witte and D. Vogt
Catalysis Science & Technology 2015 vol. 5(Issue 1) pp:176-183
Publication Date(Web):29 Aug 2014
DOI:10.1039/C4CY00790E
A supported Pt nanoparticle-based catalyst was used in the chemoselective hydrogenation of nitroarenes to N-arylhydroxylamines (N-AHA). Optimization of NB hydrogenation conditions showed that substantially higher N-PHA yields can be obtained at low temperature. Especially, the influence of an increased hydrogen pressure on selectivity is remarkable. Maximum yields increase from 55% N-PHA at 4 bar H2 to 80% at 23 bar H2 in ethanol. Further optimization led to the use of small amounts of amine additive, TMEDA, with 50 bar H2 raising the maximum yield to 97% N-PHA. The decreased N-PHA hydrogenation rate at high H2 pressure and the presence of TMEDA allow for selective transformation of a range of other nitroarenes containing electron-withdrawing and -donating (reducible) functional groups to their N-AHAs in excellent (more than 90%) yields.
Co-reporter:Sabriye Güven, Bart Hamers, Robert Franke, Markus Priske, Marc Becker and Dieter Vogt
Catalysis Science & Technology 2014 vol. 4(Issue 2) pp:524-530
Publication Date(Web):13 Dec 2013
DOI:10.1039/C3CY00676J
The kinetics of Rh-catalysed cyclooctene hydroformylation were investigated, based on the mechanism described for a single tris(2,4-di-tert-butylphenyl)phosphite ligand coordinated to a rhodium center. The rate limiting step was found to be the coordination of cyclooctene to the metal center as suggested in literature. Parameters of the corresponding rate equation were estimated by nonlinear regression. Experimental data obtained from semi-batch reactions were compared with model predictions and shown to be in good agreement. A continuous jet-loop reactor with coupled nanofiltration was designed and the kinetics were validated.
Co-reporter:Dennis Pingen and Dieter Vogt
Catalysis Science & Technology 2014 vol. 4(Issue 1) pp:47-52
Publication Date(Web):17 Sep 2013
DOI:10.1039/C3CY00513E
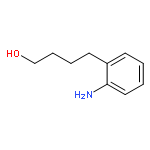
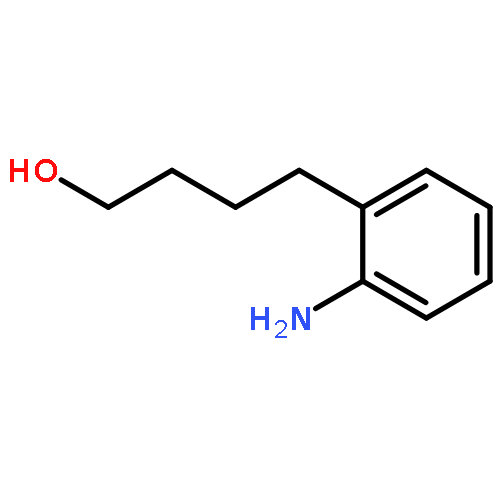
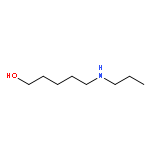
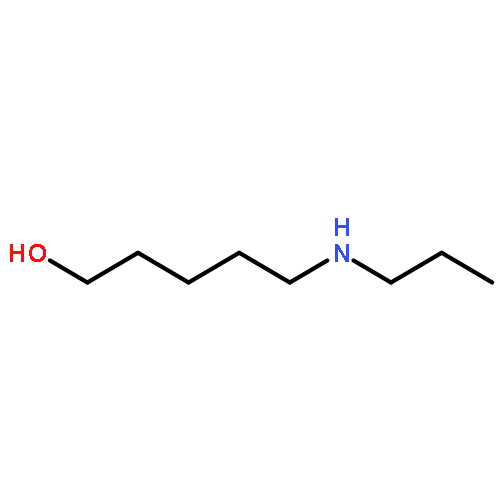
By employing an amination catalyst, previously used in the direct synthesis of amines from alcohol with ammonia, n-amino-alcohols could be selectively cyclized to either the amide or the amine. By the addition of water, the amine could be produced as the major product whereas adding a sacrificial ketone as a hydrogen acceptor resulted in the amide as the major product. Without an additive a mixture of both the amine and the amide was observed. N-substituted amino-alcohols solely gave cyclic amines under these conditions. From 2-(n-alkanol) anilines the cyclic amines were produced, where the n-propanol derivative selectively formed quinoline as the major product.
Co-reporter:Dr. Coen Hendriksen;Dr. Evgeny A. Pidko;Dr. Gang Yang;Dr. Benjamin Schäffner;Dr. Dieter Vogt
Chemistry - A European Journal 2014 Volume 20( Issue 38) pp:12037-12040
Publication Date(Web):
DOI:10.1002/chem.201404082
Abstract
With regard to sustainability, carbon dioxide (CO2) is an attractive C1 building block. However, due to thermodynamic restrictions, reactions incorporating CO2 are relatively limited so far. One of the so-called “dream reactions” in this field is the catalytic oxidative coupling of CO2 and ethene and subsequent β-H elimination to form acrylic acid. This reaction has been studied intensely for decades. However up to this date no suitable catalytic process has been established. Here we show that the catalytic conversion of ethene and CO2 to acrylate is possible in the presence of a homogeneous nickel catalyst in combination with a “hard” Lewis acid. For the first time, catalytic conversion of CO2 and ethene to acrylate with turnover numbers (TON) of up to 21 was demonstrated.
Co-reporter:Dennis Pingen, Martin Lutz, and Dieter Vogt
Organometallics 2014 Volume 33(Issue 7) pp:1623-1629
Publication Date(Web):March 17, 2014
DOI:10.1021/om4011998
The Ru-catalyzed direct amination of alcohols with ammonia was investigated for the RuHCl(CO)(PPh3)3/Xantphos system in order to gain mechanistic insight. For several Ru(II) precursor complexes the influence of different additives on catalytic performance was investigated. NMR studies revealed that the reaction of RuHCl(CO)(PPh3)3/Xantphos with the alcohol in the presence of a strong base initially formed an inactive dihydrido Ru species. However, by addition of a ketone, the dihydride was (re)activated, where the corresponding imine is the actual activator, formed by immediate condensation of the ketone with ammonia. In the absence of a base, added ketone significantly enhanced catalyst activity. Catalytically inactive RuCl2(PPh3)3 could be activated by base, demonstrating that also complexes without the CO ligand give active catalysts. On the basis of these observations a mechanism was proposed, closely related to known transfer hydrogenation mechanisms.
Co-reporter:Dennis Pingen, Tomas Lebl, Martin Lutz, Gary S. Nichol, Paul C. J. Kamer, and Dieter Vogt
Organometallics 2014 Volume 33(Issue 11) pp:2798-2805
Publication Date(Web):May 28, 2014
DOI:10.1021/om5003182
With RuHCl(CO)(PPh3)3 as the starting material, the complexes RuHCl(CO)(PPh3)(L) were prepared for L = Xantphos and closely related ligands. Their catalytic activity in the direct amination of cyclohexanol showed large differences depending on the different backbone structures. In those complexes the Xantphos-type ligand backbones are slightly bent and display fluxionality, studied by VT-NMR. This was assigned to the “flipping” of the backbone via the bridging atoms in the xanthene backbone. Via line shape analysis of the peaks, the Gibbs free energy of activation of the flipping movement was found to be around 56 kJ/mol in all cases. However, the activation enthalpy and entropy differed considerably. Employing RuCl2(PPh3)3 as the precursor resulted in the trans-coordinated complexes RuCl2(PPh3)(L) for L = Xantphos, Sixantphos. Fluxionality was no longer observed, due to the fact that in these complexes the O atom in the backbone also coordinates to the Ru.
Co-reporter:Sabriye Güven;Marko M. L. Nieuwenhuizen;Dr. Bart Hamers;Dr. Robert Franke;Dr. Markus Priske;Dr. Marc Becker;Dr. Dieter Vogt
ChemCatChem 2014 Volume 6( Issue 2) pp:603-610
Publication Date(Web):
DOI:10.1002/cctc.201300818
Abstract
The kinetics of Rh-catalyzed neohexene hydroformylation were investigated with the bulky monodentate ligand tris(2,4-di-tert-butylphenyl)phosphite. The hydrogenolysis of the Rh–acyl intermediate was identified as the rate-limiting step for both the linear and the branched aldehydes. Rate equations for both aldehydes were derived and kinetic parameters were estimated. Increased aldehyde linearity at higher temperatures, frequently observed in hydroformylation, was elucidated by deuterioformylation experiments. These showed that at 100 °C the formation of linear Rh–alkyl was more reversible than the formation of the branched derivative. The ratio of linear to branched Rh–acyl species was determined by in situ high-pressure IR spectroscopy experiments, which allowed the difference in the activation energies for the hydrogenolysis steps towards the aldehyde isomers to be quantified. The hydrogenolysis of Rh–acyl was found to be the step that caused the greatest temperature effect on the regioselectivity.
Co-reporter:Dr. Dennis Pingen;Dr. Olivier Diebolt;Dr. Dieter Vogt
ChemCatChem 2013 Volume 5( Issue 10) pp:2905-2912
Publication Date(Web):
DOI:10.1002/cctc.201300407
Abstract
A slightly adapted catalyst system has been successfully applied in the direct amination of primary and secondary alcohols. Moreover, the applicability to diols has been shown, giving high selectivity towards the primary diamines. It was found that the Ru/P ratio as well as the amount of ammonia used are highly important in this system, especially for higher substrate loadings. The catalyst was employed on a larger batch scale for the conversion of isomannide to the corresponding diamine. Additionally, it was shown that the catalyst is stable for at least six consecutive runs. No significant loss of activity and selectivity was observed.
Co-reporter:Dennis Pingen and Dieter Vogt
Catalysis Science & Technology (2011-Present) 2014 - vol. 4(Issue 1) pp:NaN52-52
Publication Date(Web):2013/09/17
DOI:10.1039/C3CY00513E
By employing an amination catalyst, previously used in the direct synthesis of amines from alcohol with ammonia, n-amino-alcohols could be selectively cyclized to either the amide or the amine. By the addition of water, the amine could be produced as the major product whereas adding a sacrificial ketone as a hydrogen acceptor resulted in the amide as the major product. Without an additive a mixture of both the amine and the amide was observed. N-substituted amino-alcohols solely gave cyclic amines under these conditions. From 2-(n-alkanol) anilines the cyclic amines were produced, where the n-propanol derivative selectively formed quinoline as the major product.
Co-reporter:Sabriye Güven, Bart Hamers, Robert Franke, Markus Priske, Marc Becker and Dieter Vogt
Catalysis Science & Technology (2011-Present) 2014 - vol. 4(Issue 2) pp:NaN530-530
Publication Date(Web):2013/12/13
DOI:10.1039/C3CY00676J
The kinetics of Rh-catalysed cyclooctene hydroformylation were investigated, based on the mechanism described for a single tris(2,4-di-tert-butylphenyl)phosphite ligand coordinated to a rhodium center. The rate limiting step was found to be the coordination of cyclooctene to the metal center as suggested in literature. Parameters of the corresponding rate equation were estimated by nonlinear regression. Experimental data obtained from semi-batch reactions were compared with model predictions and shown to be in good agreement. A continuous jet-loop reactor with coupled nanofiltration was designed and the kinetics were validated.
Co-reporter:Evert H. Boymans, P. T. Witte and D. Vogt
Catalysis Science & Technology (2011-Present) 2015 - vol. 5(Issue 1) pp:NaN183-183
Publication Date(Web):2014/08/29
DOI:10.1039/C4CY00790E
A supported Pt nanoparticle-based catalyst was used in the chemoselective hydrogenation of nitroarenes to N-arylhydroxylamines (N-AHA). Optimization of NB hydrogenation conditions showed that substantially higher N-PHA yields can be obtained at low temperature. Especially, the influence of an increased hydrogen pressure on selectivity is remarkable. Maximum yields increase from 55% N-PHA at 4 bar H2 to 80% at 23 bar H2 in ethanol. Further optimization led to the use of small amounts of amine additive, TMEDA, with 50 bar H2 raising the maximum yield to 97% N-PHA. The decreased N-PHA hydrogenation rate at high H2 pressure and the presence of TMEDA allow for selective transformation of a range of other nitroarenes containing electron-withdrawing and -donating (reducible) functional groups to their N-AHAs in excellent (more than 90%) yields.
Co-reporter:Dennis Pingen, Çiğdem Altıntaş, Max Rudolf Schaller and Dieter Vogt
Dalton Transactions 2016 - vol. 45(Issue 29) pp:NaN11771-11771
Publication Date(Web):2016/06/10
DOI:10.1039/C6DT01525E
A Ru-based half sandwich complex used in amine and alcohol racemization reactions was found to be active in the splitting of secondary amines to primary amines using NH3. Conversions up to 80% along with very high selectivities were achieved. However, after about 80% conversion the catalyst lost activity. Similar to Shvo's catalyst, the complex might deactivate under the influence of ammonia. It was revealed that not NH3 but mainly the primary amine is responsible for the deactivation.



![Phosphine, benzo[kl]xanthene-6,8-diylbis[diphenyl-](http://img.cochemist.com/ccimg/261800/261733-20-4.png)
![Phosphine, benzo[kl]xanthene-6,8-diylbis[diphenyl-](http://img.cochemist.com/ccimg/261800/261733-20-4_b.png)
![1,2,3,4,5,6,8,9,10,11,12,13-dodecahydro-Dicycloocta[b,e]pyrazine](http://img.cochemist.com/ccimg/4500/4427-67-2.png)
![1,2,3,4,5,6,8,9,10,11,12,13-dodecahydro-Dicycloocta[b,e]pyrazine](http://img.cochemist.com/ccimg/4500/4427-67-2_b.png)







![Hexahydro-furo[3,2-b]furan-3,6-diamine](http://img.cochemist.com/ccimg/125400/125335-70-8.png)
![Hexahydro-furo[3,2-b]furan-3,6-diamine](http://img.cochemist.com/ccimg/125400/125335-70-8_b.png)
![Benzo[b]thiophene, 2-ethenyl-](http://img.cochemist.com/ccimg/78700/78646-50-1.png)
![Benzo[b]thiophene, 2-ethenyl-](http://img.cochemist.com/ccimg/78700/78646-50-1_b.png)


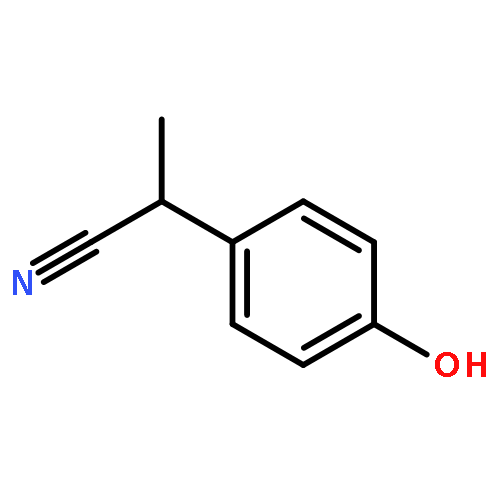
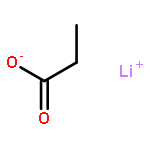
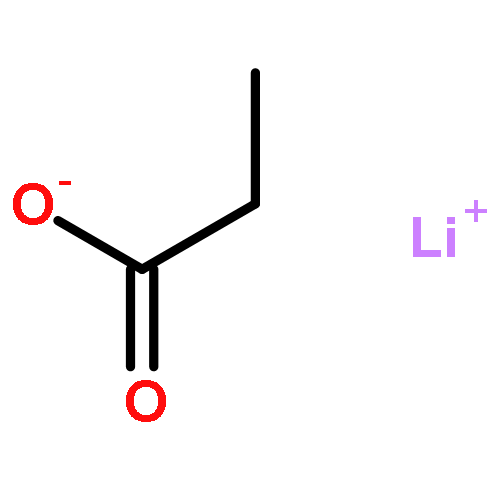
![Bicyclo[2.2.1]heptan-2-amine, 1,3,3-trimethyl-](http://img.cochemist.com/ccimg/21300/21252-46-0.png)
![Bicyclo[2.2.1]heptan-2-amine, 1,3,3-trimethyl-](http://img.cochemist.com/ccimg/21300/21252-46-0_b.png)




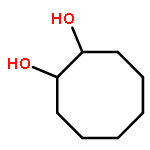



![Bicyclo[2.2.1]heptan-2-amine,1,7,7-trimethyl-, (1R,2S,4R)-rel-](http://img.cochemist.com/ccimg/500/464-42-6.png)
![Bicyclo[2.2.1]heptan-2-amine,1,7,7-trimethyl-, (1R,2S,4R)-rel-](http://img.cochemist.com/ccimg/500/464-42-6_b.png)
















![Bicyclo[3.1.1]hept-3-en-2-ol,4,6,6-trimethyl-](http://img.cochemist.com/ccimg/500/473-67-6.png)
![Bicyclo[3.1.1]hept-3-en-2-ol,4,6,6-trimethyl-](http://img.cochemist.com/ccimg/500/473-67-6_b.png)





