Co-reporter:Dr. Molly M. Lee;Zhe Gao; Dr. Blake R. Peterson
Angewandte Chemie International Edition 2017 Volume 56(Issue 24) pp:6927-6931
Publication Date(Web):2017/06/06
DOI:10.1002/anie.201703298
AbstractThe anticancer drug paclitaxel (Taxol) exhibits paradoxical and poorly understood effects against slow-growing tumors. To investigate its biological activity, fluorophores such as Oregon Green have been linked to this drug. However, this modification increases its polarity by approximately 1000-fold and reduces the toxicity of Taxol towards cancer cell lines by over 200-fold. To construct more drug-like fluorescent probes suitable for imaging by confocal microscopy and analysis by flow cytometry, we synthesized derivatives of Taxol linked to the drug-like fluorophore Pacific Blue (PB). We found that PB-Gly-Taxol bound the target protein β-tubulin with both high affinity in vitro and high specificity in living cells, exhibited substantial cytotoxicity towards HeLa cells, and was a highly sensitive substrate of the multidrug resistance transporter P-glycoprotein (P-gp).
Co-reporter:Dr. Molly M. Lee;Zhe Gao; Dr. Blake R. Peterson
Angewandte Chemie 2017 Volume 129(Issue 24) pp:7031-7035
Publication Date(Web):2017/06/06
DOI:10.1002/ange.201703298
AbstractThe anticancer drug paclitaxel (Taxol) exhibits paradoxical and poorly understood effects against slow-growing tumors. To investigate its biological activity, fluorophores such as Oregon Green have been linked to this drug. However, this modification increases its polarity by approximately 1000-fold and reduces the toxicity of Taxol towards cancer cell lines by over 200-fold. To construct more drug-like fluorescent probes suitable for imaging by confocal microscopy and analysis by flow cytometry, we synthesized derivatives of Taxol linked to the drug-like fluorophore Pacific Blue (PB). We found that PB-Gly-Taxol bound the target protein β-tubulin with both high affinity in vitro and high specificity in living cells, exhibited substantial cytotoxicity towards HeLa cells, and was a highly sensitive substrate of the multidrug resistance transporter P-glycoprotein (P-gp).
Co-reporter:Sahishna Phaniraj, Zhe Gao, Digamber Rane, Blake R. Peterson
Dyes and Pigments 2016 Volume 135() pp:127-133
Publication Date(Web):December 2016
DOI:10.1016/j.dyepig.2016.05.007
•Resorufin was used to synthesize resorufamines as a novel red fluorescent chemotype.•The resorufamine HRA localizes in the endoplasmic reticulum (ER) of mammalian cells.•HRA is a potent, selective, non-toxic, and cost-effective fluorescent probe of the ER.The endoplasmic reticulum (ER) of eukaryotic cells plays critical roles in the processing of secreted and transmembrane proteins. Defects in these functions are associated with a wide range of pathologies. To image this organelle, cells are often treated with fluorescent ER-Tracker dyes. Although these compounds are selective, existing red fluorescent probes of the ER are costly glibenclamide derivatives that inhibit ER-associated sulphonylurea receptors. To provide simpler and more cost-effective red fluorescent probes of the ER, we synthesized amino analogues of the fluorophore resorufin. By varying the polarity of linked substituents, we identified hexyl resorufamine (HRA) as a novel hydrophobic (cLogD (pH 7.4) = 3.8) red fluorescent (Ex. 565 nm; Em. 614 nm in ethanol) molecular probe. HRA is exceptionally bright in organic solvents (quantum yield = 0.70), it exclusively localizes to the ER of living HeLa cells as imaged by confocal microscopy, it is effective at concentrations as low as 100 nM, and it is non-toxic under these conditions. To examine its utility, we used HRA to facilitate visualization of small molecule-mediated release of a GFP-GPI fusion protein from the ER into the secretory pathway. HRA represents a potent, selective, and cost-effective probe for imaging and labeling the ER.
Co-reporter:David Hymel, Sutang Cai, Qi Sun, Rebecca S. Henkhaus, Chamani Perera and Blake R. Peterson
Chemical Communications 2015 vol. 51(Issue 78) pp:14624-14627
Publication Date(Web):13 Aug 2015
DOI:10.1039/C5CC06325F
Mammalian cells acquire cholesterol, a critical membrane constituent, through multiple mechanisms. We synthesized mimics of cholesterol, fluorescent N-alkyl-3β-cholesterylamine-glutamic acids, that are rapidly incorporated into cellular plasma membranes compared with analogous cholesteryl amides, ethers, esters, carbamates, and a sitosterol analogue. This process was inhibited by ezetimibe, indicating a receptor-mediated uptake pathway.
Co-reporter:J. Matthew Meinig and Blake R. Peterson
ACS Chemical Biology 2015 Volume 10(Issue 2) pp:570
Publication Date(Web):November 21, 2014
DOI:10.1021/cb500856c
Inhibitors of the PI3-kinase/Akt (protein kinase B) pathway are under investigation as anticancer and antiviral agents. Akt inhibitor-IV (ChemBridge 5233705, CAS 681281-88-9, AKTIV), a small molecule reported to inhibit this pathway, exhibits potent anticancer and broad-spectrum antiviral activity. However, depending on concentration, this cationic benzimidazole derivative exhibits paradoxical positive or negative effects on the phosphorylation of Akt that are not well understood. To elucidate its mechanism of action, we investigated its spectroscopic properties. This compound proved to be sufficiently fluorescent (excitation λmax = 388 nm, emission λmax = 460 nm) to enable examination of its uptake and distribution in living mammalian cells. Despite a low quantum yield of 0.0016, imaging of HeLa cells treated with AKTIV (1 μM, 5 min) by confocal laser scanning microscopy, with excitation at 405 nm, revealed extensive accumulation in mitochondria. Treatment of Jurkat lymphocytes with 1 μM AKTIV for 15 min caused accumulation to over 250 μM in these organelles, whereas treatment with 5 μM AKTIV yielded concentrations of over 1 mM in mitochondria, as analyzed by flow cytometry. This massive loading resulted in swelling of these organelles, followed by their apparent disintegration. These effects were associated with profound disruption of cellular bioenergetics including mitochondrial depolarization, diminished mitochondrial respiration, and release of reactive oxygen species. Because mitochondria play key roles in both cancer proliferation and viral replication, we conclude that the anticancer and antiviral activities of AKTIV predominantly result from its direct and immediate effects on the structure and function of mitochondria.
Co-reporter:J. Matthew Meinig;Dr. Liqiang Fu;Dr. Blake R. Peterson
Angewandte Chemie International Edition 2015 Volume 54( Issue 33) pp:9696-9699
Publication Date(Web):
DOI:10.1002/anie.201504156
Abstract
The endoplasmic reticulum (ER) plays critical roles in the processing of secreted and transmembrane proteins. To deliver small molecules to this organelle, we synthesized fluorinated hydrophobic analogues of the fluorophore rhodol. These cell-permeable fluorophores are exceptionally bright, with quantum yields of around 0.8, and they were found to specifically accumulate in the ER of living HeLa cells, as imaged by confocal laser scanning microscopy. To target a biological pathway controlled by the ER, we linked a fluorinated hydrophobic rhodol to 5-nitrofuran-2-acrylaldehyde. In contrast to an untargeted nitrofuran warhead, delivery of this electrophilic nitrofuran to the ER by the rhodol resulted in cytotoxicity comparable to the ER-targeted cytotoxin eeyarestatin I, and specifically inhibited protein processing by the ubiquitin–proteasome system. Fluorinated hydrophobic rhodols are outstanding fluorophores that enable the delivery of small molecules for targeting ER-associated proteins and pathways.
Co-reporter:J. Matthew Meinig;Dr. Liqiang Fu;Dr. Blake R. Peterson
Angewandte Chemie 2015 Volume 127( Issue 33) pp:9832-9835
Publication Date(Web):
DOI:10.1002/ange.201504156
Abstract
The endoplasmic reticulum (ER) plays critical roles in the processing of secreted and transmembrane proteins. To deliver small molecules to this organelle, we synthesized fluorinated hydrophobic analogues of the fluorophore rhodol. These cell-permeable fluorophores are exceptionally bright, with quantum yields of around 0.8, and they were found to specifically accumulate in the ER of living HeLa cells, as imaged by confocal laser scanning microscopy. To target a biological pathway controlled by the ER, we linked a fluorinated hydrophobic rhodol to 5-nitrofuran-2-acrylaldehyde. In contrast to an untargeted nitrofuran warhead, delivery of this electrophilic nitrofuran to the ER by the rhodol resulted in cytotoxicity comparable to the ER-targeted cytotoxin eeyarestatin I, and specifically inhibited protein processing by the ubiquitin–proteasome system. Fluorinated hydrophobic rhodols are outstanding fluorophores that enable the delivery of small molecules for targeting ER-associated proteins and pathways.
Co-reporter:David Hymel ; Zachary R. Woydziak
Journal of the American Chemical Society 2014 Volume 136(Issue 14) pp:5241-5244
Publication Date(Web):March 24, 2014
DOI:10.1021/ja501253b
Critical protein–protein interactions are ubiquitous in biology. To provide a new method to detect these interactions, we designed and synthesized fluorinated bromopyronins as molecular probes. These electrophilic compounds rapidly react with amines via a SNAr mechanism to form modestly electrophilic aminopyronin fluorophores. To investigate whether proteins modified with aminopyronins might selectively transfer these fluorophores between proximal lysine residues at protein–protein interfaces, immunoglobulin-G (IgG) was conjugated to fluorinated pyronins and added to unlabeled Protein A (SpA) from S. aureus. Analysis by gel electrophoresis and mass spectrometry revealed transfer of this fluorophore from IgG to specific lysines of its binding partner SpA but not to bovine serum albumin (BSA) as a nonbinding control. Examination of an X-ray structure of IgG bound to SpA revealed that the fluorophore was selectively transferred between amino groups of lysines that reside within ∼10 Å at the protein–protein interface. To evaluate whether this approach could be used to identify interactions with endogenous cellular proteins, pyronin-modified Rnase A was added to crude extracts of human HeLa cells. Analysis of interacting proteins by gel electrophoresis revealed the endogenous ribonuclease inhibitor as the primary cellular target. Given that proximal lysine residues frequently reside at protein–protein interfaces, this method may facilitate identification of diverse protein–protein interactions present in complex biological matrices.
Co-reporter:Aaron Bender, Zachary R. Woydziak, Liqiang Fu, Michael Branden, Zhenguo Zhou, Brian D. Ackley, and Blake R. Peterson
ACS Chemical Biology 2013 Volume 8(Issue 3) pp:636
Publication Date(Web):December 21, 2012
DOI:10.1021/cb300396j
Unlike the digestive systems of vertebrate animals, the lumen of the alimentary canal of Caenorhabditis elegans is unsegmented and weakly acidic (pH ∼4.4), with ultradian fluctuations to pH > 6 every 45–50 s. To probe the dynamics of this acidity, we synthesized novel acid-activated fluorophores termed Kansas Reds. These dicationic derivatives of rhodamine B become concentrated in the lumen of the intestine of living C. elegans and exhibit tunable pKa values (2.3–5.4), controlled by the extent of fluorination of an alkylamine substituent, that allow imaging of a range of acidic fluids in vivo. Fluorescence video microscopy of animals freely feeding on these fluorophores revealed that acidity in the C. elegans intestine is discontinuous; the posterior intestine contains a large acidic segment flanked by a smaller region of higher pH at the posterior-most end. Remarkably, during the defecation motor program, this hot spot of acidity rapidly moves from the posterior intestine to the anterior-most intestine where it becomes localized for up to 7 s every 45–50 s. Studies of pH-insensitive and base-activated fluorophores as well as mutant and transgenic animals revealed that this dynamic wave of acidity requires the proton exchanger PBO-4, does not involve substantial movement of fluid, and likely involves the sequential activation of proton transporters on the apical surface of intestinal cells. Lacking a specific organ that sequesters low pH, C. elegans compartmentalizes acidity by producing of a dynamic hot spot of protons that rhythmically migrates from the posterior to anterior intestine.
Co-reporter:Zachary R. Woydziak, Liqiang Fu, and Blake R. Peterson
The Journal of Organic Chemistry 2012 Volume 77(Issue 1) pp:473-481
Publication Date(Web):November 23, 2011
DOI:10.1021/jo202062f
Fluorination of fluorophores can substantially enhance their photostability and improve spectroscopic properties. To facilitate access to fluorinated fluorophores, bis(2,4,5-trifluorophenyl)methanone was synthesized by treatment of 2,4,5-trifluorobenzaldehyde with a Grignard reagent derived from 1-bromo-2,4,5-trifluorobenzene, followed by oxidation of the resulting benzyl alcohol. This hexafluorobenzophenone was subjected to sequential nucleophilic aromatic substitution reactions, first at one or both of the more reactive 4,4′-fluorines, and second by cyclization through substitution of the less reactive 2,2′-fluorines, using a variety of oxygen, nitrogen, and sulfur nucleophiles, including hydroxide, methoxide, amines, and sulfide. This method yields symmetrical and asymmetrical fluorinated benzophenones, xanthones, acridones, and thioxanthones and provides scalable access to known and novel precursors to fluorinated analogues of fluorescein, rhodamine, and other derivatives. Spectroscopic studies revealed that several of these precursors are highly fluorescent, with tunable absorption and emission spectra, depending on the substituents. This approach should allow access to a wide variety of novel fluorinated fluorophores and related compounds.
Co-reporter:Qi Sun ; Runzhi Wu ; Sutang Cai ; Yuan Lin ; Llewlyn Sellers ; Kaori Sakamoto ; Biao He
Journal of Medicinal Chemistry 2011 Volume 54(Issue 5) pp:1126-1139
Publication Date(Web):February 14, 2011
DOI:10.1021/jm100912b
Inhibitors of the PI3-kinase/AKT (protein kinase B) pathway are under investigation as anticancer and antiviral agents. The benzimidazole derivative AKT inhibitor-IV (ChemBridge 5233705) affects this pathway and exhibits potent anticancer and antiviral activity. To probe its biological activity, we synthesized AKT inhibitor-IV and 21 analogues using a novel six-step route based on ZrCl4-catalyzed cyclization of 1,2-arylenediamines with α,β-unsaturated aldehydes. We examined effects on viability of HeLa carcinoma cells, viability of normal human cells (NHBE), replication of recombinant parainfluenza virus 5 (PIV5) in HeLa cells, and replication of the intracellular bacterium Mycobacterium fortuitum in HeLa cells. Replacement of the benzimidazole N-ethyl substitutent of AKT inhibitor-IV with N-hexyl and N-dodecyl groups enhanced antiviral activity and cytotoxicity against the cancer cell line, but these compounds showed substantially lower toxicity (from 6-fold to >20-fold) against NHBE cells and no effect on M. fortuitum, suggesting inhibition of one or more host protein(s) required for proliferation of cancer cells and PIV5. The key structural elements identified here may facilitate identification of targets of this highly biologically active scaffold.
Co-reporter:Runzhi Wu ; Eric D. Smidansky ; Hyung Suk Oh ; Ratree Takhampunya ; Radhakrishnan Padmanabhan ; Craig E. Cameron
Journal of Medicinal Chemistry 2010 Volume 53(Issue 22) pp:7958-7966
Publication Date(Web):October 22, 2010
DOI:10.1021/jm100593s
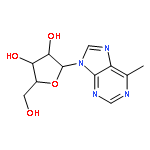
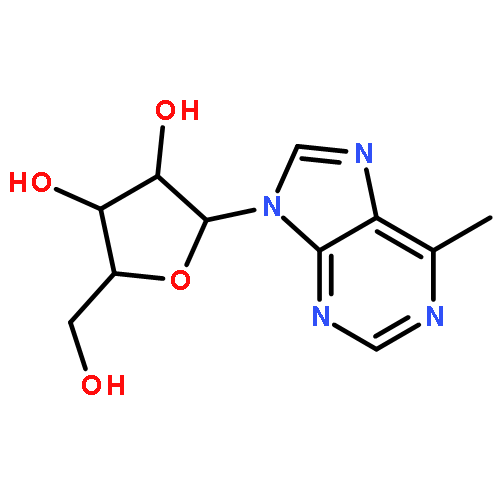
Bioisosteric deaza analogues of 6-methyl-9-β-d-ribofuranosylpurine, a hydrophobic analogue of adenosine, were synthesized and evaluated for antiviral activity. Whereas the 1-deaza and 3-deaza analogues were essentially inactive in plaque assays of infectivity, a novel 7-deaza-6-methyl-9-β-d-ribofuranosylpurine analogue, structurally related to the natural product tubercidin, potently inhibited replication of poliovirus (PV) in HeLa cells (IC50 = 11 nM) and dengue virus (DENV) in Vero cells (IC50 = 62 nM). Selectivity against PV over cytotoxic effects to HeLa cells was >100-fold after incubation for 7 h. Mechanistic studies of the 5′-triphosphate of 7-deaza-6-methyl-9-β-d-ribofuranosylpurine revealed that this compound is an efficient substrate of PV RNA-dependent RNA polymerase (RdRP) and is incorporated into RNA mimicking both ATP and GTP.
Co-reporter:David Hymel, Blake R. Peterson
Advanced Drug Delivery Reviews (15 June 2012) Volume 64(Issue 9) pp:797-810
Publication Date(Web):15 June 2012
DOI:10.1016/j.addr.2012.02.007
Receptor-mediated endocytosis is a highly efficient mechanism for cellular uptake of membrane-impermeant ligands. Cells use this process to acquire nutrients, initiate signal transduction, promote development, regulate neurotransmission, and maintain homeostasis. Natural receptors that participate in receptor-mediated endocytosis are structurally diverse, ranging from large transmembrane proteins to small glycolipids embedded in the outer leaflet of cellular plasma membranes. Despite their vast structural differences, these receptors share common features of binding to extracellular ligands, clustering in dynamic membrane regions that pinch off to yield intracellular vesicles, and accumulation of receptor-ligand complexes in membrane-sealed endosomes. Receptors typically dissociate from ligands in endosomes and cycle back to the cell surface, whereas internalized ligands are usually delivered into lysosomes, where they are degraded, but some can escape and penetrate into the cytosol. Here, we review efforts to develop synthetic cell surface receptors, defined as nonnatural compounds, exemplified by mimics of cholesterol, that insert into plasma membranes, bind extracellular ligands including therapeutics, probes, and endogenous proteins, and engage endocytic membrane trafficking pathways. By mimicking natural mechanisms of receptor-mediated endocytosis, synthetic cell surface receptors have the potential to function as prosthetic molecules capable of seamlessly augmenting the endocytic uptake machinery of living mammalian cells.Download high-res image (358KB)Download full-size image
Co-reporter:David Hymel, Sutang Cai, Qi Sun, Rebecca S. Henkhaus, Chamani Perera and Blake R. Peterson
Chemical Communications 2015 - vol. 51(Issue 78) pp:NaN14627-14627
Publication Date(Web):2015/08/13
DOI:10.1039/C5CC06325F
Mammalian cells acquire cholesterol, a critical membrane constituent, through multiple mechanisms. We synthesized mimics of cholesterol, fluorescent N-alkyl-3β-cholesterylamine-glutamic acids, that are rapidly incorporated into cellular plasma membranes compared with analogous cholesteryl amides, ethers, esters, carbamates, and a sitosterol analogue. This process was inhibited by ezetimibe, indicating a receptor-mediated uptake pathway.
.jpg)
![1H-Thieno[3,4-d]imidazole-6-pentanoicacid, 2-amino-3a,4,6,6a-tetrahydro-, (3aR,6S,6aS)-](http://img.cochemist.com/ccimg/13400/13395-35-2.png)
![1H-Thieno[3,4-d]imidazole-6-pentanoicacid, 2-amino-3a,4,6,6a-tetrahydro-, (3aR,6S,6aS)-](http://img.cochemist.com/ccimg/13400/13395-35-2_b.png)


















![9H-XANTHEN-9-ONE, 2,7-DIFLUORO-3,6-BIS[(2-METHOXYETHOXY)METHOXY]-](http://img.cochemist.com/ccimg/430500/430459-53-3.png)
![9H-XANTHEN-9-ONE, 2,7-DIFLUORO-3,6-BIS[(2-METHOXYETHOXY)METHOXY]-](http://img.cochemist.com/ccimg/430500/430459-53-3_b.png)













![6-Bromo-7-methyl-1H-imidazo[4,5-b]pyridine](http://img.cochemist.com/ccimg/92000/91996-63-3.png)
![6-Bromo-7-methyl-1H-imidazo[4,5-b]pyridine](http://img.cochemist.com/ccimg/92000/91996-63-3_b.png)
![3-[3BETA-CHOLEST-5-EN-3-YL)OXY]-1-PROPANAMINE](http://img.cochemist.com/ccimg/71600/71507-49-8.png)
![3-[3BETA-CHOLEST-5-EN-3-YL)OXY]-1-PROPANAMINE](http://img.cochemist.com/ccimg/71600/71507-49-8_b.png)



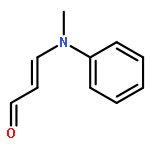



![7-Methyl-3H-imidazo[4,5-b]pyridine](http://img.cochemist.com/ccimg/27600/27582-20-3.png)
![7-Methyl-3H-imidazo[4,5-b]pyridine](http://img.cochemist.com/ccimg/27600/27582-20-3_b.png)
![7H-Pyrrolo[2,3-d]pyrimidine,4-chloro-7-b-D-ribofuranosyl-](http://img.cochemist.com/ccimg/16800/16754-80-6.png)
![7H-Pyrrolo[2,3-d]pyrimidine,4-chloro-7-b-D-ribofuranosyl-](http://img.cochemist.com/ccimg/16800/16754-80-6_b.png)
![(2R,3R,4R,5R)-2-(Acetoxymethyl)-5-(4-chloro-7H-pyrrolo[2,3-d]pyrimidin-7-yl)tetrahydrofuran-3,4-diyl diacetate](http://img.cochemist.com/ccimg/16800/16754-79-3.png)
![(2R,3R,4R,5R)-2-(Acetoxymethyl)-5-(4-chloro-7H-pyrrolo[2,3-d]pyrimidin-7-yl)tetrahydrofuran-3,4-diyl diacetate](http://img.cochemist.com/ccimg/16800/16754-79-3_b.png)




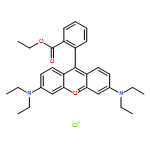
![Spiro[isobenzofuran-1(3H),9'-[9H]xanthen]-3-one,3'-amino-6'-hydroxy-](http://img.cochemist.com/ccimg/3100/3086-44-0.png)
![Spiro[isobenzofuran-1(3H),9'-[9H]xanthen]-3-one,3'-amino-6'-hydroxy-](http://img.cochemist.com/ccimg/3100/3086-44-0_b.png)


![[(3-chlorophenyl)hydrazono]malononitrile](http://img.cochemist.com/ccimg/600/555-60-2.png)
![[(3-chlorophenyl)hydrazono]malononitrile](http://img.cochemist.com/ccimg/600/555-60-2_b.png)
![7H-Pyrrolo[2,3-d]pyrimidin-4-amine,7-b-D-ribofuranosyl-](http://img.cochemist.com/ccimg/100/69-33-0.png)
![7H-Pyrrolo[2,3-d]pyrimidin-4-amine,7-b-D-ribofuranosyl-](http://img.cochemist.com/ccimg/100/69-33-0_b.png)


![3,6-diamino-9-[2-(methoxycarbonyl)phenyl]xanthylium chloride](/data/chemimg/622800/62669-70-9.png)
![3,6-diamino-9-[2-(methoxycarbonyl)phenyl]xanthylium chloride](/data/chemimg/622800/62669-70-9_b.png)