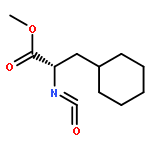
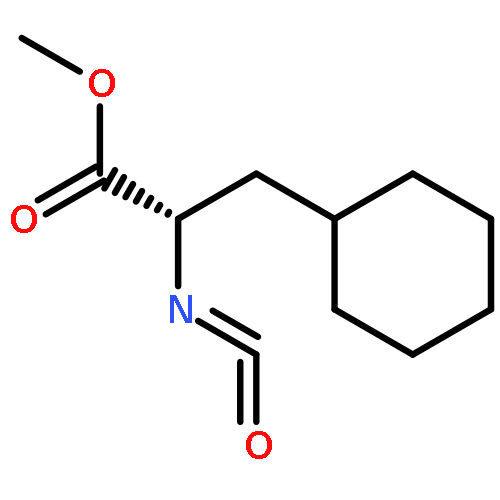
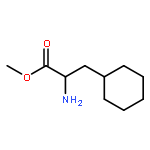
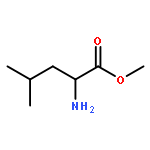
Co-reporter:Anushka C. Galasiti Kankanamalage, Yunjeong Kim, Athri D. Rathnayake, Kevin R. Alliston, Michelle M. Butler, Steven C. Cardinale, Terry L. Bowlin, William C. Groutas, and Kyeong-Ok Chang
Journal of Medicinal Chemistry July 27, 2017 Volume 60(Issue 14) pp:6239-6239
Publication Date(Web):July 3, 2017
DOI:10.1021/acs.jmedchem.7b00497
Ester and carbamate prodrugs of aldehyde bisulfite adduct inhibitors were synthesized in order to improve their pharmacokinetic and pharmacodynamic properties. The inhibitory activity of the compounds against norovirus 3C-like protease in enzyme and cell-based assays was determined. The ester and carbamate prodrugs displayed equivalent potency to those of the precursor aldehyde bisulfite adducts and precursor aldehydes. Furthermore, the rate of ester cleavage was found to be dependent on alkyl chain length. The generated prodrugs exhibited low cytotoxicity and satisfactory liver microsomes stability and plasma protein binding. The methodology described herein has wide applicability and can be extended to the bisulfite adducts of common warheads employed in the design of transition state inhibitors of serine and cysteine proteases of medical relevance.
Co-reporter:Anushka C. Galasiti Kankanamalage, Yunjeong Kim, Athri D. Rathnayake, Vishnu C. Damalanka, Pathum M. Weerawarna, Sean T. Doyle, Amer F. Alsoudi, D.M. Padmasankha Dissanayake, Gerald H. Lushington, Nurjahan Mehzabeen, Kevin P. Battaile, Scott Lovell, Kyeong-Ok Chang, William C. Groutas
European Journal of Medicinal Chemistry 2017 Volume 126(Volume 126) pp:
Publication Date(Web):27 January 2017
DOI:10.1016/j.ejmech.2016.11.027
•The structure-guided design of norovirus 3CL protease inhibitors was performed.•Potency was modulated by exploiting interactions with the S4 subsite of the enzyme.•X-ray crystal structures of enzyme-inhibitor complexes confirmed the mechanism.•The inhibitor design rationale was validated by x-ray crystallographic studies.Human noroviruses are the primary cause of epidemic and sporadic acute gastroenteritis. The worldwide high morbidity and mortality associated with norovirus infections, particularly among the elderly, immunocompromised patients and children, constitute a serious public health concern. There are currently no approved human vaccines or norovirus-specific small-molecule therapeutics or prophylactics. Norovirus 3CL protease has recently emerged as a potential therapeutic target for the development of anti-norovirus agents. We hypothesized that the S4 subsite of the enzyme may provide an effective means of designing potent and cell permeable inhibitors of the enzyme. We report herein the structure-guided exploration and exploitation of the S4 subsite of norovirus 3CL protease in the design and synthesis of effective inhibitors of the protease.Download high-res image (267KB)Download full-size image
Co-reporter:Vishnu C. Damalanka, Yunjeong Kim, Anushka C. Galasiti Kankanamalage, Gerald H. Lushington, Nurjahan Mehzabeen, Kevin P. Battaile, Scott Lovell, Kyeong-Ok Chang, William C. Groutas
European Journal of Medicinal Chemistry 2017 Volume 127(Volume 127) pp:
Publication Date(Web):15 February 2017
DOI:10.1016/j.ejmech.2016.12.033
•A novel series of macrocyclic inhibitors of norovirus 3CL protease inhibitors is reported.•High resolution crystal structures of enzyme-inhibitor complexes have confirmed the mechanism of action.•The structural determinants involved in binding have been delineated.•The inhibitor design rationale was validated by x-ray crystallographic studies.Norovirus infections have a major impact on public health worldwide, yet there is a current dearth of norovirus-specific therapeutics and prophylactics. This report describes the discovery of a novel class of macrocyclic inhibitors of norovirus 3C-like protease, a cysteine protease that is essential for virus replication. SAR, structural, and biochemical studies were carried out to ascertain the effect of structure on pharmacological activity and permeability. Insights gained from these studies have laid a solid foundation for capitalizing on the therapeutic potential of the series of inhibitors described herein.Download high-res image (211KB)Download full-size image
Co-reporter:Vishnu C. Damalanka; Yunjeong Kim; Kevin R. Alliston; Pathum M. Weerawarna; Anushka C. Galasiti Kankanamalage; Gerald H. Lushington; Nurjahan Mehzabeen; Kevin P. Battaile; Scott Lovell; Kyeong-Ok Chang;William C. Groutas
Journal of Medicinal Chemistry 2016 Volume 59(Issue 5) pp:1899-1913
Publication Date(Web):January 29, 2016
DOI:10.1021/acs.jmedchem.5b01464
Human noroviruses are the primary causative agents of acute gastroenteritis and a pressing public health burden worldwide. There are currently no vaccines or small molecule therapeutics available for the treatment or prophylaxis of norovirus infections. Norovirus 3CL protease plays a vital role in viral replication by generating structural and nonstructural proteins via the cleavage of the viral polyprotein. Thus, molecules that inhibit the viral protease may have potential therapeutic value. We describe herein the structure-based design, synthesis, and in vitro and cell-based evaluation of the first class of oxadiazole-based, permeable macrocyclic inhibitors of norovirus 3CL protease.
Co-reporter:Pathum M. Weerawarna, Yunjeong Kim, Anushka C. Galasiti Kankanamalage, Vishnu C. Damalanka, Gerald H. Lushington, Kevin R. Alliston, Nurjahan Mehzabeen, Kevin P. Battaile, Scott Lovell, Kyeong-Ok Chang, William C. Groutas
European Journal of Medicinal Chemistry 2016 Volume 119() pp:300-318
Publication Date(Web):25 August 2016
DOI:10.1016/j.ejmech.2016.04.013
•Novel triazole-based macrocyclic inhibitors of norovirus 3CL protease were synthesized.•The interplay of conformation and activity was probed using NMR and X-ray crystallography.•Bound inhibitors assume a β-strand conformation according to X-ray crystal structure.•Loss of critical hydrogen bonding interactions was revealed by X-ray crystallography.Outbreaks of acute gastroenteritis caused by noroviruses constitute a public health concern worldwide. To date, there are no approved drugs or vaccines for the management and prophylaxis of norovirus infections. A potentially effective strategy for the development of norovirus therapeutics entails the discovery of inhibitors of norovirus 3CL protease, an enzyme essential for noroviral replication. We describe herein the structure-based design of the first class of permeable, triazole-based macrocyclic inhibitors of norovirus 3C-like protease, as well as pertinent X-ray crystallographic, biochemical, spectroscopic, and antiviral studies.Figure optionsDownload full-size imageDownload as PowerPoint slide
Co-reporter:Anushka C. Galasiti Kankanamalage; Yunjeong Kim; Pathum M. Weerawarna; Roxanne Adeline Z. Uy; Vishnu C. Damalanka; Sivakoteswara Rao Mandadapu; Kevin R. Alliston; Nurjahan Mehzabeen; Kevin P. Battaile; Scott Lovell; Kyeong-Ok Chang;William C. Groutas
Journal of Medicinal Chemistry 2015 Volume 58(Issue 7) pp:3144-3155
Publication Date(Web):March 12, 2015
DOI:10.1021/jm5019934
Norovirus infection constitutes the primary cause of acute viral gastroenteritis. There are currently no vaccines or norovirus-specific antiviral therapeutics available for the management of norovirus infection. Norovirus 3C-like protease is essential for viral replication, consequently, inhibition of this enzyme is a fruitful avenue of investigation that may lead to the emergence of antinorovirus therapeutics. We describe herein the optimization of dipeptidyl inhibitors of norovirus 3C-like protease using iterative SAR, X-ray crystallographic, and enzyme and cell-based studies. We also demonstrate herein in vivo efficacy of an inhibitor using the murine model of norovirus infection.
Co-reporter:Yunjeong Kim; Anushka C. Galasiti Kankanamalage; Kyeong-Ok Chang;William C. Groutas
Journal of Medicinal Chemistry 2015 Volume 58(Issue 24) pp:9438-9450
Publication Date(Web):August 10, 2015
DOI:10.1021/acs.jmedchem.5b00762
Noroviruses are members of the family Caliciviridae. Norovirus infections are a global health burden that impacts >20 million individuals annually in the U.S. alone. Noroviruses are associated with high morbidity among vulnerable populations, particularly immunocompromised patients. This perspective highlights recent developments related to the discovery and development of norovirus-specific small-molecule therapeutics as well as recent advances in our understanding of norovirus biology and pathogenesis. Most of the work in this area is at the early discovery stage and has been primarily focused on inhibitors of norovirus 3C-like protease and RNA dependent RNA polymerase. However, recent discoveries emanating from basic studies in norovirus research have resulted in the identification of new host-related drug targets that can be exploited. A repurposed compound has been advanced to human clinical studies.
Co-reporter:Huiguo Lai, Dengfeng Dou, Sridhar Aravapalli, Tadahisa Teramoto, Gerald H. Lushington, Tom M. Mwania, Kevin R. Alliston, David M. Eichhorn, Radhakrishnan Padmanabhan, William C. Groutas
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 1) pp:102-113
Publication Date(Web):1 January 2013
DOI:10.1016/j.bmc.2012.10.058
1,2-Benzisothiazol-3(2H)-ones and 1,3,4-oxadiazoles individually have recently attracted considerable interest in drug discovery, including as antibacterial and antifungal agents. In this study, a series of functionalized 1,2-benzisothiazol-3(2H)-one—1,3,4-oxadiazole hybrid derivatives were synthesized and subsequently screened against Dengue and West Nile virus proteases. Ten out of twenty-four compounds showed greater than 50% inhibition against DENV2 and WNV proteases ([I] = 10 μM). The IC50 values of compound 7n against DENV2 and WNV NS2B/NS3 were found to be 3.75 ± 0.06 and 4.22 ± 0.07 μM, respectively. The kinetics data support a competitive mode of inhibition by compound 7n. Molecular modeling studies were performed to delineate the putative binding mode of this series of compounds. This study reveals that the hybrid series arising from the linking of the two scaffolds provides a suitable platform for conducting a hit-to-lead optimization campaign via iterative structure–activity relationship studies, in vitro screening and X-ray crystallography.A series of functionalized 1,2-benzisothiazol-3(2H)-one—1,3,4-oxadiazole hybrid derivatives (I) were synthesized and shown to inhibit Dengue and West Nile virus proteases. The IC50 values of compound 7n against DENV2 and WNV proteases were determined to be 3.75 ± 0.06 and 4.22 ± 0.07 μM, respectively. The kinetics data support a competitive mode of inhibition by compound 7n. The results of docking studies corroborate some general activity trends observed in the tested compounds.
Co-reporter:Sivakoteswara Rao Mandadapu, Mallikarjuna Reddy Gunnam, Anushka C. Galasiti Kankanamalage, Roxanne Adeline Z. Uy, Kevin R. Alliston, Gerald H. Lushington, Yunjeong Kim, Kyeong-Ok Chang, William C. Groutas
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 21) pp:5941-5944
Publication Date(Web):1 November 2013
DOI:10.1016/j.bmcl.2013.08.073
The design, synthesis, and evaluation of a series of dipeptidyl α-hydroxyphosphonates is reported. The synthesized compounds displayed high anti-norovirus activity in a cell-based replicon system, as well as high enzyme selectivity.The design, synthesis, and evaluation of the anti-norovirus activity of a series of dipeptidyl α-hydroxyphosphonates is reported. The synthesized compounds displayed high anti-norovirus activity in a cell-based replicon system.
Co-reporter:Sivakoteswara Rao Mandadapu, Pathum M. Weerawarna, Allan M. Prior, Roxanne Adeline Z. Uy, Sridhar Aravapalli, Kevin R. Alliston, Gerald H. Lushington, Yunjeong Kim, Duy H. Hua, Kyeong-Ok Chang, William C. Groutas
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 13) pp:3709-3712
Publication Date(Web):1 July 2013
DOI:10.1016/j.bmcl.2013.05.021
The design, synthesis, and in vitro evaluation of the first macrocyclic inhibitor of 3C and 3C-like proteases of picornavirus, norovirus, and coronavirus are reported. The in vitro inhibitory activity (50% effective concentration) of the macrocyclic inhibitor toward enterovirus 3C protease (CVB3 Nancy strain), and coronavirus (SARS-CoV) and norovirus 3C-like proteases, was determined to be 1.8, 15.5 and 5.1 μM, respectively.The design, synthesis, and in vitro inhibitory activity of the first series of macrocyclic inhibitors of 3C and 3C-like proteases of picornavirus, norovirus, and coronavirus, are reported.
Co-reporter:Sivakoteswara Rao Mandadapu, Mallikarjuna Reddy Gunnam, Kok-Chuan Tiew, Roxanne Adeline Z. Uy, Allan M. Prior, Kevin R. Alliston, Duy H. Hua, Yunjeong Kim, Kyeong-Ok Chang, William C. Groutas
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 1) pp:62-65
Publication Date(Web):1 January 2013
DOI:10.1016/j.bmcl.2012.11.026
Noroviruses are the most common cause of acute viral gastroenteritis, accounting for >21 million cases annually in the US alone. Norovirus infections constitute an important health problem for which there are no specific antiviral therapeutics or vaccines. In this study, a series of bisulfite adducts derived from representative transition state inhibitors (dipeptidyl aldehydes and α-ketoamides) was synthesized and shown to exhibit anti-norovirus activity in a cell-based replicon system. The ED50 of the most effective inhibitor was 60 nM. This study demonstrates for the first time the utilization of bisulfite adducts of transition state inhibitors in the inhibition of norovirus 3C-like protease in vitro and in a cell-based replicon system. The approach described herein can be extended to the synthesis of the bisulfite adducts of other classes of transition state inhibitors of serine and cysteine proteases, such as α-ketoheterocycles and α-ketoesters.A series of bisulfite adducts derived from representative transition state inhibitors (dipeptidyl aldehydes and α-ketoamides) was synthesized and shown to exhibit inhibitory activity against Norwalk virus 3C protease in vitro, as well as anti-norovirus activity in a cell-based replicon system.
Co-reporter:Sridhar Aravapalli, Huiguo Lai, Tadahisa Teramoto, Kevin R. Alliston, Gerald H. Lushington, Eron L. Ferguson, R. Padmanabhan, William C. Groutas
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 13) pp:4140-4148
Publication Date(Web):1 July 2012
DOI:10.1016/j.bmc.2012.04.055
Dengue and West Nile viruses (WNV) are mosquito-borne members of flaviviruses that cause significant morbidity and mortality. There is no approved vaccine or antiviral drugs for human use to date. In this study, a series of functionalized meta and para aminobenzamide derivatives were synthesized and subsequently screened in vitro against Dengue virus and West Nile virus proteases. Four active compounds were identified which showed comparable activity toward the two proteases and shared in common a meta or para(phenoxy)phenyl group. The inhibition constants (Ki) for the most potent compound 7n against Dengue and West Nile virus proteases were 8.77 and 5.55 μM, respectively. The kinetics data support a competitive mode of inhibition of both proteases by compound 7n. This conclusion is further supported by molecular modeling. This study reveals a new chemical scaffold which is amenable to further optimization to yield potent inhibitors of the viral proteases via the combined utilization of iterative medicinal chemistry/structure–activity relationship studies and in vitro screening.A series of functionalized meta and para aminobenzamide derivatives (I) was synthesized and subsequently screened in vitro against Dengue virus and West Nile virus proteases. Four active compounds were identified which showed comparable activity toward the two proteases and shared in common a meta or para(phenoxy)phenyl group. The inhibition constants for the most potent compound 7n against Dengue and West Nile virus proteases were 8.77 and 5.55 μM, respectively.
Co-reporter:Kok-Chuan Tiew, Dengfeng Dou, Tadahisa Teramoto, Huiguo Lai, Kevin R. Alliston, Gerald H. Lushington, R. Padmanabhan, William C. Groutas
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 3) pp:1213-1221
Publication Date(Web):1 February 2012
DOI:10.1016/j.bmc.2011.12.047
Two click chemistry-derived focused libraries based on the benz[d]isothiazol-3(2H)-one scaffold were synthesized and screened against Dengue virus and West Nile virus NS2B-NS3 proteases. Several compounds (4l, 7j–n) displayed noteworthy inhibitory activity toward Dengue virus NS2B-NS3 protease in the absence and presence of added detergent. These compounds could potentially serve as a launching pad for a hit-to-lead optimization campaign.Two click chemistry-derived focused libraries based on the benz[d]isothiazol-3(2H)-one scaffold were synthesized and screened against Dengue virus and West Nile virus NS2B-NS3 proteases. Several compounds (4l, 7j–n) displayed noteworthy inhibitory activity toward Dengue virus NS2B-NS3 protease in the absence and presence of added detergent.
Co-reporter:Dengfeng Dou, Kok-Chuan Tiew, Sivakoteswara Rao Mandadapu, Mallikarjuna Reddy Gunnam, Kevin R. Alliston, Yunjeong Kim, Kyeong-Ok Chang, William C. Groutas
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 6) pp:2111-2118
Publication Date(Web):15 March 2012
DOI:10.1016/j.bmc.2012.01.030
The development of small molecule therapeutics to combat norovirus infection is of considerable interest from a public health perspective because of the highly contagious nature of noroviruses. A series of amino acid-derived acyclic sulfamide-based norovirus inhibitors has been synthesized and evaluated using a cell-based replicon system. Several compounds were found to display potent anti-norovirus activity, low toxicity, and good aqueous solubility. These compounds are suitable for further optimization of pharmacological and ADMET properties.A series of amino acid-derived acyclic sulfamide-based norovirus inhibitors has been synthesized and evaluated using a cell-based replicon system. Several compounds were found to display potent anti-norovirus activity, low toxicity, and good aqueous solubility. These compounds are suitable for further optimization of in vivo potency and ADMET properties.
Co-reporter:Dengfeng Dou, Guijia He, Sivakoteswara Rao Mandadapu, Sridhar Aravapalli, Yunjeong Kim, Kyeong-Ok Chang, William C. Groutas
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 1) pp:377-379
Publication Date(Web):1 January 2012
DOI:10.1016/j.bmcl.2011.10.122
There is currently an unmet need for the development of small-molecule therapeutics for norovirus infection. The piperazine scaffold, a privileged structure embodied in many pharmacological agents, was used to synthesize an array of structurally-diverse derivatives which were screened for anti-norovius activity in a cell-based replicon system. The studies described herein demonstrate for the first time that functionalized piperazine derivatives possess anti-norovirus activity. Furthermore, these studies have led to the identification of two promising compounds (6a and 9l) that can be used as a launching pad for the optimization of potency, cytotoxicity, and drug-like characteristics.The piperazine scaffold, a privileged structure embodied in a range of pharmacological agents, was used to synthesize an array of structurally-diverse derivatives which were screened for anti-norovius activity in a cell-based replicon system. The studies described herein have demonstrated that piperazine derivatives posses anti-norovirus activity. Furthermore, these studies have resulted in the identification of two promising compounds (6a and 9l) that can be used as a launching pad for the optimization of potency, cytotoxicity, and drug-like characteristics.
Co-reporter:Sivakoteswara Rao Mandadapu, Pathum M. Weerawarna, Mallikarjuna Reddy Gunnam, Kevin R. Alliston, Gerald H. Lushington, Yunjeong Kim, Kyeong-Ok Chang, William C. Groutas
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 14) pp:4820-4826
Publication Date(Web):15 July 2012
DOI:10.1016/j.bmcl.2012.05.055
A series of structurally-diverse α-ketoamides and α-ketoheterocycles was synthesized and subsequently investigated for inhibitory activity against norovirus 3CL protease in vitro, as well as anti-norovirus activity in a cell-based replicon system. The synthesized compounds were found to inhibit norovirus 3CL protease in vitro and to also exhibit potent anti-norovirus activity in a cell-based replicon system.Series of structurally-diverse α-ketoamides and α-ketoheterocycles was synthesized and subsequently investigated for inhibitory activity against Norwalk virus 3C protease in vitro, as well as anti-norovirus activity in a cell-based replicon system.
Co-reporter:Dengfeng Dou, Kok-Chuan Tiew, Guijia He, Sivakoteswara Rao Mandadapu, Sridhar Aravapalli, Kevin R. Alliston, Yunjeong Kim, Kyeong-Ok Chang, William C. Groutas
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 20) pp:5975-5983
Publication Date(Web):15 October 2011
DOI:10.1016/j.bmc.2011.08.054
A new class of compounds that exhibit anti-norovirus activity in a cell-based system and embody in their structure a cyclosulfamide scaffold has been identified. The structure of the initial hit (compound 2a, ED50 4 μM, TD50 50 μM) has been prospected by exploiting multiple points of diversity and generating appropriate structure–activity relationships.A new class of compounds that exhibit anti-norovirus activity in a cell-based system and embody in their structure a cyclosulfamide scaffold, has been identified. The structure of the initial hit (compound 2a (structure (I) where R = H), ED50 4 μM, TD50 50 μM) has been prospected by exploiting multiple points of diversity and generating appropriate structure–activity relationships.
Co-reporter:Dengfeng Dou, Deepu Alex, Bingfan Du, Kok-Chuan Tiew, Sridhar Aravapalli, Sivakoteswara Rao Mandadapu, Richard Calderone, William C. Groutas
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 19) pp:5782-5787
Publication Date(Web):1 October 2011
DOI:10.1016/j.bmc.2011.08.029
A series of broad-spectrum antifungal agents based on the 1,2-benzisothiazol-3(2H)-one scaffold is reported. Preliminary structure–activity relationship studies have established the importance of the presence of the heterocyclic ring, a methyl group, and a phenyl ring for optimal manifestation of antifungal activity.A series of broad-spectrum antifungal agents based on the 1,2-benzisothiazol-3(2H)-one scaffold is reported.
Co-reporter:Dengfeng Dou, Sivakoteswara Rao Mandadapu, Kevin R. Alliston, Yunjeong Kim, Kyeong-Ok Chang, William C. Groutas
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 19) pp:5749-5755
Publication Date(Web):1 October 2011
DOI:10.1016/j.bmc.2011.08.032
A scaffold hopping strategy was employed to identify new chemotypes that inhibit noroviruses. The replacement of the cyclosulfamide scaffold by an array of heterocyclic scaffolds lead to the identification of additional series of compounds that possessed anti-norovirus activity in a cell-based replicon system.A scaffold hopping strategy was employed to identify new chemotypes that inhibit noroviruses. The replacement of the cyclosulfamide scaffold by an array of heterocyclic scaffolds lead to the identification of additional series of compounds that possessed anti-norovirus activity in a cell-based replicon system.
Co-reporter:Kok-Chuan Tiew, Guijia He, Sridhar Aravapalli, Sivakoteswara Rao Mandadapu, Mallikarjuna Reddy Gunnam, Kevin R. Alliston, Gerald H. Lushington, Yunjeong Kim, Kyeong-Ok Chang, William C. Groutas
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 18) pp:5315-5319
Publication Date(Web):15 September 2011
DOI:10.1016/j.bmcl.2011.07.016
The first series of peptidyl aldehyde inhibitors that incorporate in their structure a glutamine surrogate has been designed and synthesized based on the known substrate specificity of Norwalk virus 3C protease. The inhibitory activity of the compounds with the protease and with a norovirus cell-based replicon system was investigated. Members of this class of compounds exhibited noteworthy activity both in vitro and in a cell-based replicon system.Peptidyl aldehydes were synthesized and used to inhibit Norwalk virus 3C protease.
Co-reporter:Dengfeng Dou, Guijia He, Kevin R. Alliston, William C. Groutas
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 10) pp:3177-3180
Publication Date(Web):15 May 2011
DOI:10.1016/j.bmcl.2010.12.033
The general strategy and rationale underlying the design of COPD therapeutics that possess protease inhibitory activity and are also capable of releasing a species that attenuates inflammation by inhibiting caspase-1, are described. The synthesis and in vitro biochemical evaluation of a dual function molecule that sequentially inhibits HNE and caspase-1 in a time-dependent manner is reported.A designed dual functional inhibitor was found to sequentially inhibit human neutrophil elastase and caspase-1 in vitro.
Co-reporter:Guijia He, Dengfeng Dou, Liuqing Wei, Kevin R. Alliston, William C. Groutas
European Journal of Medicinal Chemistry 2010 Volume 45(Issue 9) pp:4280-4287
Publication Date(Web):September 2010
DOI:10.1016/j.ejmech.2010.06.028
A series of compounds based on the N-amino-4-imidazolidinone scaffold was synthesized and screened against human neutrophil elastase (HNE). These studies lead to the identification of a selective, low micromolar reversible competitive inhibitor of HNE.A series of derivatives based on the N-amino-4-imidazolidinone scaffold (I) was synthesized and shown to inhibit human neutrophil elastase.
Co-reporter:Dengfeng Dou, Guijia He, Rongze Kuang, Qingfong Fu, Radhika Venkataraman, William C. Groutas
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 18) pp:6646-6650
Publication Date(Web):15 September 2010
DOI:10.1016/j.bmc.2010.07.071
A structurally-diverse series of carboxylate derivatives based on the 1,2,5-thiadiazolidin-one 1,1 dioxide scaffold were synthesized and used to probe the S′ subsites of human neutrophil elastase (HNE) and neutrophil proteinase 3 (Pr 3). Several compounds are potent inhibitors of HNE but devoid of inhibitory activity toward Pr 3, suggesting that the S′ subsites of HNE exhibit significant plasticity and can, unlike Pr 3, tolerate various large hydrophobic groups. The results provide a promising framework for the design of highly selective inhibitors of the two enzymes.A series of compounds based on the 1,2,5-thiadiazolidin-3-one 1,1 dioxide scaffold were synthesized and used to probe the S′ subsites of human neutrophil elastase and proteinase 3.
Co-reporter:Dengfeng Dou, Prasanth Viwanathan, Yi Li, Guijia He, Kevin R. Alliston, Gerald H. Lushington, Joshua D. Brown-Clay, R. Padmanabhan, and William C. Groutas
ACS Combinatorial Science 2010 Volume 12(Issue 6) pp:836
Publication Date(Web):September 30, 2010
DOI:10.1021/cc100091h
The 1-oxo-1, 2, 3, 4-tetrahydroisoquinoline and 1-Oxo-1, 2-dihydroisoquinoline scaffolds were utilized in the design and solution phase synthesis of focused libraries of compounds for screening against West Nile Virus (WNV) protease. Exploratory studies have led to the identification of a WNV protease inhibitor (a 1-oxo-1, 2-dihydroisoquinoline-based derivative, 12j) which could potentially serve as a launching pad for a hit-to-lead optimization campaign. The identified hit was devoid of any inhibitory activity toward a panel of mammalian serine proteases.
Co-reporter:Dengfeng Dou, Guijia He, Yi Li, Zhong Lai, Liuqing Wei, Kevin R. Alliston, Gerald H. Lushington, David M. Eichhorn, William C. Groutas
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 3) pp:1093-1102
Publication Date(Web):1 February 2010
DOI:10.1016/j.bmc.2009.12.057
The S′ subsites of human neutrophil proteinase 3 (Pr 3) were probed by constructing diverse libraries of compounds based on the 1,2,3,5-thiatriazolidin-3-one 1,1-dioxide using combinational and click chemistry methods. The multiple points of diversity embodied in the heterocyclic scaffold render it well-suited to the exploration of the S′ subsites of Pr 3. Molecular modeling studies suggest that further exploration of the S′ subsites of Pr 3 using the aforementioned heterocyclic scaffold may lead to the identification of highly selective, reversible competitive inhibitors of Pr 3.A series of 1,2,3,5-thiatriazolidin-3-one 1,1-dioxide derivatives were synthesized and used to probe the S′ subsites of human neutrophil proteinase 3.
Co-reporter:Yi Li, Dengfeng Dou, Guijia He, Gerald H. Lushington, William C. Groutas
Bioorganic & Medicinal Chemistry 2009 Volume 17(Issue 10) pp:3536-3542
Publication Date(Web):15 May 2009
DOI:10.1016/j.bmc.2009.04.011
A series of mechanism-based inhibitors designed to interact with the S′ subsites of serine proteases was synthesized and their inhibitory activity toward the closely-related serine proteases human neutrophil elastase (HNE) and proteinase 3 (PR 3) was investigated. The compounds were found to be time-dependent inhibitors of HNE and were devoid of any inhibitory activity toward PR 3. The results suggest that highly selective inhibitors of serine proteases whose primary substrate specificity and active sites are similar can be identified by exploiting differences in their S′ subsites. The best inhibitor (compound 16) had a kinact/KI value of 4580 M−1 s−1.A series of 1,2,5-thiadiazolidin-3-one 1,1-dioxide derivatives were found to be potent and selective inhibitors of human neutrophil elastase but not proteinase 3.
Co-reporter:Dengfeng Dou;Erach R. Talaty;Curtis E. Moore;John C. Bullinger;David M. Eichhorn ;William C. Groutas
Journal of Heterocyclic Chemistry 2009 Volume 46( Issue 4) pp:669-673
Publication Date(Web):
DOI:10.1002/jhet.136
Co-reporter:Weijun Huang ; Yasufumi Yamamoto ; Yi Li ; Dengfeng Dou ; Kevin R. Alliston ; Robert P. Hanzlik ; Todd D. Williams ;William C. Groutas
Journal of Medicinal Chemistry 2008 Volume 51(Issue 7) pp:2003-2008
Publication Date(Web):March 5, 2008
DOI:10.1021/jm700966p
The mechanism of action of a general class of mechanism-based inhibitors of serine proteases, including human neutrophil elastase (HNE), has been elucidated by determining the X-ray crystal structure of an enzyme–inhibitor complex. The captured intermediate indicates that processing of inhibitor by the enzyme generates an N-sulfonyl imine functionality that is tethered to Ser195, in accordance with the postulated mechanism of action of this class of inhibitors. The identity of the HNE-N-sulfonyl imine species was further corroborated using electrospray ionization mass spectrometry.
Co-reporter:Jiaying Zhong, Zhong Lai, Christopher S. Groutas, Tzutshin Wong, Xiangdong Gan, Kevin R. Alliston, David Eichhorn, John R. Hoidal, William C. Groutas
Bioorganic & Medicinal Chemistry 2004 Volume 12(Issue 23) pp:6249-6254
Publication Date(Web):1 December 2004
DOI:10.1016/j.bmc.2004.08.044
The pathogenesis of a range of human diseases arises from the aberrant activity of proteolytic enzymes. Agents capable of selectively modulating the activity of these enzymes are of potential therapeutic value. Thus, there is a continuing need for the design of scaffolds that can be used in the development of new classes of protease inhibitors. We describe herein the serendipitous discovery of an unexpected rearrangement that leads to the formation of two novel templates that can be used in the design of protease inhibitors.
Co-reporter:Yunjeong Kim, Anushka C. Galasiti Kankanamalage, Vishnu C. Damalanka, Pathum M. Weerawarna, William C. Groutas, Kyeong-Ok Chang
Antiviral Research (January 2016) Volume 125() pp:
Publication Date(Web):1 January 2016
DOI:10.1016/j.antiviral.2015.11.010
•Series of dipeptidyl aldehyde or ketomide compounds were highly effective against enterovirus-D68 and human rhinoviruses.•The highly effective ketoamide compound is shown to have favorable pharmacokinetics profiles.•These may have a high potential for further antiviral development against emerging enterovirus-D68 and human rhinoviruses.Enterovirus D68 (EV-D68) is an emerging pathogen responsible for mild to severe respiratory infections that occur mostly in infants, children and teenagers. EV-D68, one of more than 100 non-polio enteroviruses, is acid-labile and biologically similar to human rhinoviruses (HRV) (originally classified as HRV87). However, there is no approved preventive or therapeutic measure against EV-D68, HRV, or other enteroviruses. In this study, we evaluated the antiviral activity of series of dipeptidyl compounds against EV-D68 and HRV strains, and demonstrated that several peptidyl aldehyde and α-ketoamide peptidyl compounds are potent inhibitors of EV-D68 and HRV strains with high in-vitro therapeutic indices (>1000). One of the α-ketoamide compounds is shown to have favorable pharmacokinetics profiles, including a favorable oral bioavailability in rats. Recent successful development of α-ketoamide protease inhibitors against hepatitis C virus suggests these compounds may have a high potential for further optimization and development against emerging EV-D68, as well as HRV.
Co-reporter:Qingliang Yang, Yi Li, Dengfeng Dou, Xiangdong Gan, Swathi Mohan, Christopher S. Groutas, Laura E. Stevenson, Zhong Lai, Kevin R. Alliston, Jiaying Zhong, Todd D. Williams, William C. Groutas
Archives of Biochemistry and Biophysics (15 July 2008) Volume 475(Issue 2) pp:115-120
Publication Date(Web):15 July 2008
DOI:10.1016/j.abb.2008.04.020



![Carbamic acid, [4-[(methylsulfonyl)oxy]butyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/174700/174626-25-6.png)
![Carbamic acid, [4-[(methylsulfonyl)oxy]butyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/174700/174626-25-6_b.png)







![Carbamic acid, [3-[(methylsulfonyl)oxy]propyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/112700/112663-43-1.png)
![Carbamic acid, [3-[(methylsulfonyl)oxy]propyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/112700/112663-43-1_b.png)
![Hydrazinecarboxylic acid,2-[1-oxo-3-[[(phenylmethoxy)carbonyl]amino]propyl]-, 1,1-dimethylethylester](http://img.cochemist.com/ccimg/110600/110503-79-2.png)
![Hydrazinecarboxylic acid,2-[1-oxo-3-[[(phenylmethoxy)carbonyl]amino]propyl]-, 1,1-dimethylethylester](http://img.cochemist.com/ccimg/110600/110503-79-2_b.png)