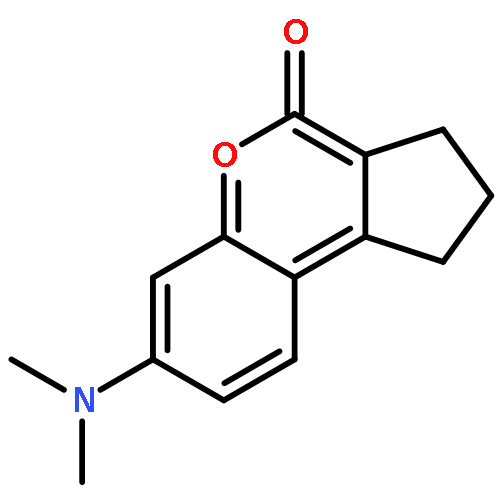
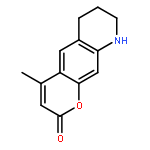
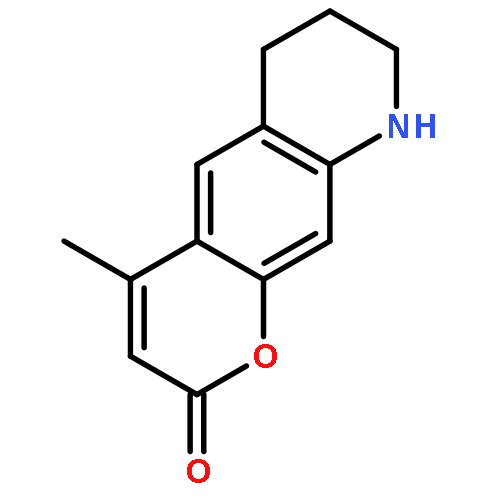
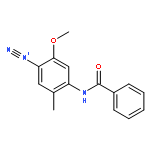
Co-reporter:Shuo Chai, Jin-Dou Huang
Computational and Theoretical Chemistry 2015 Volume 1069() pp:48-55
Publication Date(Web):1 October 2015
DOI:10.1016/j.comptc.2015.07.008
•Halogenated substituent enhances the charge transport abilities.•Intermolecular interaction influences the molecular packing and electronic coupling.•The hole mobility is improved by proper functionalization of organic semiconductors.The electronic and charge transport properties of the derivatives based on tetracene with aryl and halogenated aryl substituents have been investigated theoretically. This kind of functionalization is demonstrated to have a significant effect to stabilize the molecular orbital, densify the molecular packing, enhance the electronic coupling, and further lead to a high mobility, though it would also cause some increases in the reorganization energy. The packing modes of FPPT and PPT crystals are analyzed in details and effective coupling projected areas are put forward to understand the intermolecular interactions. Interestingly, FPPT is found to have a well-ordered packing as well as the improved hole mobility of 4.67 cm2 V−1 s−1. In addition, the contributions of different frequencies of vibration to the total reorganization energies are also discussed with the normal mode analysis. This study clarifies the halogenated substituent effect on transport properties and provides the guide for molecular design of novel functional materials.
Co-reporter:Shuo Chai, Shu-Lin Cong
Computational and Theoretical Chemistry 2014 Volume 1034() pp:80-84
Publication Date(Web):15 April 2014
DOI:10.1016/j.comptc.2014.02.019
•Intramolecular proton transfer of HBQ is investigated by DFT and TDDFT methods.•Substituent effect on photophysical properties of HBQ compounds are discussed.•Potential energy curve calculation is performed to show reaction mechanism of ESIPT.•HBQ-a is preferable in terms of molecular orbital, Stokes shift and proton transfer.The excited state intramolecular proton transfer (ESIPT) and the substituent effect of 10-hydroxybenzo[h]quinoline (HBQ) compounds are investigated using the time-dependent density functional theory (TDDFT) method. With the spectra and potential energy curve calculations we have demonstrated the occurrence of an ultrafast excited state intramolecular proton transfer reaction in HBQ compounds. The HBQ-a and HBQ-b in the enol conformations can convert into the keto tautomers in the excited state S1. The significant Stokes shift of HBQ-a is observed, as large as 300 nm, which is much larger than that of HBQ-b. The calculated molecular orbital gap between the lowest unoccupied molecular orbital (LUMO) and the highest occupied molecular orbital (HOMO) for HBQ-a is 3.33 eV, smaller than the 3.79 eV for HBQ-b. The calculations demonstrate that it is much easier to take place the ESIPT reaction for HBQ-a than HBQ-b. The reaction mechanism of ESIPT is analyzed with theoretical potential energy curves and the ESIPT reaction energy barriers of 3.73 kcal/mol for HBQ-a and 17.75 kcal/mol for HBQ-b are obtained. These results clearly indicate that the substituent with electron-withdrawing groups in the hydrogen-accepting moiety in HBQ-a facilitate the proton transfer in the excited state.
Co-reporter:Shuo Chai, Shu-Lin Cong
Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2014 Volume 125() pp:67-72
Publication Date(Web):5 May 2014
DOI:10.1016/j.saa.2014.01.092
•The twisted intramolecular charge transfer of FVB is investigated by TDDFT method.•The hydrogen bonding strengthening in the excited state is demonstrated.•The mechanism of TICT is clarified.The excited state hydrogen bonding dynamics and corresponding photophysical processes of fast violet B (FVB) in hydrogen-donating methanol (MeOH) solution are investigated by using time-dependent density functional theory (TDDFT) method. In the FVB molecule, there are CO, NH groups which could act as hydrogen acceptor and donor. It is demonstrated that both the intramolecular hydrogen bond O⋯HN in FVB and intermolecular hydrogen bond CO⋯HO between FVB and MeOH are formed in the ground state S0 and strengthened in the excited state S1. The absorption spectra are obviously red shifted for the hydrogen-bonded complex in comparison with FVB monomer in the low energy range. The theoretical investigation demonstrates that the twisted intramolecular charge transfer takes place in the excited states for both isolated FVB and hydrogen-bonded complex, and the dominant twisting is along N2C3 bond. The potential energy curve is investigated to understand the photophysics process of FVB and hydrogen-bonded complex.Graphical abstract