Co-reporter:Franz Bracher;Tim Tremmel
Archiv der Pharmazie 2017 Volume 350(Issue 7) pp:
Publication Date(Web):2017/07/01
DOI:10.1002/ardp.201600236
Natural products are a rich source of bioactive compounds, and numerous natural compounds have found application in cancer chemotherapy. However, unfavorable physicochemical properties often prevent the use of the original natural product as a drug. A prominent example is camptothecin from the Chinese tree Camptotheca acuminata, which shows extraordinary cytotoxic activity based on a specific molecular mode of action (inhibition of human topoisomerase I). Due to its extremely poor solubility, the original natural product cannot be used as a drug. The marketed drug topotecan was developed from this lead structure by semi-synthesis utilizing a Mannich aminomethylation as the crucial step. In this review, the long-distance run leading to this drug and further perspectives are summarized.
Co-reporter:Duc Nghia Ong, Sebastian Dittrich, Sören Swyter, Manfred Jung, Franz Bracher
Tetrahedron 2017 Volume 73, Issue 38(Issue 38) pp:
Publication Date(Web):21 September 2017
DOI:10.1016/j.tet.2017.08.005
3-Arylideneindolin-2-ones are an attractive structural class in drug development, including registered drugs and drug candidates for treatment of cancer, Alzheimer's disease, infections and others. While residues at C-5 and C-7 are easily introduced by electrophilic substitution reactions, position 4 is much less accessible. Here we describe a novel and effective synthesis of highly substituted 3-arylideneindolin-2-ones with flexible modification at C-4. Starting from 1,2-dichloro-3-nitrobenzene a central building block, a trihalogenated indolin-2-one, could be prepared in 6 high yielding steps. Subsequent modification of position 4 and 3 of this building block provided a library of highly substituted 4-substituted 3-arylidene-6,7-dichloroindolin-2-ones. This 8 step synthetic sequence utilized preferably precipitation and washing processes as method of purification.Download high-res image (110KB)Download full-size image
Co-reporter:Christoph Müller, Sandra Hemmers, Nicholas Bartl, Alois Plodek, Andreas Körner, Valbona Mirakaj, Martin Giera, Franz Bracher
European Journal of Medicinal Chemistry 2017 Volume 140(Volume 140) pp:
Publication Date(Web):10 November 2017
DOI:10.1016/j.ejmech.2017.08.011
•New chemotype of mammalian Δ24-dehydrocholesterol reductase (DHCR24) inhibitors.•Characterization of highly active, selective and non-toxic steroidal inhibitors.•Compound 27 (SH-42) has submicromolar activity on human DHCR24.•Significant accumulation of desmosterol in mice under SH-42 treatment after 5 days.The enzyme Δ24-dehydrocholesterol reductase (DHCR24) catalyzes the reduction of the Δ24-double bond in the side chain of cholesterol precursors. Recent biochemical investigations fuel the hope that inhibition of DHCR24, resulting in an accumulation of desmosterol, can open new therapeutic options for treating hepatitis C virus infections, certain forms of cancer and atherosclerosis. In turn, there is a high need for selective, potent and non-toxic inhibitors of DHCR24. Previous reports as well as our re-evaluation showed that established DHCR24 inhibitors are not suitable for this purpose. Based on the lathosterol-derived amide MGI-21 (IC50 823 nM for inhibition of overall cholesterol biosynthesis in HL-60 cells) we performed a systematic variation of the side chain functionality and identified the steroidal 3,22-diols 29 and 30, as well as several esters thereof, as extremely potent (IC50 < 5 nM), selective, and non-toxic DHCR24 inhibitors. In mice, diester 27 (SH-42) led to a significant increase in plasma desmosterol levels. The new inhibitors described here are valuable tools for investigating the therapeutic potential of DHCR24 inhibition.Download high-res image (121KB)Download full-size image
Co-reporter:Tobias A. Popp, Cynthia Tallant, Catherine Rogers, Oleg Fedorov, Paul E. Brennan, Susanne Müller, Stefan Knapp, and Franz Bracher
Journal of Medicinal Chemistry 2016 Volume 59(Issue 19) pp:8889-8912
Publication Date(Web):September 27, 2016
DOI:10.1021/acs.jmedchem.6b00774
CBP (CREB (cAMP responsive element binding protein) binding protein (CREBBP)) and P300 (adenovirus E1A-associated 300 kDa protein) are two closely related histone acetyltransferases (HATs) that play a key role in the regulation of gene transcription. Both proteins contain a bromodomain flanking the HAT catalytic domain that is important for the targeting of CBP/P300 to chromatin and which offeres an opportunity for the development of protein–protein interaction inhibitors. Here we present the development of CBP/P300 bromodomain inhibitors with 2,3,4,5-tetrahydro-1,4-benzoxazepine backbone, an N-acetyl-lysine mimetic scaffold that led to the recent development of the chemical probe I-CBP112. We present comprehensive SAR of this inhibitor class as well as demonstration of cellular on target activity of the most potent and selective inhibitor TPOP146, which showed 134 nM affinity for CBP with excellent selectivity over other bromodomains.
Co-reporter:Marco Keller, Annette Wolfgardt, Christoph Müller, Rainer Wilcken, Frank M. Böckler, Simonetta Oliaro-Bosso, Terenzio Ferrante, Gianni Balliano, Franz Bracher
European Journal of Medicinal Chemistry 2016 Volume 109() pp:13-22
Publication Date(Web):15 February 2016
DOI:10.1016/j.ejmech.2015.12.025
•Arylpiperidines are a new class of oxidosqualene cyclase inhibitors.•They are steroidomimetic compounds mimicking cationic high energy intermediates.•Compound 29 has submicromolar activity on human OSC and high species selectivity.•This compound effectively reduces total cholesterol biosynthesis in a cellular assay.•A plausible binding mode is suggested on the basis of docking experiments.The cyclization of oxidosqualene to lanosterol, catalyzed by the enzyme oxidosqualene cyclase (OSC), goes through a number of carbocationic high energy intermediates (HEI), and mimicking these intermediates is a promising approach for the development of OSC inhibitors. 3-Arylpiperidines (or tetrahydropyridines) were designed as steroidomimetic rings A + C equivalents containing two protonable amino groups for mimicking both the pro-C4 HEI and the pro-C20 HEI of the OSC-mediated cyclization cascade. Inhibitory activity is strongly dependent on the nature of the lipophilic substituent representing an equivalent of the sterol side chain. Here aromatic residues (substituted benzyl, cinnamyl, naphthylmethyl) were found to be most suitable. Docking experiments on a first optimized 3-arylpiperidine compound led to an isomeric 4-arylpiperidine with submicromolar activity on human OSC. This inhibitor reduced total cholesterol biosynthesis in a cellular assay with an IC50 value of 0.26 μM.
Co-reporter:Alexandra Kamlah, Florian Lirk, Franz Bracher
Tetrahedron 2016 Volume 72(Issue 6) pp:837-845
Publication Date(Web):11 February 2016
DOI:10.1016/j.tet.2015.12.049
Starting from readily available indole-2-carboxylic acids, 1-substituted β-carbolines (among them the alkaloids harmane and isoharmine) are readily obtained via the corresponding 2-acylindoles, bromination at C-3, followed by a one-pot Stille cross-coupling with tributyl[(Z)-2-ethoxyvinyl]stannane, and ring closure with ammonium acetate. 1-Substituted isoquinolines are available in an analogous manner starting from 2-bromobenzoic acid.
Co-reporter:Tobias Alexander Popp, Edgar Uhl, Duc Nghia Ong, Sebastian Dittrich, Franz Bracher
Tetrahedron 2016 Volume 72(Issue 13) pp:1668-1674
Publication Date(Web):31 March 2016
DOI:10.1016/j.tet.2016.02.019
A novel approach to 2,3,4,5-tetrahydro-1H-1,4-benzodiazepines starting from N-Boc-protected 2-aminobenzyl alcohols and N-nosyl-protected 2-aminoacetaldehyde dimethyl acetal is presented here. After connection of both building blocks under Mitsunobu conditions, a one-pot cyclization is accomplished with triethylsilane and trifluoroacetic acid. This conversion involves Boc deprotection and an intramolecular reductive N-alkylation. A consecutive methylation at N-1 can be accomplished by adding 1,3,5-trioxane to the reaction mixture. The resulting N4-monoprotected 1,4-benzodiazepines are versatile building blocks for the synthesis of variously substituted drug candidates.
Co-reporter:Benedikt Melzer and Franz Bracher
Organic & Biomolecular Chemistry 2015 vol. 13(Issue 28) pp:7664-7672
Publication Date(Web):09 Jun 2015
DOI:10.1039/C5OB00926J
Methoxy- and benzyloxy-substituted isoquinolines are regioselectively metalated at C-1 with the Knochel–Hauser base, subsequent trapping with aromatic aldehydes gives aryl(isoquinolin-1-yl)carbinols as building blocks for divergent syntheses of different types of benzylisoquinoline alkaloids. Photochemical cyclization of ortho-bromo analogues under reductive conditions gives oxoaporphine alkaloids. Nine benzylisoquinoline alkaloids and two oxoaporphine alkaloids were obtained in two or three steps from appropriate isoquinolines.
Co-reporter:Alois Plodek;Mathias König
European Journal of Organic Chemistry 2015 Volume 2015( Issue 6) pp:1302-1308
Publication Date(Web):
DOI:10.1002/ejoc.201403502
Abstract
We report the synthesis of the azaoxoaporphine alkaloid sampangine (4) and a series of ring A analogues and isomers of the marine pyridoacridine alkaloid ascididemin (2). This approach starts from readily available 1-bromo[2,7]naphthyridine (12) or 4-bromobenzo[c][2,7]naphthyridine (5), and the ring A scaffold bearing an ester moiety is introduced by a Suzuki or Negishi cross-coupling reaction. The final cyclization step was achieved through a directed remote ring metallation with the Knochel–Hauser base (TMPMgCl·LiCl; TMP = 2,2,6,6-tetramethylpiperidinyl), followed by intramolecular trapping of the ester group.
Co-reporter:Sebastian Dittrich
European Journal of Organic Chemistry 2015 Volume 2015( Issue 36) pp:8024-8033
Publication Date(Web):
DOI:10.1002/ejoc.201501180
Abstract
A synthesis of terminal vinylsilanes by triflimide-catalysed rearrangement of N-(1-trimethylsilyl)allylhydrazones is reported. This protocol provides a convenient access to versatile olefinic building blocks through a traceless bond construction. Hydrazones derived from aromatic aldehydes give cis-cyclopropanes in an unexpected side-reaction.
Co-reporter:Alois Plodek, Franz Bracher
Tetrahedron Letters 2015 Volume 56(Issue 11) pp:1445-1447
Publication Date(Web):11 March 2015
DOI:10.1016/j.tetlet.2015.01.176
Herein we report the total synthesis of the heptacyclic pyridoacridine alkaloid eilatin (1) and its synthetic isomer isoeilatin (2). Starting from readily available 9-methyl-3,4-dihydroacridin-1(2H)-one (5) and 2-aminoacetophenone, a divergent synthesis gave alkaloid 1 in seven, and isomer 2 in eight steps.
Co-reporter:Benjamin Strödke;André P. Gehring
Archiv der Pharmazie 2015 Volume 348( Issue 2) pp:125-131
Publication Date(Web):
DOI:10.1002/ardp.201400328
1-Acetylcarbazoles are readily converted to 3-desazacanthin-4-ones upon treatment with Bredereck's reagent, but in contrast to canthin-4-ones, these do not undergo ring transformation reactions with guanidine. Only after N-protection (methyl or 2-(trimethylsilyl)ethoxymethyl group), 2-desaza analogues of the alkaloid annomontine are accessible via the enaminoketones obtained by condensation with Bredereck's reagent. One of the annomontine analogues is an inhibitor of the Plasmodium falciparum CDC-like kinases (CLK) and shows antimalarial activity.
Co-reporter:Sebastian Dittrich, Franz Bracher
Tetrahedron 2015 Volume 71(Issue 17) pp:2530-2539
Publication Date(Web):29 April 2015
DOI:10.1016/j.tet.2015.03.021
An extension of a recently described olefin synthesis under traceless bond construction via triflimide (HNTf2)-catalyzed rearrangement of N-Boc-N-allylhydrazones is presented. This protocol opens a new approach to 1,1-disubstituted olefins. Application of this methodology to a new synthesis of episterol was hampered by undesired acid-catalyzed double bond isomerizations, but an unprecedented fragmentation of sterically hindered N-Boc-N-allylhydrazones to give nitriles was observed in this course. A reference sample of episterol was prepared utilizing new silyl-protected intermediates.
Co-reporter:Tim Tremmel, Franz Bracher
Tetrahedron 2015 Volume 71(Issue 28) pp:4640-4646
Publication Date(Web):8 July 2015
DOI:10.1016/j.tet.2015.05.002
Two novel approaches to the canthin-4-one ring system have been worked out. Claisen-type condensation of 1-acetyl-β-carboline with N-acyl benzotriazoles gives, via intermediate 1,3-diketones, 6-alkylcanthin-4-ones in one single operation, but this protocol is restricted to small alkyl substituents. An alternative approach, via 1-ethynyl-β-carboline and 1-isoxazolyl-β-carbolines, followed by reductive isoxazole cleavage and cyclization, is more versatile. 5,6-disubstituted canthin-4-ones are accessible via iodination at C-5 and subsequent Pd-catalyzed cross-coupling.
Co-reporter:Melanie Krojer;Christoph Müller
Archiv der Pharmazie 2014 Volume 347( Issue 2) pp:108-122
Publication Date(Web):
DOI:10.1002/ardp.201300296
Grundmann's ketone is converted to a spiroacetal containing a 5-hydroxymethyl-5-nitro-1,3-dioxane moiety whose hydroxymethyl group can be esterified or directly substituted with primary and secondary amines. Among the resulting aminomethyl spiroacetals, several ones bearing diamino residues were found to be inhibitors of the enzyme Δ8,7-isomerase in cholesterol biosynthesis. The complex bicyclic building block derived from Grundmann's ketone could be replaced by a properly substituted tetraline scaffold, without noteworthy loss in activity. This opens the opportunity to perform further structural modifications for the design of new steroidomimetic inhibitors of human Δ8,7-isomerase.
Co-reporter:Jürgen Krauss;Christoph Müller;Julia Kießling;Sabine Richter;Verena Staudacher
Archiv der Pharmazie 2014 Volume 347( Issue 4) pp:283-290
Publication Date(Web):
DOI:10.1002/ardp.201300338
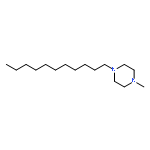
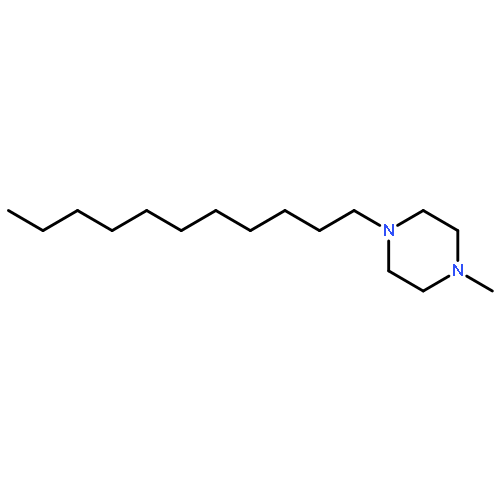
A series of N-alkyl trans-decahydroisoquinoline, 1,2,3,4-tetrahydroisoquinoline, and 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline derivatives were synthesized starting from the respective secondary amines by N-alkylation with alkyl bromides. The compounds with C11-alkyl chains showed antifungal potency comparable to clotrimazole, and inhibit enzymes of the ergosterol biosynthesis (Δ14-reductase and Δ8,7-isomerase), depending on the heterocyclic scaffold and the investigated species.
Co-reporter:Benedikt Melzer, Alois Plodek, and Franz Bracher
The Journal of Organic Chemistry 2014 Volume 79(Issue 15) pp:7239-7242
Publication Date(Web):July 11, 2014
DOI:10.1021/jo501312d
A four-step total synthesis of the marine pyridoacridine alkaloid demethyldeoxyamphimedine (5) is presented. With an overall yield of 6.4%, this pentacyclic compound has been synthesized by utilizing only two commercial building blocks, ethyl nicotinate and 2-iodoaniline. The final cyclization step was achieved via a directed remote ring metalation with Knochel–Hauser base (TMPMgCl·LiCl) followed by intramolecular trapping of an ester group.
Co-reporter:Markus Gans, Franz Bracher
Tetrahedron 2014 70(5) pp: 1084-1090
Publication Date(Web):
DOI:10.1016/j.tet.2013.11.065
Co-reporter:Stefanie Lange, Marco Keller, Christoph Müller, Simonetta Oliaro-Bosso, Gianni Balliano, Franz Bracher
European Journal of Medicinal Chemistry 2013 Volume 63() pp:758-764
Publication Date(Web):May 2013
DOI:10.1016/j.ejmech.2013.03.002
A series of aminopropylindenes, designed as mimics of a cationic high energy intermediate in the oxidosqualene cyclase1 (OSC)-mediated cyclization of 2,3-oxidosqualen to lanosterol was prepared from Grundmann's ketone. Screening on OSCs from five different organisms revealed interesting activities and selectivities of some of the compounds. A N,N-dimethylaminopropyl derivative showed promising inhibition of Trypanosoma cruzi OSC in combination with low cytotoxicity, and showed significant reduction of cholesterol biosynthesis in a human cell line.Graphical abstractHighlights► Aminopropylindenes are a new chemotype of oxidosqualene cyclase inhibitors. ► Depending on the amino group the compounds show selectivity for oxidosqualene cyclases from different organisms. ► A N,N-dimethylaminopropyl derivative showed promising inhibition of Trypanosoma cruzi oxidosqualene cyclase. ► For two of the inhibitors significant reduction of cholesterol biosynthesis was demonstrated in a whole cell assay.
Co-reporter:Mathias König, Christoph Müller, Franz Bracher
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 7) pp:1925-1943
Publication Date(Web):1 April 2013
DOI:10.1016/j.bmc.2013.01.041
Starting from Grundmann′s ketone a new chemotype of inhibitors of the post-squalene part of cholesterol biosynthesis was developed. Stereoselective introduction of an angular methyl group at C-3a, followed by a plethora of functionalisations at C-4 and C-5 led to cis-configured amino alcohols as a new chemotype of inhibitors of cholesterol biosynthesis. In cell-based screening systems these compounds were identified to be selective inhibitors of human Δ8,7-sterol isomerase, inhibiting total cholesterol biosynthesis with IC50 values in the low nanomolar range. The most active compounds did not affect fungal Δ8,7-sterol isomerase (in ergosterol biosynthesis), neither showed noteworthy antimicrobial and cytotoxic effects.Secosteroid-type cis-configured amino alcohols are potent and selective inhibitors of human Δ8,7-sterol isomerase in cholesterol biosynthesis, whereas epimeric trans-aminoalcohols and aminoketones are inactive.
Co-reporter:Alois Plodek, Stephan Raeder, Franz Bracher
Tetrahedron 2013 69(46) pp: 9857-9864
Publication Date(Web):
DOI:10.1016/j.tet.2013.08.085
Co-reporter:Kilian Huber ; Laurent Brault ; Oleg Fedorov ; Christelle Gasser ; Panagis Filippakopoulos ; Alex N. Bullock ; Doriano Fabbro ; Jörg Trappe ; Jürg Schwaller ; Stefan Knapp
Journal of Medicinal Chemistry 2012 Volume 55(Issue 1) pp:403-413
Publication Date(Web):December 5, 2011
DOI:10.1021/jm201286z
Development of both potent and selective kinase inhibitors is a challenging task in modern drug discovery. The innate promiscuity of kinase inhibitors largely results from ATP-mimetic binding to the kinase hinge region. We present a novel class of substituted 7,8-dichloro-1-oxo-β-carbolines based on the distinct structural features of the alkaloid bauerine C whose kinase inhibitory activity does not rely on canonical ATP-mimetic hinge interactions. Intriguingly, cocrystal structures revealed an unexpected inverted binding mode and the presence of halogen bonds with kinase backbone residues. The compounds exhibit excellent selectivity over a comprehensive panel of human protein kinases while inhibiting selected kinases such as the oncogenic PIM1 at low nanomolar concentrations. Together, our biochemical and structural data suggest that this scaffold may serve as a valuable template for the design and development of specific inhibitors of various kinases including the PIM family of kinases, CLKs, DAPK3 (ZIPK), BMP2K (BIKE), and others.
Co-reporter:Panagis Filippakopoulos, Sarah Picaud, Oleg Fedorov, Marco Keller, Matthias Wrobel, Olaf Morgenstern, Franz Bracher, Stefan Knapp
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 6) pp:1878-1886
Publication Date(Web):15 March 2012
DOI:10.1016/j.bmc.2011.10.080
Benzodiazepines are psychoactive drugs with anxiolytic, sedative, skeletal muscle relaxant and amnestic properties. Recently triazolo-benzodiazepines have been also described as potent and highly selective protein interaction inhibitors of bromodomain and extra-terminal (BET) proteins, a family of transcriptional co-regulators that play a key role in cancer cell survival and proliferation, but the requirements for high affinity interaction of this compound class with bromodomains has not been described. Here we provide insight into the structure–activity relationship (SAR) and selectivity of this versatile scaffold. In addition, using high resolution crystal structures we compared the binding mode of a series of benzodiazepine (BzD) and related triazolo-benzotriazepines (BzT) derivatives including clinically approved drugs such as alprazolam and midazolam. Our analysis revealed the importance of the 1-methyl triazolo ring system for BET binding and suggests modifications for the development of further high affinity bromodomain inhibitors.A number of triazolo-benzodiazepines including drugs such as alprazolam have been developed as protein interaction inhibitors that target bromodomains of the BET family.Figure optionsDownload full-size imageDownload as PowerPoint slide
Co-reporter:Stephan Raeder
Archiv der Pharmazie 2012 Volume 345( Issue 10) pp:822-826
Publication Date(Web):
DOI:10.1002/ardp.201200019
Abstract
A novel approach to the pyridoacridine ring system of the ascididemin-type marine alkaloids is presented. This approach allows for the introduction of the ring A of the alkaloids by using a simple aromatic aldehyde building block. The viability of this approach is demonstrated with the synthesis of AK37, a bioactive deaza analogue of the alkaloid ascididemin. Starting from 3-cyano-4-methylquinoline, a sequence of regioselective homolytic benzoylation, annulation of a bromopyridine ring, and radical cyclization leads to the pentacyclic ring system.
Co-reporter:Alois Plodek, Stephan Raeder, Franz Bracher
Tetrahedron 2012 68(24) pp: 4693-4700
Publication Date(Web):
DOI:10.1016/j.tet.2012.04.023
Co-reporter:Oleg Fedorov, Kilian Huber, Andreas Eisenreich, Panagis Filippakopoulos, Oliver King, Alex N. Bullock, Damian Szklarczyk, Lars J. Jensen, Doriano Fabbro, Jörg Trappe, Ursula Rauch, Franz Bracher, Stefan Knapp
Chemistry & Biology 2011 Volume 18(Issue 1) pp:67-76
Publication Date(Web):28 January 2011
DOI:10.1016/j.chembiol.2010.11.009
There is a growing recognition of the importance of protein kinases in the control of alternative splicing. To define the underlying regulatory mechanisms, highly selective inhibitors are needed. Here, we report the discovery and characterization of the dichloroindolyl enaminonitrile KH-CB19, a potent and highly specific inhibitor of the CDC2-like kinase isoforms 1 and 4 (CLK1/CLK4). Cocrystal structures of KH-CB19 with CLK1 and CLK3 revealed a non-ATP mimetic binding mode, conformational changes in helix αC and the phosphate binding loop and halogen bonding to the kinase hinge region. KH-CB19 effectively suppressed phosphorylation of SR (serine/arginine) proteins in cells, consistent with its expected mechanism of action. Chemical inhibition of CLK1/CLK4 generated a unique pattern of splicing factor dephosphorylation and had at low nM concentration a profound effect on splicing of the two tissue factor isoforms flTF (full-length TF) and asHTF (alternatively spliced human TF).Highlights► We report a highly selective nanomolar inhibitor KH-CB19 for the kinases CLK1/4 and DYRK1 ► KH-CB19 cocrystal structures revealed an ATP competitive but not ATP mimetic binding mode ► KH-CB19 formed halogen bonds with the kinase hinge region ► KH-CB19 led to dephosphorylation of SR proteins and effected splicing of TF isoforms in cells
Co-reporter:Kilian Huber ; Jörg Schemies ; Urszula Uciechowska ; Julia M. Wagner ; Tobias Rumpf ; Felicitas Lewrick ; Regine Süss ; Wolfgang Sippl ; Manfred Jung
Journal of Medicinal Chemistry 2010 Volume 53(Issue 3) pp:1383-1386
Publication Date(Web):December 23, 2009
DOI:10.1021/jm901055u
Class III histone deacetylases (sirtuins) play pivotal roles in many cellular processes. They are linked to extended lifespan and to the pathogenesis of cancer and neuronal disorders. We present novel sirtuin inhibitors based on a 6,7-dichloro-2-oxindole scaffold with low micromolar activity. In vitro activity was rationalized by docking studies, and hyperacetylation of sirtuin targets could be demonstrated in cell culture.
Co-reporter:Andreas Puzik
Journal of Heterocyclic Chemistry 2010 Volume 47( Issue 2) pp:449-453
Publication Date(Web):
DOI:10.1002/jhet.302
Co-reporter:Isolde Wetzel, Lars Allmendinger and Franz Bracher
Journal of Natural Products 2009 Volume 72(Issue 10) pp:1908-1910
Publication Date(Web):September 21, 2009
DOI:10.1021/np900515b
In 2004, a new anti-HIV alkaloid named drymaritin was isolated from Drymaria diandra. The authors identified the alkaloid as 5-methoxycanthin-4-one on the basis of spectroscopic data. Here we describe a synthetic approach that unambiguously gave 5-methoxycanthin-4-one, but the synthetic product showed spectroscopic data significantly different from those of the Drymaria alkaloid. Extensive re-evaluation of the spectroscopic data published for this and related alkaloids has led to the conclusion that drymaritin does not have a canthin-4-one backbone, but is identical to the known alkaloid cordatanine (4-methoxycanthin-6-one).
Co-reporter:Delphine Renard, Johann Perruchon, Martin Giera, Jörg Müller, Franz Bracher
Bioorganic & Medicinal Chemistry 2009 Volume 17(Issue 23) pp:8123-8137
Publication Date(Web):1 December 2009
DOI:10.1016/j.bmc.2009.09.037
The synthesis of some novel azasteroids and thiasteroids based on a pregnan nucleus with a Δ7 double bond in two to five steps from the key aldehyde (3S,20S)-20-formylpregn-7-en-3-yl acetate has been disclosed herein. These compounds were evaluated as potential inhibitors of the enzyme Δ24-sterol methyltransferase (24-SMT), which is a key enzyme in the biosynthesis of ergosterol, and for their effects on the growth of the yeast Yarrowia lipolytica. Most of the side chain modified analogues were recognized as 24-SMT inhibitors, and in particular the 23-azasteroids 5f–5i and the 24-azasteroid 11 showed potent antifungal activity. The target enzyme could be identified unambiguously using an improved whole-cell assay combined with GC–MS analysis of the sterol pattern resulting upon incubation with the inhibitors.Novel antifungal Δ7-azasteroids and thiasteroids were prepared as inhibitors of the enzyme Δ24-sterol methyltransferase (24-SMT).
Co-reporter:Andreas Puzik
Journal of Heterocyclic Chemistry 2009 Volume 46( Issue 4) pp:770-773
Publication Date(Web):
DOI:10.1002/jhet.126
Co-reporter:Kathrin Mink
Archiv der Pharmazie 2007 Volume 340(Issue 8) pp:
Publication Date(Web):13 JUL 2007
DOI:10.1002/ardp.200700064
Hetero analogues of the alkaloids cleistopholine and sampangine were prepared in order to investigate the significance of the (aza)quinoid partial structures for antimicrobial activity. Several analogues containing amino or sulfone groups showed high antimicrobial activities, indicating that the (aza)quinoid partial structures of the alkaloids are not an indispensable requisite for antimicrobial activity.
Co-reporter:Matthias Lotter;Johannes Schilling;Eberhard Reimann
Archiv der Pharmazie 2006 Volume 339(Issue 12) pp:
Publication Date(Web):16 NOV 2006
DOI:10.1002/ardp.200600134
The one-pot reaction of 4-benzylpyridine-3-carbonitrile with Bredereck's reagent and subsequent treatment with either glacial acetic acid and sulfuric acid or ammonium acetate provided the new bioactive naphthyridine alkaloids lophocladine A and B, respectively.
Co-reporter:Nyamdari Batbayar, Shiiter Enkhzaya, Jigjidsuren Tunsag, Dulamjav Batsuren, David S Rycroft, Susanne Sproll, Franz Bracher
Phytochemistry 2003 Volume 62(Issue 4) pp:543-550
Publication Date(Web):February 2003
DOI:10.1016/S0031-9422(02)00514-9
From the aerial parts of four Delphinium species 11 known and 3 new norditerpenoid alkaloids have been isolated: from D. dissectum Huth: delavaine A/B, deoxylycoctonine, methyllycaconitine; new: 10-hydroxymethyllycaconitine; from D. excelsum Reichenb.: delcaroline, delectinine, delterine, methyllycaconitine; new: 10-hydroxymethyllycaconitine, 18-O-methyldelterine and 10-hydroxynudicaulidine; from D. grandiflorum L.: delcosine, deltatsine, grandiflorine, methyllycaconitine; from D. triste Fisch.: delcosine, macrocentridine, 14-dehydrodelcosine. The structures of the new alkaloids were established on the basis of MS, 1H, 13C, DEPT, homonuclear COSY, HMQC and HMBC NMR spectroscopic techniques.From the aerial parts of four Delphinium species 11 known and the 3 new norditerpenoid alkaloids 10-hydroxymethyllycaconitine, 18-O-methyldelterine and 10-hydroxynudicaulidine have been isolated.
Co-reporter:Franz Bracher;Jochen Daab
European Journal of Organic Chemistry 2002 Volume 2002(Issue 14) pp:
Publication Date(Web):1 JUL 2002
DOI:10.1002/1099-0690(200207)2002:14<2288::AID-EJOC2288>3.0.CO;2-G
The first total synthesis of ficuseptine [4,6-bis(4-methoxyphenyl)-1,2,3-trihydroindolizidinium chloride] (1), an alkaloid from Ficus septica, is described. The crucial steps in this five-step synthesis are a palladium-catalyzed bis(arylation) of a dibromopyridine under Suzuki conditions and a palladium-catalyzed alkynylation of an iodopyridine under Sonogashira conditions, as well as a novel Sandmeyer-type iodination of a 2-aminopyridine derivative. (© Wiley-VCH Verlag GmbH, 69451 Weinheim, Germany, 2002)
Co-reporter:Franz Bracher;Jürgen Krauß
European Journal of Organic Chemistry 2001 Volume 2001(Issue 24) pp:
Publication Date(Web):26 NOV 2001
DOI:10.1002/1099-0690(200112)2001:24<4701::AID-EJOC4701>3.0.CO;2-6
A straightforward approach to both enantiomers of zearalane is described from the enantiomerically pure alkenol (S)-4, prepared by a kinetic enzymatic resolution of the racemate. The key step is a Pd-catalyzed cross-coupling of an arene trifluoromethanesulfonate with a 9-alkyl-9-borabicyclo[3.3.1]nonane derivative. The two enantiomers 2a and 2b have been obtained in an enantiodivergent manner by macrolactonization of the hydroxy acid (S)-7 with either Gerlach’s modification of the Corey lactonization or a Mitsunobu lactonization.
Co-reporter:Franz Bracher, Karina P. Randau, Holger Lerche
Fitoterapia (April 2008) Volume 79(Issue 3) pp:236-237
Publication Date(Web):1 April 2008
DOI:10.1016/j.fitote.2007.12.001
Crototropone (3-hydroxy-5,6-dimethoxy-2-methylcyclohepta-2,4,6-trien-1-one) was isolated from roots of Croton zehntneri. The structure was established by spectroscopic methods.
Co-reporter:Benedikt Melzer and Franz Bracher
Organic & Biomolecular Chemistry 2015 - vol. 13(Issue 28) pp:NaN7672-7672
Publication Date(Web):2015/06/09
DOI:10.1039/C5OB00926J
Methoxy- and benzyloxy-substituted isoquinolines are regioselectively metalated at C-1 with the Knochel–Hauser base, subsequent trapping with aromatic aldehydes gives aryl(isoquinolin-1-yl)carbinols as building blocks for divergent syntheses of different types of benzylisoquinoline alkaloids. Photochemical cyclization of ortho-bromo analogues under reductive conditions gives oxoaporphine alkaloids. Nine benzylisoquinoline alkaloids and two oxoaporphine alkaloids were obtained in two or three steps from appropriate isoquinolines.



![Bicyclo[2.2.1]heptane-2-acetyl chloride](/data/chemimg/1843500/54057-58-8.png)
![Bicyclo[2.2.1]heptane-2-acetyl chloride](/data/chemimg/1843500/54057-58-8_b.png)