Co-reporter:Remi Patouret, Theodore M. Kamenecka
Tetrahedron Letters 2016 Volume 57(Issue 14) pp:1597-1599
Publication Date(Web):6 April 2016
DOI:10.1016/j.tetlet.2016.02.102
•Regioselective cycloaddition reaction of arenediazonium salts with trimethylsilyldiazomethane.•Best catalyst to perform this reaction was silver trifluoroacetate.•A large set of diazonium salt was used under optimized conditions.•Gram scale have been successfully accomplished.A regioselective cycloaddition reaction of arenediazonium salts with trimethylsilyldiazomethane is reported. A series of 2-aryltetrazoles were obtained in good to moderate yields with wide functional group compatibility. Furthermore, this cycloaddition reaction opens the way to build up the versatile intermediate 2-aryl-5-bromotetrazole.A regioselective cycloaddition reaction of arenediazonium salts with trimethylsilyldiazomethane is reported. A series of 2-aryltetrazoles were obtained in good to moderate yields with wide functional group compatibility. Furthermore, this cycloaddition reaction opens the way to build up the versatile intermediate 2-aryl-5-bromotetrazole.
Co-reporter:Christelle Doebelin, Yuanjun He, Theodore M. Kamenecka
Tetrahedron Letters 2016 Volume 57(Issue 50) pp:5658-5660
Publication Date(Web):14 December 2016
DOI:10.1016/j.tetlet.2016.11.012
•Enantioselective synthesis of 3,3-difluoroproline.•Short scalable synthesis.•Resolution provided both enantiomer in high ee and yield.An efficient route for the synthesis of enantiopure 3,3-difluoroproline on multigram-scale is described herein. The deoxofluorination can be achieved with DAST on the corresponding racemic pyrrolidinone in good yield. Resolution of the racemate by crystallization with D- and L-tyrosine hydrazide provides both enantiomers of 3,3-difluoroproline in high yield and ee%.
Co-reporter:Alice Asteian, Anne-Laure Blayo, Yuanjun He, Marcel Koenig, Youseung Shin, Dana S. Kuruvilla, Cesar A. Corzo, Michael D. Cameron, Li Lin, Claudia Ruiz, Susan Khan, Naresh Kumar, Scott Busby, David P. Marciano, Ruben D. Garcia-Ordonez, Patrick R. Griffin, and Theodore M. Kamenecka
ACS Medicinal Chemistry Letters 2015 Volume 6(Issue 9) pp:998
Publication Date(Web):August 4, 2015
DOI:10.1021/acsmedchemlett.5b00218
The thiazolidinediones (TZD) typified by rosiglitazone are the only approved therapeutics targeting PPARγ for the treatment of type-2 diabetes (T2DM). Unfortunately, despite robust insulin sensitizing properties, they are accompanied by a number of severe side effects including congestive heart failure, edema, weight gain, and osteoporosis. We recently identified PPARγ antagonists that bind reversibly with high affinity but do not induce transactivation of the receptor, yet they act as insulin sensitizers in mouse models of diabetes (SR1664).1 This Letter details our synthetic exploration around this novel series of PPARγ antagonists based on an N-biphenylmethylindole scaffold. Structure–activity relationship studies led to the identification of compound 46 as a high affinity PPARγ antagonist that exhibits antidiabetic properties following oral administration in diet-induced obese mice.Keywords: diabetes; indole; nuclear receptor; PPARγ;
Co-reporter:Zhuang Jin, Pasha Khan, Youseung Shin, Jingyi Wang, Li Lin, Michael D. Cameron, Jon M. Lindstrom, Paul J. Kenny, Theodore M. Kamenecka
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 2) pp:674-678
Publication Date(Web):15 January 2014
DOI:10.1016/j.bmcl.2013.11.049
The design and synthesis of a series of substituted heteroaromatic α4β2α5 positive allosteric modulators is reported. The optimization and development of the heteroaromatic series was carried out from NS9283, and several potent analogues, such as 3-(5-(pyridin-3-yl)-2H-tetrazol-2-yl)benzonitrile (5k) and 3,3′-(2H-tetrazole-2,5-diyl)dipyridine (12h) with good in vitro efficacy were discovered.
Co-reporter:Paolo Di Fruscia, Yuanjun He, Marcel Koenig, Sahba Tabrizifard, Ainhoa Nieto, Patricia H. McDonald, Theodore M. Kamenecka
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 16) pp:3974-3978
Publication Date(Web):15 August 2014
DOI:10.1016/j.bmcl.2014.06.033
Neurotensin (NT) is an endogenous tridecapeptide found in the central nervous system (CNS) and in peripheral tissues. Neurotensin exerts a wide range of physiological effects and it has been found to play a critical role in a number of human diseases, such as schizophrenia, Parkinson’s disease and drug addiction. The discovery of small-molecule non-peptide neurotensin receptor (NTSR) modulators would represent an important breakthrough as such compounds could be used as pharmacological tools, to further decipher the cellular functions of neurotensin, and potentially as therapeutic agents to treat human disease. Herein, we report the identification of non-peptide low-micromolar neurotensin receptor 1 (NTSR1) full agonists, discovered through structural optimization of the known NTSR1 partial agonist 1. In vitro cellular screenings, based on an intracellular Ca2+ mobilization assay, revealed our best hit molecule 8 (SR-12062) to have an EC50 of 2 μM at NTSR1 with full agonist behaviour (Emax = 100%), showing a higher efficacy and ∼90-fold potency improvement compared to parent compound 1 (EC50 = 178 μM; Emax = 17%).SAR investigation of the weak neurotensin receptor 1 (NTSR1) partial agonist 1, identified by researchers at Wyeth, led to the discovery of single-digit micromolar NTSR1 full agonists.
Co-reporter:Pasha M. Khan, Bahaa El-Dien M. El-Gendy, Naresh Kumar, Ruben Garcia-Ordonez, Li Lin, Claudia H. Ruiz, Michael D. Cameron, Patrick R. Griffin, Theodore M. Kamenecka
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 2) pp:532-536
Publication Date(Web):15 January 2013
DOI:10.1016/j.bmcl.2012.11.025
The structure–activity relationship study of a diphenylpropanamide series of ROR-γ selective modulators is reported. Compounds were screened using chimeric receptor Gal4 DNA-binding domain (DBD)-NR ligand binding domain cotransfection assay in a two-step format. Three different regions of the scaffold were modified to assess the effects on repression of ROR-γ transcriptional activity and potency. The lead compound 1 exhibits modest mouse pharmacokinetics and an acceptable in vitro profile which makes it a suitable in vivo probe to interrogate the functions of ROR-γ in animal models of disease.The structure–activity relationship study of a diphenylpropanamide series of ROR-γ selective modulators is reported. Compounds were screened using chimeric receptor Gal4 DNA-binding domain (DBD)-NR ligand binding domain cotransfection assay in a two-step format. The lead compound 1 exhibits modest mouse pharmacokinetics and an acceptable in vitro profile which makes it a suitable in vivo probe to interrogate the functions of ROR-γ in animal models of disease.
Co-reporter:Rong Jiang, Bozena Frackowiak, Youseung Shin, Xinyi Song, Weimin Chen, Li Lin, Michael D. Cameron, Derek R. Duckett, Theodore M. Kamenecka
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 9) pp:2683-2687
Publication Date(Web):1 May 2013
DOI:10.1016/j.bmcl.2013.02.082
Starting from pyrazole HTS hit (1), a series of 1-aryl-1H-indazoles have been synthesized as JNK3 inhibitors with moderate selectivity against JNK1. SAR studies led to the synthesis of 5r as double digital nanomolar JNK3 inhibitor with good in vivo exposure.The structure–activity relationship study of a 5-aminoindazole series of JNK3 kinase inhibitors is reported. Starting from HTS screening hit 1, cyclization led to an indazole series with improved in vitro activity. The lead compound identified (5r) is a potent JNK3 inhibitor with good exposure in mouse which makes it a suitable in vivo probe to interrogate the functions of JNK3 kinase in animal models of disease.
Co-reporter:Romain Noel, Xinyi Song, Youseung Shin, Subhashis Banerjee, Douglas Kojetin, Li Lin, Claudia H. Ruiz, Michael D. Cameron, Thomas P. Burris, Theodore M. Kamenecka
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 11) pp:3739-3742
Publication Date(Web):1 June 2012
DOI:10.1016/j.bmcl.2012.04.023
The design and synthesis of a novel series of Rev-erbα agonists is described. The development and optimization of the tetrahydroisoquinoline series was carried out from an earlier acyclic series of Rev-erbα agonists. Through the optimization of the scaffold 1, several potent compounds with good in vivo profiles were discovered.The design and synthesis of a novel series of Rev-erbα agonists is described. The development of N-acylated tetrahydroisoquinolines from an earlier acyclic series of Rev-erbα agonists has led to potent and efficacious compounds like 6j with good in vivo exposure.
Co-reporter:Rong Jiang, Xinyi Song, Purva Bali, Anthony Smith, Claudia Ruiz Bayona, Li Lin, Michael D. Cameron, Patricia H. McDonald, Paul J. Kenny, Theodore M. Kamenecka
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 12) pp:3890-3894
Publication Date(Web):15 June 2012
DOI:10.1016/j.bmcl.2012.04.122
A series of orexin receptor antagonists was synthesized based on a substituted piperidine scaffold. Through traditional medicinal chemistry structure–activity relationships (SAR), installation of various groups at the 3–6-positions of the piperidine led to modest enhancement in receptor selectivity. Compounds were profiled in vivo for plasma and brain levels in order to identify candidates suitable for efficacy in a model of drug addiction.A series of orexin receptor antagonists was synthesized based on a substituted piperidine scaffold. Through traditional medicinal chemistry structure–activity relationships (SAR), installation of various groups at the 3–6-positions of the piperidine led to modest enhancement in receptor selectivity. Compounds were profiled in vivo for plasma and brain levels in order to identify candidates suitable for efficacy in a model of drug addiction.
Co-reporter:Youseung Shin, Romain Noel, Subhashis Banerjee, Douglas Kojetin, Xinyi Song, Yuanjun He, Li Lin, Michael D. Cameron, Thomas P. Burris, Theodore M. Kamenecka
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 13) pp:4413-4417
Publication Date(Web):1 July 2012
DOI:10.1016/j.bmcl.2012.04.126
The structure–activity relationship study of a small molecule Rev-erbα agonist is reported. The potency and efficacy of the agonists in a cell-based assay were optimized as compared to the initial lead. Modest mouse pharmacokinetics coupled with an improved in vitro profile make 12e a suitable in vivo probe to interrogate the functions of Rev-erbα in animal models of disease.The synthesis and optimization of a series of small molecule Rev-erbα agonists is described. Compounds with good cell-based potency and efficacy and good mouse exposure provides for in vivo probes to interrogate the function of Rev-erbα in animal models of disease.
Co-reporter:Xinyi Song, Xiaohai Li, Claudia H. Ruiz, Yan Yin, Yangbo Feng, Theodore M. Kamenecka, Michael D. Cameron
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 4) pp:1611-1614
Publication Date(Web):15 February 2012
DOI:10.1016/j.bmcl.2011.12.125
Cytochrome P450s are the major family of enzymes responsible for the oxidative metabolism of pharmaceuticals and xenobiotics. CYP3A4 and CYP3A5 have been shown to have overlapping substrate and inhibitor profiles and their inhibition has been demonstrated to be involved in numerous pharmacokinetic drug–drug interactions. Here we report the first highly selective CYP3A4 inhibitor optimized from an initial lead with ≈30-fold selectivity over CYP3A5 to yield a series of compounds with greater than 1000-fold selectivity.
Co-reporter:Romain Noël, Youseung Shin, Xinyi Song, Yuanjun He, Marcel Koenig, Weimin Chen, Yuan Yuan Ling, Li Lin, Claudia H. Ruiz, Phil LoGrasso, Michael D. Cameron, Derek R. Duckett, Theodore M. Kamenecka
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 9) pp:2732-2735
Publication Date(Web):1 May 2011
DOI:10.1016/j.bmcl.2010.11.104
The design and synthesis of a novel series of c-jun N-terminal kinase (JNK) inhibitors is described. The development of the 4-(pyrazol-3-yl)-pyridine series was discovered from an earlier pyrimidine series of JNK inhibitors. Through the optimization of the scaffold 2, several potent compounds with good in vivo profiles were discovered.The design and synthesis of a novel series of c-jun N-terminal kinase (JNK) inhibitors is described. The development of the 4-(pyrazol-3-yl)-pyridine series was discovered from an earlier pyrimidine series of JNK inhibitors. Through the optimization of the scaffold 2, several potent compounds with good in vivo profiles were discovered.
Co-reporter:Xinyi Song, Weimin Chen, Li Lin, Claudia H. Ruiz, Michael D. Cameron, Derek R. Duckett, Theodore M. Kamenecka
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 23) pp:7072-7075
Publication Date(Web):1 December 2011
DOI:10.1016/j.bmcl.2011.09.090
The design and synthesis of a novel series of c-jun N-terminal kinase (JNK3) inhibitors is described. The development and optimization of the 2-phenoxypyridine series was carried out from an earlier pyrimidine series of JNK1 inhibitors. Through the optimization of the scaffold 2, several potent compounds with good in vivo profiles were discovered.The design and synthesis of a novel series of c-jun N-terminal kinase (JNK) inhibitors is described. The development of the 2-phenoxypyridine series was discovered from an earlier pyrimidine series of JNK inhibitors. Through the optimization of the scaffold 5, several potent compounds with good in vivo profiles were developed.
Co-reporter:Youseung Shin, Weiming Chen, Jeff Habel, Derek Duckett, Yuan Yuan Ling, Marcel Koenig, Yuanjun He, Tomas Vojkovsky, Philip LoGrasso, Theodore M. Kamenecka
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 12) pp:3344-3347
Publication Date(Web):15 June 2009
DOI:10.1016/j.bmcl.2009.03.086
A novel series of c-jun N-terminal kinase (JNK) inhibitors were designed and developed from a high-throughput-screening hit. Through the optimization of the piperazine amide 1, several potent compounds were discovered. The X-ray crystal structure of 4g showed a unique binding mode different from other well known JNK3 inhibitors.A novel series of c-jun N-terminal kinase (JNK) inhibitors were designed and developed from high-throughput-screening lead 1.
Co-reporter:Theodore M. Kamenecka, Céline Bonnefous, Steven Govek, Jean-Michel Vernier, John Hutchinson, Janice Chung, Grace Reyes-Manalo, Jeffery J. Anderson
Bioorganic & Medicinal Chemistry Letters 2005 Volume 15(Issue 19) pp:4350-4353
Publication Date(Web):1 October 2005
DOI:10.1016/j.bmcl.2005.06.059
Modulation of the metabotropic glutamate subtype 5 (mGlu5) receptor may be useful in the treatment of a variety of central nervous system disorders. Herein, we report on the discovery, synthesis, and biological evaluation of dipyridyl amines as small molecule mGlu5 antagonists.The metabotropic glutamate subtype 5 (mGlu5) receptor has been implicated in a number of CNS disorders. Herein, we report on the discovery, synthesis, and biological evaluation of dipyridyl amines as small molecule mGlu5 antagonists.
Co-reporter:Zhuang Jin, Hua Lin, Sathish Srinivasan, Jerome C. Nwachukwu, Nelson Bruno, Patrick R. Griffin, Kendall W. Nettles, Theodore M. Kamenecka
Bioorganic & Medicinal Chemistry Letters (15 January 2017) Volume 27(Issue 2) pp:
Publication Date(Web):15 January 2017
DOI:10.1016/j.bmcl.2016.11.007
Adverse effects of glucocorticoids could be limited by developing new compounds that selectively modulate anti-inflammatory activity of the glucocorticoid receptor (GR). We have synthesized a novel series of steroidal GR ligands, including potent agonists, partial agonists and antagonists with a wide range of effects on inhibiting secretion of interleukin-6. Some of these new ligands were designed to directly impact conformational stability of helix-12, in the GR ligand-binding domain (LBD). These compounds modulated GR activity and glucocorticoid-induced gene expression in a manner that was inversely correlated to the degree of inflammatory response. In contrast, compounds designed to directly modulate LBD epitopes outside helix-12, led to dissociated levels of GR-mediated gene expression and inflammatory response. Therefore, these new series of compounds and their derivatives will be useful to dissect the ligand-dependent features of GR signaling specificity.Adverse effects of glucocorticoids could be limited by developing new compounds that selectively modulate anti-inflammatory activity of the glucocorticoid receptor (GR). We have synthesized a novel series of steroidal GR ligands, including potent agonists, partial agonists and antagonists with a wide range of effects on inhibiting secretion of interleukin-6. Some of these new ligands were designed to directly impact conformational stability of helix-12, in the GR ligand-binding domain (LBD). These compounds modulated GR activity and glucocorticoid-induced gene expression in a manner that was inversely correlated to the degree of inflammatory response. In contrast, compounds designed to directly modulate LBD epitopes outside helix-12, led to dissociated levels of GR-mediated gene expression and inflammatory response. Therefore, these new series of compounds and their derivatives will be useful to dissect the ligand-dependent features of GR signaling specificity.
.jpg)
![2-Fluoro-4'-[[4-(4-pyridinylmethyl)-1-piperazinyl]methyl]-α,α-bis(trifluoromethyl)-[1,1'-biphenyl]-4-methanol](http://img.cochemist.com/ccimg/1359200/1359164-11-6.png)
![2-Fluoro-4'-[[4-(4-pyridinylmethyl)-1-piperazinyl]methyl]-α,α-bis(trifluoromethyl)-[1,1'-biphenyl]-4-methanol](http://img.cochemist.com/ccimg/1359200/1359164-11-6_b.png)
![4'-[(5-{[(1s)-1-(4-bromophenyl)ethyl]carbamoyl}-2,3-dimethyl-1h-i Ndol-1-yl)methyl]-2-biphenylcarboxylic Acid](http://img.cochemist.com/ccimg/1338300/1338259-06-5.png)
![4'-[(5-{[(1s)-1-(4-bromophenyl)ethyl]carbamoyl}-2,3-dimethyl-1h-i Ndol-1-yl)methyl]-2-biphenylcarboxylic Acid](http://img.cochemist.com/ccimg/1338300/1338259-06-5_b.png)












![Benzamide, N-[1,1'-biphenyl]-2-yl-2-chloro-5-nitro-](http://img.cochemist.com/ccimg/372100/372093-22-6.png)
![Benzamide, N-[1,1'-biphenyl]-2-yl-2-chloro-5-nitro-](http://img.cochemist.com/ccimg/372100/372093-22-6_b.png)
![Benzamide, N-[1,1'-biphenyl]-3-yl-2-chloro-5-nitro-](http://img.cochemist.com/ccimg/372100/372093-17-9.png)
![Benzamide, N-[1,1'-biphenyl]-3-yl-2-chloro-5-nitro-](http://img.cochemist.com/ccimg/372100/372093-17-9_b.png)


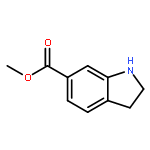
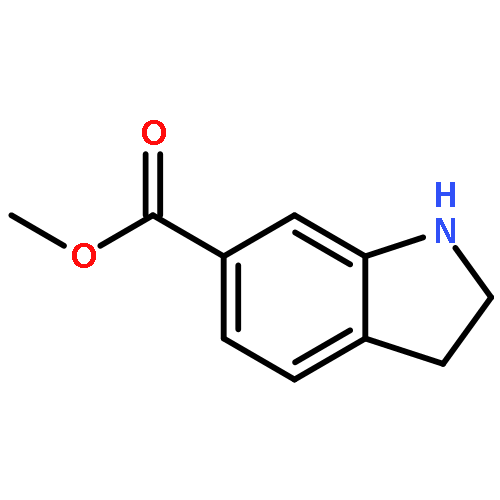






![Benzamide, N-[1,1'-biphenyl]-4-yl-2-chloro-5-nitro-](http://img.cochemist.com/ccimg/315200/315194-71-9.png)
![Benzamide, N-[1,1'-biphenyl]-4-yl-2-chloro-5-nitro-](http://img.cochemist.com/ccimg/315200/315194-71-9_b.png)


![2-chloro-N-[4-(1,1,1,3,3,3-hexafluoro-2-hydroxypropan-2-yl)phenyl]benzamide](http://img.cochemist.com/ccimg/303200/303126-97-8.png)
![2-chloro-N-[4-(1,1,1,3,3,3-hexafluoro-2-hydroxypropan-2-yl)phenyl]benzamide](http://img.cochemist.com/ccimg/303200/303126-97-8_b.png)


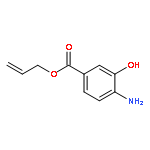
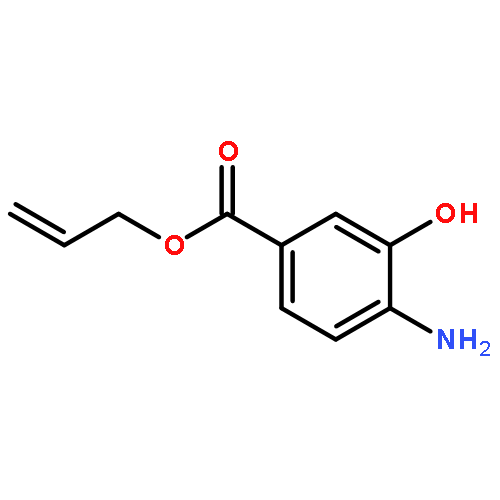








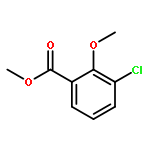
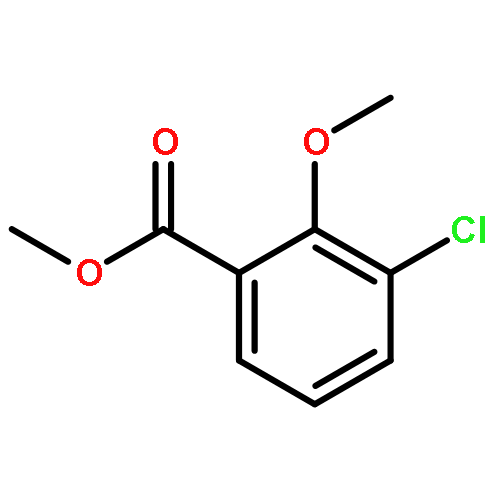
![[1-(4-ISOPROPYLPHENYL)ETHYL]HYDRAZINE](http://img.cochemist.com/ccimg/76300/76283-12-0.png)
![[1-(4-ISOPROPYLPHENYL)ETHYL]HYDRAZINE](http://img.cochemist.com/ccimg/76300/76283-12-0_b.png)
![BENZO[A]PYRENE-7-D(9CI)](http://img.cochemist.com/ccimg/76300/76279-30-6.png)
![BENZO[A]PYRENE-7-D(9CI)](http://img.cochemist.com/ccimg/76300/76279-30-6_b.png)
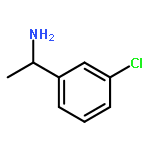
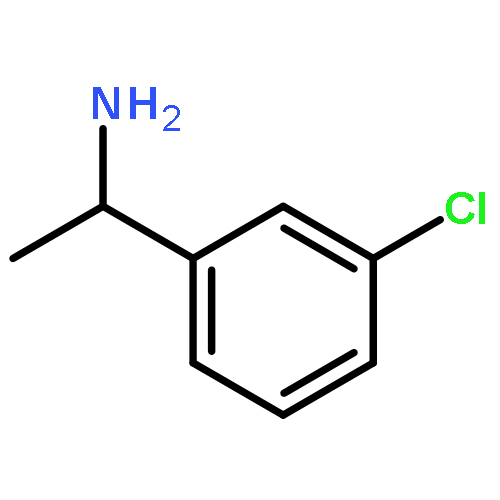
![[1,1'-Biphenyl]-3-sulfonyl chloride](http://img.cochemist.com/ccimg/65700/65685-01-0.png)
![[1,1'-Biphenyl]-3-sulfonyl chloride](http://img.cochemist.com/ccimg/65700/65685-01-0_b.png)































![4'-[(2,3-dimethyl-5-{[(1s)-1-(4-nitrophenyl)ethyl]carbamoyl}-1h-i Ndol-1-yl)methyl]-2-biphenylcarboxylic Acid](http://img.cochemist.com/ccimg/1338300/1338259-05-4.png)
![4'-[(2,3-dimethyl-5-{[(1s)-1-(4-nitrophenyl)ethyl]carbamoyl}-1h-i Ndol-1-yl)methyl]-2-biphenylcarboxylic Acid](http://img.cochemist.com/ccimg/1338300/1338259-05-4_b.png)