Co-reporter:Yang Gao, De-Yong Ye, Wei-Cheng Zhou, Yong Chu
European Journal of Medicinal Chemistry 2017 Volume 135(Volume 135) pp:
Publication Date(Web):28 July 2017
DOI:10.1016/j.ejmech.2017.04.039
•Novel benzothiazinones were synthesized and proved to be potent and selective GSK 3β inhibitors.•The mechanism of inhibition to GSK 3β was proved to be non-ATP competitive.•Most of these inhibitors showed good antitumor activity against ovarian cancer cell lines.•Compound 20g could arrest G0/G1 cell cycle and induce apoptosis of A2780 cells.•Compound 20g also exhibited moderate antitumor activity in a female BALB/C nude mice model.Glycogen synthase kinase 3β (GSK 3β) is a highly conserved serine/threonine kinase, and its roles in cancer remain controversial. Cumulative evidence supported that GSK 3β inhibitors could suppress ovarian cancer (OC) development in vitro and made a new direction for ovarian cancer treatment. Here, we reported a series of novel substituted benzothiazinones as non-ATP competitive inhibitors of GSK 3β. Further studies showed that most of them had antiproliferative activities in ovarian cancer cell lines in vitro. As the most promising candidate of them, compound 20g induced cells apoptosis, arrested the cell cycle at the G1 phase in the A2780 cell line and showed moderate suppression efficacy in a female BALB/C nude mice model. All of the results demonstrated that compound 20g, as the first reported non-ATP competitive small molecule inhibitor of GSK 3β with suppression efficacy on ovarian cancer both in vitro and in vivo, might represent a potential candidate for the treatment of OC.Download high-res image (134KB)Download full-size image
Co-reporter:Xiang-Yu Qi, Yang Cao, Ya-Li Li, Ming-Guang Mo, Lu Zhou, De-Yong Ye
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 15(Issue 15) pp:
Publication Date(Web):1 August 2017
DOI:10.1016/j.bmcl.2017.05.074
Sphingomyelin synthase (SMS) is a key enzyme in sphingomyelin biosynthetic pathway, whose activity is highly related to the atherosclerosis progression. SMS2 could serve as a promising therapeutic target for atherosclerosis. Based on the structure of lead compound D2, a series of oxazolopyridine derivatives were designed, synthesized, and their inhibitory activities against purified SMS1 and SMS2 enzymes were evaluated respectively. The representative molecules QY4 and QY16 possess micromolar inhibitory activities against SMS2 and excellent isoform preferences over SMS1, qualified to be selected as potential molecules in further discovery of specific SMS2 inhibitors.Download high-res image (136KB)Download full-size image
Co-reporter:Ya-li Li, Xiang-yu Qi, Hui Jiang, Xiao-dong Deng, Yan-ping Dong, Ting-bo Ding, Lu Zhou, Peng Men, Yong Chu, Ren-xiao Wang, Xian-cheng Jiang, De-yong Ye
Bioorganic & Medicinal Chemistry 2015 23(18) pp: 6173-6184
Publication Date(Web):
DOI:10.1016/j.bmc.2015.07.060
Co-reporter:Xiaodong Deng, Fu Lin, Ya Zhang, Yan Li, Lu Zhou, Bin Lou, Yue Li, Jibin Dong, Tingbo Ding, Xiancheng Jiang, Renxiao Wang, Deyong Ye
European Journal of Medicinal Chemistry 2014 Volume 73() pp:1-7
Publication Date(Web):12 February 2014
DOI:10.1016/j.ejmech.2013.12.002
•Sphingomyelin synthase is a novel potential drug target for metabolic diseases.•The small-molecule SMS inhibitors discovered by rational design are firstly revealed.•The hit D2 was demonstrated to be an effective SMS inhibitor both in vitro and in vivo.Sphingomyelin synthase (SMS), which catalyzes ceramide as one of the substrates to produce sphingomyelin, is a critical factor in the sphingolipid biosynthesis pathway. Recent studies indicated that SMS could serve as a novel potential drug target for the treatment of various metabolic diseases such as insulin resistance and atherosclerosis. However, very few small-molecule inhibitors of SMS are known. In this study, we performed structure-based virtual screening in combination with chemical synthesis and bioassay and discovered a class of small-molecule SMS inhibitors. The most potent compound exhibited an IC50 value lower than 20 μM in an in vitro enzymatic assay. To the best of our knowledge, this is the first time that small-molecule SMS inhibitors with potency close to the micromolar range are publicly revealed. The structure–activity relationship demonstrated by this class of compounds provides insights into the structural features that are essential for effective SMS inhibition.The hit D2 was supposed be a potential molecular tool in SMS bio-function studies as well as for developing a new class of therapeutic drug treating various metabolic diseases.
Co-reporter:Peng Zhang, Hai-Rong Hu, Shi-Hui Bian, Zhao-Hui Huang, Yong Chu, De-Yong Ye
European Journal of Medicinal Chemistry 2013 Volume 61() pp:95-103
Publication Date(Web):March 2013
DOI:10.1016/j.ejmech.2012.09.021
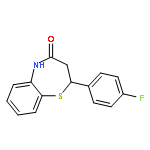
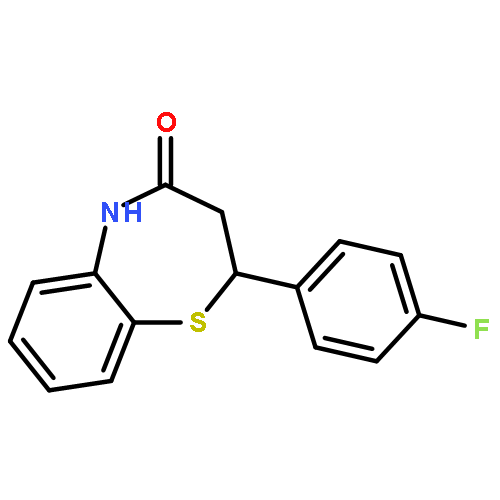
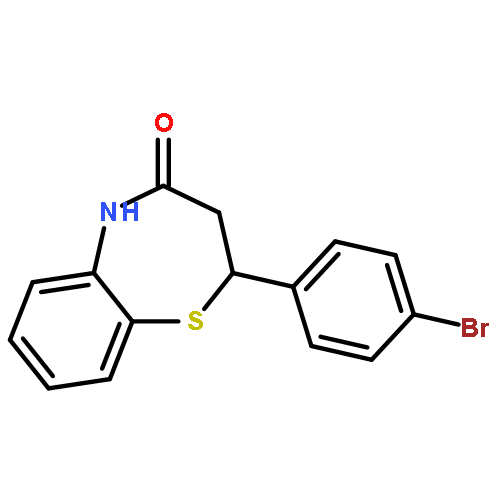
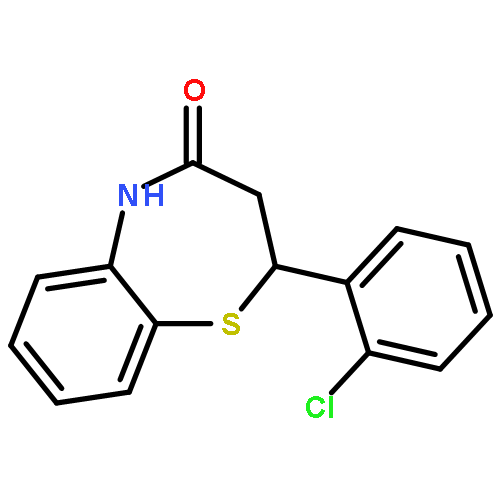
Glycogen synthase kinase-3β (GSK-3β) plays a key role in type II diabetes and Alzheimer's diseases, to which non-ATP competitive inhibitors represent an effectively therapeutical approach due to their good specificity. Herein, a series of small molecules benzothiazepinones (BTZs) as novel non-ATP competitive inhibitors of GSK-3β have been designed and synthesized. The in vitro evaluation performed by luminescent assay showed most BTZ derivatives have inhibitory effects in micromolar scale. Among them compounds 6l, 6t and 6v have the IC50 values of 25.0 μM, 27.8 μM and 23.0 μM, respectively. Moreover 6v is devoid of any inhibitory activity in the assays to other thirteen protein kinases. Besides, SAR is analyzed and a hypothetical enzymatic binding mode is proposed by molecular docking study, which would be useful for new candidates design.Graphical abstract2,3-dihydro-2,5-disubstituted-1,5-benzothiazepin-4(5H)-one.Highlights► A series of 2,3-dihydro-2,5-disubstituted-1,5-benzothiazepin-4(5H)-one were synthesized. ► These compounds showed GSK-3β inhibitory activities in vitro. ► Kinetic analysis showed compound 6v is non-ATP GSK-3β inhibitor. ► Molecular docking studies suggested the putative binding modes.
Co-reporter:Xing Gao;Haojun Gong;Peng Men;Lu Zhou
Chinese Journal of Chemistry 2013 Volume 31( Issue 9) pp:1164-1170
Publication Date(Web):
DOI:10.1002/cjoc.201300079
Abstract
A novel series of eight SMS and sPLA2 dual inhibitors containing indole and α-amino cyanide fragments of different length and substitution position was synthesized and evaluated by three different in vitro assays. Biological evaluation showed that all compounds provided inhibitory effects against SMS (about 50% inhibition at 100 µmol/L) and sPLA2 (14–32 µmol/L). All the compounds had the SMS activity better than the positive control compound D609 in SMS2 homogenate, with compounds 5b and 5e ideal for liver homogenate and SMS2 high expression cell homogenate, respectively.
Co-reporter:Wu-Hui Song, Ming-Ming Liu, Dong-Wei Zhong, Ye-lin Zhu, Mike Bosscher, Lu Zhou, De-Yong Ye, Zheng-Hong Yuan
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 16) pp:4528-4531
Publication Date(Web):15 August 2013
DOI:10.1016/j.bmcl.2013.06.045
A series of diketo tetrazoles and diketo triazoles were designed and synthesized as bioisosteres of α,γ-diketo acid, the active site inhibitor of HCV (Hepatitis C virus) polymerase NS5B. Among the synthesized compounds, 4-(4-fluorobenzyloxy)phenyl diketo triazole (30) exhibited anti-HCV activity with an EC50 value of 3.9 μM and an SI value more than 128. The reduction of viral protein and mRNA levels were also validated, supporting the anti-HCV activity of compound 30. These results provide convincing evidence that the diketo tetrazoles and diketo triazoles can be developed as bioisosteres of α,γ-diketo acid to exhibit potent inhibitory activity against HCV.A series of diketo tetrazoles and diketo triazoles were designed and synthesized as bioisosteres of α,γ-diketo acid. Among them, compound 30 exhibited potent anti-HCV activity with an EC50 value of 3.9 μM and an SI value more than 128. The reduction of viral protein and mRNA levels were also validated.
Co-reporter:Ming-Ming Liu, Lu Zhou, Pei-Lan He, Yi-Nan Zhang, Jia-Yi Zhou, Qing Shen, Xin-Wen Chen, Jian-Ping Zuo, Wei Li, De-Yong Ye
European Journal of Medicinal Chemistry 2012 Volume 52() pp:33-43
Publication Date(Web):June 2012
DOI:10.1016/j.ejmech.2012.03.002
Common feature based pharmacophore and structure-based docking approaches have been employed in the identification of novel anti-HCV candidates from our in-house database. A total of 31 hits identified in silico were screened in vitro assay. 20 Compounds demonstrated anti-HCV activities (EC50 < 50 μM), including two naturally occurring flavones apigenin (21) and luteolin (22) with low micromole EC50 values and three compounds (23, 24 and 25) of novel scaffolds with moderate potencies. In addition, pharmacophore refinement was also conducted based on the current knowledge of flavone-derived anti-HCV candidates and the results of combined in silico and in vitro assays.Combined pharmacophore and structure-based approaches were employed in screening novel anti-HCV candidates and 20 compounds were found to be active in vitro including two naturally occurring flavones.Highlights► The HipHop pharmacophore model was generated for NS5B inhibitors. ► Virtual screening was performed based on both pharmacophore and docking models. ► Several selected compounds were discovered to possess anti-HCV activities.
Co-reporter:Peng Zhang, Hai-Rong Hu, Zhao-Hui Huang, Jia-Yi Lei, Yong Chu, De-Yong Ye
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 23) pp:7232-7236
Publication Date(Web):1 December 2012
DOI:10.1016/j.bmcl.2012.09.043
Glycogen synthase kinase-3β (GSK-3β) is an important serine/threonine kinase that has been proved as a key target for neurodegenerative diseases and diabetes. Up to date, most of known inhibitors are bound to the ATP-binding pocket of GSK-3β, which might lead widespread effects due to the high homology between kinases. Recently, some of its non-ATP competitive inhibitors had been confirmed having therapeutical effects owing to their high selectivity. This finding opens a new pathway to study hopeful drugs for treatment of these diseases. However, it is still a challenge nowadays on how to efficiently find non-ATP competitors. Here, we successfully discovered a novel scaffold of benzothiazepinones (BTZs) as selective non-ATP competitive GSK-3β inhibitors through virtual screening approach. A 3D receptor model of substrate binding site of GSK-3β was constructed and applied to screen against drug-like Maybridge database through Autodock program. BTZ compounds were top ranked as efficient hits and were then synthesized for further screening. Among them, the representative compound 4j showed activity to GSK-3β (IC50: 25 μM) in non-ATP competitive mechanism, and nearly no inhibitory effect on other 10 related protein kinases. Overall, the results point out that BTZ compounds might be useful in treatment of Alzheimer’s disease and diabetes mellitus as novel GSK-3β inhibitors. It also suggests, on the other hand, that virtual screening would provide a valuable tool in combination with in vitro assays for the identification of novel selective and potent inhibitors.
Co-reporter:Ya Zhang;Fu Lin;Xiaodong Deng;Renxiao Wang
Chinese Journal of Chemistry 2011 Volume 29( Issue 8) pp:1567-1575
Publication Date(Web):
DOI:10.1002/cjoc.201180282
Abstract
Sphingomyelin synthase (SMS) produces sphingomyelin and diacylglycerol from ceramide and phosphatidylcholine. It plays an important role in cell survival and apoptosis, inflammation, and lipid homeostasis, and therefore has been noticed in recent years as a novel potential drug target. In this study, we combined homology modeling, molecular docking, molecular dynamics simulation, and normal mode analysis to derive a three-dimensional structure of human sphingomyelin synthase (hSMS1) in complex with sphingomyelin. Our model provides a reasonable explanation on the catalytic mechanism of hSMS1. It can also explain the high selectivity of hSMS1 towards phosphocholine and sphingomyelin as well as some other known experimental results about hSMS1. Moreover, we also derived a complex model of D609, the only known small-molecule inhibitor of hSMS1 so far. Our hSMS1 model may serve as a reasonable structural basis for the discovery of more effective small-molecule inhibitors of hSMS1.
Co-reporter:Kaiqing Ma, Chunbo Zhang, Mingming Liu, Yong Chu, Lu Zhou, Changqi Hu, Deyong Ye
Tetrahedron Letters 2010 Volume 51(Issue 14) pp:1870-1872
Publication Date(Web):7 April 2010
DOI:10.1016/j.tetlet.2010.02.001
The first, stereospecific, and elegant synthesis of the natural product (+)-Carainterol A was developed by using the Robinson annulation reaction as a key step to build the eudesmane sketeton.The first, stereospecific, and elegant synthesis of the natural product (+)-Carainterol A was developed by using the Robinson annulation reaction as a key step to build the eudesmane sketeton.
Co-reporter:Yan Wei Hu, Li Zuo, De Yong Ye, Wen Hu Duan
Chinese Chemical Letters 2009 Volume 20(Issue 10) pp:1157-1160
Publication Date(Web):October 2009
DOI:10.1016/j.cclet.2009.04.028
A new debenzylation of benzyl esters by silica-supported sodium hydrogen sulfate is described. The debenzylation could be achieved selectively and efficiently in good to excellent yields without affecting sensitive functional groups such as nitro, unsaturated bonds, and ethyl ester.
Co-reporter:Ya-Ju Zhou, Li-Ping Zhu, Yun Tang, De-Yong Ye
European Journal of Medicinal Chemistry 2007 Volume 42(Issue 7) pp:977-984
Publication Date(Web):July 2007
DOI:10.1016/j.ejmech.2006.12.029
Structure-based 3D-QSAR studies were performed on a series of novel heteroarylpiperazine derivatives as 5-HT3 receptor antagonists with comparative molecular field analysis (CoMFA) and comparative molecular similarity indices analysis (CoMSIA) methods. The compounds were initially docked into the binding pocket of the homology model of 5-HT3 receptor using GOLD program. The docked conformations with the highest score were then extracted and used to build the 3D-QSAR models, with cross-validated rcv2 values 0.716 and 0.762 for CoMFA and CoMSIA, respectively. The CoMFA and CoMSIA contour plots were also fitted into the 3D structural model of the receptor to identify the key interactions between them, which might be helpful for designing new potent 5-HT3 receptor antagonists.3D-QSAR studies were performed on 28 heteroarylpiperazine derivatives as 5-HT3 receptor ligands by CoMFA and CoMSIA methods to discover new potent and selective 5-HT3R antagonists.
Co-reporter:Li-Ping Zhu;De-Yong Ye;Yun Tang
Journal of Molecular Modeling 2007 Volume 13( Issue 1) pp:121-131
Publication Date(Web):2007 January
DOI:10.1007/s00894-006-0131-1
Structure-based 3D-QSAR studies were performed on 20 thiazoles against their binding affinities to the 5-HT3 receptor with comparative molecular field analysis (CoMFA) and comparative molecular similarity indices analysis (CoMSIA). The thiazoles were initially docked into the binding pocket of a human 5-HT3A receptor homology model, constructed on the basis of the crystal structure of the snail acetylcholine binding protein (AChBP), using the GOLD program. The docked conformations were then extracted and used to build the 3D-QSAR models, with cross-validated \( r^{2}_{{cv}} \) values 0.785 and 0.744 for CoMFA and CoMSIA, respectively. An additional five molecules were used to validate the models further, giving satisfactory predictive \( r^{2} \) values of 0.582 and 0.804 for CoMFA and CoMSIA, respectively. The results would be helpful for the discovery of new potent and selective 5-HT3 receptor antagonists.






















![Hexanamide,N-[(1S,2R,3E)-2-hydroxy-1-(hydroxymethyl)-3-heptadecenyl]-6-[(7-nitro-2,1,3-benzoxadiazol-4-yl)amino]-](/data/chemimg/996400/94885-02-6.png)
![Hexanamide,N-[(1S,2R,3E)-2-hydroxy-1-(hydroxymethyl)-3-heptadecenyl]-6-[(7-nitro-2,1,3-benzoxadiazol-4-yl)amino]-](/data/chemimg/996400/94885-02-6_b.png)
![Hexanamide,N-[2-hydroxy-1-(hydroxymethyl)-3-heptadecen-1-yl]-6-[(7-nitro-2,1,3-benzoxadiazol-4-yl)amino]-](/data/chemimg/985200/86701-10-2.png)
![Hexanamide,N-[2-hydroxy-1-(hydroxymethyl)-3-heptadecen-1-yl]-6-[(7-nitro-2,1,3-benzoxadiazol-4-yl)amino]-](/data/chemimg/985200/86701-10-2_b.png)
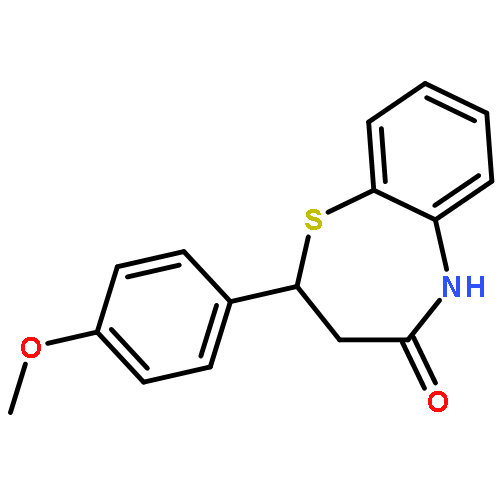
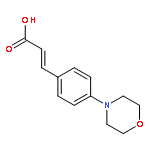
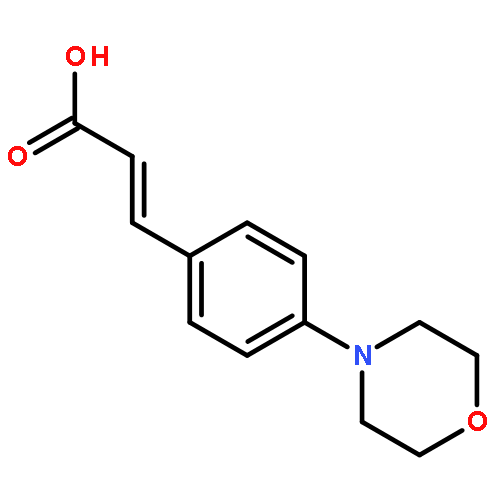
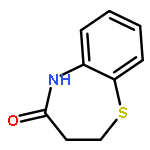
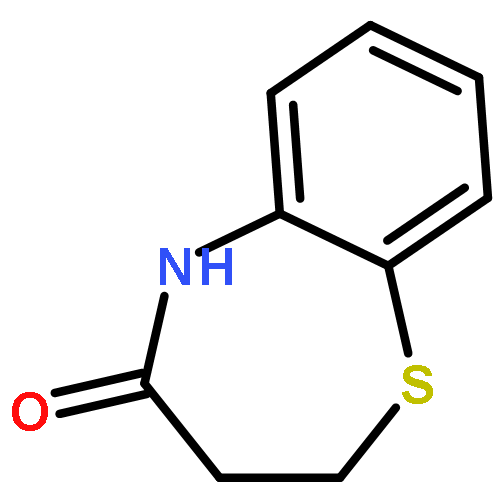
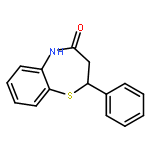
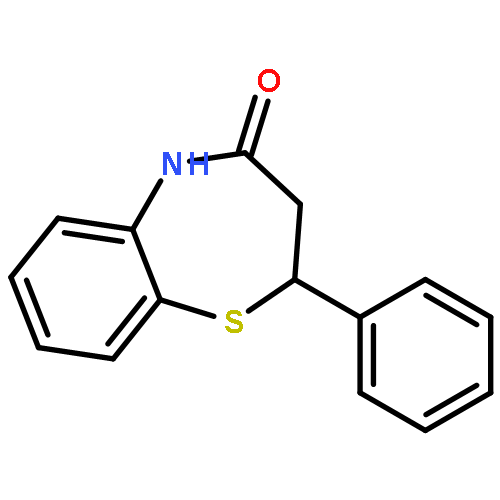
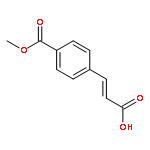
![Acetamide, 2-chloro-N-[2-(3,4-dihydroxyphenyl)ethyl]-](http://img.cochemist.com/ccimg/17700/17639-51-9.png)
![Acetamide, 2-chloro-N-[2-(3,4-dihydroxyphenyl)ethyl]-](http://img.cochemist.com/ccimg/17700/17639-51-9_b.png)


![2-(Thiophen-2-yl)-2,3-dihydrobenzo[b][1,4]thiazepin-4(5H)-one](http://img.cochemist.com/ccimg/5900/5871-68-1.png)
![2-(Thiophen-2-yl)-2,3-dihydrobenzo[b][1,4]thiazepin-4(5H)-one](http://img.cochemist.com/ccimg/5900/5871-68-1_b.png)



![4H-1-Benzopyran-4-one,3,5,7-tris(acetyloxy)-2-[3,4-bis(acetyloxy)phenyl]-](http://img.cochemist.com/ccimg/1100/1064-06-8.png)
![4H-1-Benzopyran-4-one,3,5,7-tris(acetyloxy)-2-[3,4-bis(acetyloxy)phenyl]-](http://img.cochemist.com/ccimg/1100/1064-06-8_b.png)


![Acetamide, 2-chloro-N-[2-(trifluoromethoxy)phenyl]-](http://img.cochemist.com/ccimg/656300/656227-27-9.png)
![Acetamide, 2-chloro-N-[2-(trifluoromethoxy)phenyl]-](http://img.cochemist.com/ccimg/656300/656227-27-9_b.png)
![2-chloro-n-[(4-fluorophenyl)methyl]acetamide](http://img.cochemist.com/ccimg/257300/257279-75-7.png)
![2-chloro-n-[(4-fluorophenyl)methyl]acetamide](http://img.cochemist.com/ccimg/257300/257279-75-7_b.png)
![Acetamide,2-chloro-N-[(4-chlorophenyl)methyl]-](http://img.cochemist.com/ccimg/99600/99585-88-3.png)
![Acetamide,2-chloro-N-[(4-chlorophenyl)methyl]-](http://img.cochemist.com/ccimg/99600/99585-88-3_b.png)
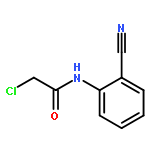
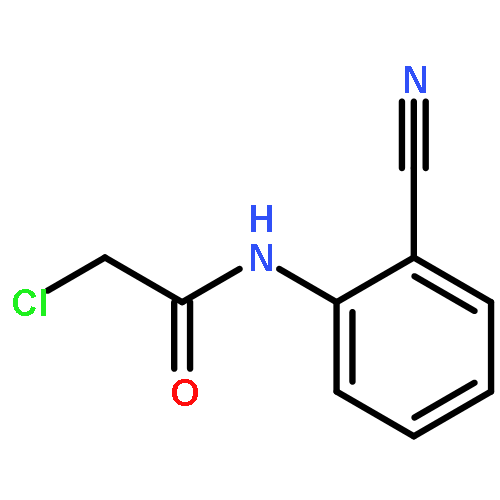
![Acetamide, N-[(4-bromophenyl)methyl]-2-chloro-](http://img.cochemist.com/ccimg/25000/24942-06-1.png)
![Acetamide, N-[(4-bromophenyl)methyl]-2-chloro-](http://img.cochemist.com/ccimg/25000/24942-06-1_b.png)