Co-reporter:Mam Y. Mboge, Brian P. Mahon, Nicole Lamas, Lillien Socorro, Fabrizio Carta, Claudiu T. Supuran, Susan C. Frost, Robert McKenna
European Journal of Medicinal Chemistry 2017 Volume 132(Volume 132) pp:
Publication Date(Web):26 May 2017
DOI:10.1016/j.ejmech.2017.03.026
•USBs preferentially bind to tumor associated CA IX and CA XII over CA II.•USBs have flexible and rotatable tail moieties.•USBs adopt a range of conformations and participate in diverse interactions within the active sites of CA II and CA IX.•Active site residue 131 is important for the isoform specificity and selectivity of USBs.Ureido-substituted benzenesulfonamides (USBs) show great promise as selective and potent inhibitors for human carbonic anhydrase hCA IX and XII, with one such compound (SLC-0111/U-F) currently in clinical trials (clinical trials.gov, NCT02215850). In this study, the crystal structures of both hCA II (off-target) and an hCA IX-mimic (target) in complex with selected USBs (U-CH3, U-F, and U-NO2), at resolutions of 1.9 Å or better, are presented, and demonstrate differences in the binding modes within the two isoforms. The presence of residue Phe 131 in hCA II causes steric hindrance (U-CH3, 1765 nM; U-F, 960 nM; U-NO2, 15 nM) whereas in hCA IX (U-CH3, 7 nM; U-F, 45 nM; U-NO2, 1 nM) and hCA XII (U-CH3, 6 nM; U-F, 4 nM; U-NO2, 6 nM), 131 is a Val and Ala, respectively, allows for more favorable binding. Our results provide insight into the mechanism of USB selective inhibition and useful information for structural design and drug development, including synthesis of hybrid USB compounds with improved physiochemical properties.Download high-res image (197KB)Download full-size image
Co-reporter:Brian P. Mahon, Justin J. Kurian, Carrie L. Lomelino, Ian R. Smith, Lilien Socorro, Antonette Bennett, Alex M. Hendon, Paul Chipman, Daniel A. Savin, Mavis Agbandje-McKenna, and Robert McKenna
Crystal Growth & Design 2016 Volume 16(Issue 11) pp:6214
Publication Date(Web):September 28, 2016
DOI:10.1021/acs.cgd.6b00643
The advances of serial crystallography techniques at synchrotron and X-ray free electron laser facilities have made possible the acquisition of useable data sets to determine 3-dimensional structures of macromolecules from micro- to nanosized crystals. In addition, the same technological hallmarks have contributed significantly to the field of time-resolved crystallography. However, the production of usable crystalline slurries for serial crystallographic experiments has been one of the limiting factors and contributes to an alternative sample “bottleneck” in crystal growth. In this study, we propose a method: labeled microbatch mixing (MBM), which has the capability to produce large quantities of microcrystals of macromolecules suitable for serial crystallographic experiments. This is shown to be successful for producing lysozyme, carbonic anhydrase, and adeno-associated virus crystals. MBM takes advantage of secondary nucleation induced by mixing via the application of steady agitation during the crystallization process. This leads to excessive nucleation, resulting in large quantities of well-diffracting microcrystals. MBM therefore presents a method that can potentially be applied to a range of macromolecules and a possible simple protocol to produce microcrystals for serial crystallographic experiments.
Co-reporter:Brian P. Mahon, Avni Bhatt, Lilien Socorro, Jenna M. Driscoll, Cynthia Okoh, Carrie L. Lomelino, Mam Y. Mboge, Justin J. Kurian, Chingkuang Tu, Mavis Agbandje-McKenna, Susan C. Frost, and Robert McKenna
Biochemistry 2016 Volume 55(Issue 33) pp:4642
Publication Date(Web):July 20, 2016
DOI:10.1021/acs.biochem.6b00243
Human carbonic anhydrase IX (hCA IX) expression in many cancers is associated with hypoxic tumors and poor patient outcome. Inhibitors of hCA IX have been used as anticancer agents with some entering Phase I clinical trials. hCA IX is transmembrane protein whose catalytic domain faces the extracellular tumor milieu, which is typically associated with an acidic microenvironment. Here, we show that the catalytic domain of hCA IX (hCA IX-c) exhibits the necessary biochemical and biophysical properties that allow for low pH stability and activity. Furthermore, the unfolding process of hCA IX-c appears to be reversible, and its catalytic efficiency is thought to be correlated directly with its stability between pH 3.0 and 8.0 but not above pH 8.0. To rationalize this, we determined the X-ray crystal structure of hCA IX-c to 1.6 Å resolution. Insights from this study suggest an understanding of hCA IX-c stability and activity in low-pH tumor microenvironments and may be applicable to determining pH-related effects on enzymes.
Co-reporter:Carrie L. Lomelino, Brian P. Mahon, Robert McKenna, Fabrizio Carta, Claudiu T. Supuran
Bioorganic & Medicinal Chemistry 2016 Volume 24(Issue 5) pp:976-981
Publication Date(Web):1 March 2016
DOI:10.1016/j.bmc.2016.01.019
SLC-0111 (4-(4-fluorophenylureido)-benzenesulfonamide) is the first carbonic anhydrase (CA, EC 4.2.1.1) IX inhibitor to reach phase I clinical trials as an antitumor/antimetastatic agent. Here we report a kinetic and X-ray crystallographic study of a congener of SLC-0111 which incorporates a thioureido instead of ureido linker between the two aromatic rings as inhibitor of four physiologically relevant CA isoforms. Similar to SLC-0111, the thioureido derivative was a weak hCA I and II inhibitor and a potent one against hCA IX and XII. X-ray crystallography of its adduct with hCA II and comparison of the structure with that of other five hCA II—sulfonamide adducts belonging to the SLC-0111 series, afforded us to understand the particular inhibition profile of the new sulfonamide. Similar to SLC-0111, the thioureido sulfonamide primarily interacted with the hydrophobic side of the hCA II active site, with the tail participating in van der Waals interactions with Phe131 and Pro202, in addition to the coordination of the deprotonated sulfonamide to the active site metal ion. On the contrary, the tail of other sulfonamides belonging to the SLC-0111 series (2-isopropyl-phenyl; 3-nitrophenyl) were orientated towards the hydrophilic half of the active site, which was correlated with orders of magnitude better inhibitory activity against hCA II, and a loss of selectivity for the inhibition of the tumor-associated CAs.
Co-reporter:Chae Un Kim;Balendu Sankara Avvaru;Sol M. Gruner;SangYoun Park;HyoJin Song
PNAS 2016 Volume 113 (Issue 19 ) pp:5257-5262
Publication Date(Web):2016-05-10
DOI:10.1073/pnas.1520786113
Carbonic anhydrases are mostly zinc metalloenzymes that catalyze the reversible hydration/dehydration of CO2/HCO3−. Previously, the X-ray crystal structures of CO2-bound holo (zinc-bound) and apo (zinc-free) human carbonic anhydrase IIs (hCA IIs) were captured at high resolution. Here,
we present sequential timeframe structures of holo- [T = 0 s (CO2-bound), 50 s, 3 min, 10 min, 25 min, and 1 h] and apo-hCA IIs [T = 0 s, 50 s, 3 min, and 10 min] during the “slow” release of CO2. Two active site waters, WDW (deep water) and WDW′ (this study), replace the vacated space created on CO2 release, and another water, WI (intermediate water), is seen to translocate to the proton wire position W1. In addition, on the rim of the active site pocket,
a water W2′ (this study), in close proximity to residue His64 and W2, gradually exits the active site, whereas His64 concurrently
rotates from pointing away (“out”) to pointing toward (“in”) active site rotameric conformation. This study provides for the
first time, to our knowledge, structural “snapshots” of hCA II intermediate states during the formation of the His64-mediated
proton wire that is induced as CO2 is released. Comparison of the holo- and apo-hCA II structures shows that the solvent network rearrangements require the
presence of the zinc ion.
Co-reporter:Melissa A. Pinard;Justin J. Kurian;Mayank Aggarwal;Mavis Agbje-McKenna
Acta Crystallographica Section F 2016 Volume 72( Issue 7) pp:573-577
Publication Date(Web):
DOI:10.1107/S2053230X16009286
Cryoannealing has been demonstrated to improve the diffraction quality and resolution of crystals of the β-carbonic anhydrase psCA3 concomitant with a change in space group. After initial flash-cooling in a liquid-nitrogen cryostream an X-ray diffraction data set from a psCA3 crystal was indexed in space group P21212 and was scaled to 2.6 Å resolution, but subsequent cryoannealing studies revealed induced protein rearrangements in the crystal contacts, which transformed the space group to I222, with a corresponding improvement of 0.7 Å in resolution. Although the change in diffraction resolution was significant, only minor changes in the psCA3 structure, which retained its catalytic `open' conformation, were observed. These findings demonstrate that cryoannealing can be successfully utilized to induce higher diffraction-quality crystals while maintaining enzymatically relevant conformations and may be useful as an experimental tool for structural studies of other enzymes where the initial diffraction quality is poor.
Co-reporter:Brian P. Mahon; Carrie L. Lomelino; Janina Ladwig; Gregory M. Rankin; Jenna M. Driscoll; Antonieta L. Salguero; Melissa A. Pinard; Daniela Vullo; Claudiu T. Supuran; Sally-Ann Poulsen
Journal of Medicinal Chemistry 2015 Volume 58(Issue 16) pp:6630-6638
Publication Date(Web):July 23, 2015
DOI:10.1021/acs.jmedchem.5b00845
Inhibition of human carbonic anhydrase IX (hCA IX) has shown to be therapeutically advantageous for treating many types of highly aggressive cancers. However, designing selective inhibitors for hCA IX has been difficult due to its high structural homology and sequence similarity with off-target hCAs. Recently, the use of glucosyl sulfamate inhibitors has shown promise as selective inhibitors for hCA IX. In this study, we present five X-ray crystal structures, determined to a resolution of 1.7 Å or better, of both hCA II (a ubiquitous CA) and an engineered hCA IX-mimic in complex with selected glucosyl sulfamates and structurally rationalize mechanisms for hCA IX selectivity. Results from this study have allowed us, for the first time, to empirically “map” key interactions of the hCA IX active site in order to establish parameters needed to design novel hCA IX selective inhibitors.
Co-reporter:Mayank Aggarwal, Teck Khiang Chua, Melissa A. Pinard, Doletha M. Szebenyi, and Robert McKenna
Biochemistry 2015 Volume 54(Issue 43) pp:6631-6638
Publication Date(Web):October 12, 2015
DOI:10.1021/acs.biochem.5b00987
Carbonic anhydrases (CAs) are enzymes that catalyze the hydration/dehydration of CO2/HCO3– with rates approaching diffusion-controlled limits (kcat/KM ∼ 108 M–1 s–1). This family of enzymes has evolved disparate protein folds that all perform the same reaction at near catalytic perfection. Presented here is a structural study of a β-CA (psCA3) expressed in Pseudomonas aeruginosa, in complex with CO2, using pressurized cryo-cooled crystallography. The structure has been refined to 1.6 Å resolution with Rcryst and Rfree values of 17.3 and 19.9%, respectively, and is compared with the α-CA, human CA isoform II (hCA II), the only other CA to have CO2 captured in its active site. Despite the lack of structural similarity between psCA3 and hCA II, the CO2 binding orientation relative to the zinc-bound solvent is identical. In addition, a second CO2 binding site was located at the dimer interface of psCA3. Interestingly, all β-CAs function as dimers or higher-order oligomeric states, and the CO2 bound at the interface may contribute to the allosteric nature of this family of enzymes or may be a convenient alternative binding site as this pocket has been previously shown to be a promiscuous site for a variety of ligands, including bicarbonate, sulfate, and phosphate ions.
Co-reporter:Brian P. Mahon, Alex M. Hendon, Jenna M. Driscoll, Gregory M. Rankin, Sally-Ann Poulsen, Claudiu T. Supuran, Robert McKenna
Bioorganic & Medicinal Chemistry 2015 Volume 23(Issue 4) pp:849-854
Publication Date(Web):15 February 2015
DOI:10.1016/j.bmc.2014.12.030
Carbonic anhydrase IX (CA IX) is a key modulator of aggressive tumor behavior and a prognostic marker and target for several cancers. Saccharin (SAC) based compounds may provide an avenue to overcome CA isoform specificity, as they display both nanomolar affinity and preferential binding, for CA IX compared to CA II (>50-fold for SAC and >1000-fold when SAC is conjugated to a carbohydrate moiety). The X-ray crystal structures of SAC and a SAC-carbohydrate conjugate bound to a CA IX-mimic are presented and compared to CA II. The structures provide substantial new insight into the mechanism of SAC selective CA isoform inhibition.
Co-reporter:Melissa A. Pinard, Shalaka R. Lotlikar, Christopher D. Boone, Daniela Vullo, Claudiu T. Supuran, Marianna A. Patrauchan, Robert McKenna
Bioorganic & Medicinal Chemistry 2015 Volume 23(Issue 15) pp:4831-4838
Publication Date(Web):1 August 2015
DOI:10.1016/j.bmc.2015.05.029
Carbonic anhydrases (CAs) are metallo-enzymes that catalyze the reversible hydration of carbon dioxide into bicarbonate and a proton. The β-class CAs (β-CAs) are expressed in prokaryotes, fungi, plants, and more recently have been isolated in some animals. The β-CA class is divided into two subclasses, termed type I and II, defined by pH catalytic activity profile and active site structural configuration. Type I β-CAs display catalytic activity over a broad pH range (6.5–9.0) with the active site zinc tetrahedrally coordinated by three amino acids and a hydroxide/water. In contrast, type II β-CAs are catalytically active only at a pH 8 and higher where they adopt a functional active site configuration like that of type I. However, below pH 8 they are conformationally self-inactivated by the addition of a fourth amino acid coordinating the zinc and thereby displacing the zinc bound solvent. We have determined the structure of psCA3, a type II β-CA, isolated from Pseudomonas aeruginosa (P. aeruginosa) PAO1 at pH 8.3, in its open active state to a resolution of 1.9 Å. The active site zinc is coordinated by Cys42, His98, Cys101 and a water/hydroxide molecule. P. aeruginosa is a multi-drug resistant bacterium and displays intrinsic resistance to most of the currently used antibiotics; therefore, there is a need for new antibacterial targets. Kinetic data confirm that psCA3 belongs to the type II subclass and that sulfamide, sulfamic acid, phenylboronic acid and phenylarsonic acid are micromolar inhibitors. In vivo studies identified that among six tested inhibitors representing sulfonamides, inorganic anions, and small molecules, acetazolamide has the most significant dose-dependent inhibitory effect on P. aeruginosa growth.
Co-reporter:Brian P. Mahon, Natalia A. Díaz-Torres, Melissa A. Pinard, Chingkuang Tu, David N. Silverman, Kathleen M. Scott, Robert McKenna
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 21) pp:4937-4940
Publication Date(Web):1 November 2015
DOI:10.1016/j.bmcl.2015.05.001
Thiomicrospira crunogena XCL-2 expresses an α-carbonic anhydrase (TcruCA). Sequence alignments reveal that TcruCA displays a high sequence identity (>30%) relative to other α-CAs. This includes three conserved histidines that coordinate the active site zinc, a histidine proton shuttling residue, and opposing hydrophilic and hydrophobic sides that line the active site. The catalytic efficiency of TcruCA is considered moderate relative to other α-CAs (kcat/KM = 1.1 × 107 M−1 s−1), being a factor of ten less efficient than the most active α-CAs. TcruCA is also inhibited by anions with Cl−, Br−, and I−, all showing Ki values in the millimolar range (53–361 mM). Hydrogen sulfide (HS−) revealed the highest affinity for TcruCA with a Ki of 1.1 μM. It is predicted that inhibition of TcruCA by HS− (an anion commonly found in the environment where Thiomicrospira crunogena is located) is a way for Thiomicrospira crunogena to regulate its carbon-concentrating mechanism (CCM) and thus the organism’s metabolic functions. Results from this study provide preliminary insights into the role of TcruCA in the general metabolism of Thiomicrospira crunogena.The α-carbonic anhydrase (α-CA) from Thiomicrospira crunogena XCL-2 Gammaproteobacterium is an active CA that is readily inhibited by anions. Anionic inhibition of TcCA suggests a means for Thiomicrospira crunogena to shift its general metabolism.
Co-reporter:Melissa A. Pinard;Mayank Aggarwal;Brian P. Mahon;Chingkuang Tu
Acta Crystallographica Section F 2015 Volume 71( Issue 10) pp:1352-1358
Publication Date(Web):
DOI:10.1107/S2053230X1501239X
Human carbonic anhydrase (CA; EC 4.2.1.1) isoform IX (CA IX) is an extracellular zinc metalloenzyme that catalyzes the reversible hydration of CO2 to HCO3−, thereby playing a role in pH regulation. The majority of normal functioning cells exhibit low-level expression of CA IX. However, in cancer cells CA IX is upregulated as a consequence of a metabolic transition known as the Warburg effect. The upregulation of CA IX for cancer progression has drawn interest in it being a potential therapeutic target. CA IX is a transmembrane protein, and its purification, yield and crystallization have proven challenging to structure-based drug design, whereas the closely related cytosolic soluble isoform CA II can be expressed and crystallized with ease. Therefore, we have utilized structural alignments and site-directed mutagenesis to engineer a CA II that mimics the active site of CA IX. In this paper, the X-ray crystal structure of this CA IX mimic in complex with sucrose is presented and has been refined to a resolution of 1.5 Å, an Rcryst of 18.0% and an Rfree of 21.2%. The binding of sucrose at the entrance to the active site of the CA IX mimic, and not CA II, in a non-inhibitory mechanism provides a novel carbohydrate moiety binding site that could be further exploited to design isoform-specific inhibitors of CA IX.
Co-reporter:Christopher D. Boone;Guo Zhong;Marci Smeltz;Margaret O. James
Acta Crystallographica Section F 2014 Volume 70( Issue 2) pp:187-189
Publication Date(Web):
DOI:10.1107/S2053230X13033591
Glutathione transferase zeta 1 (GSTZ1-1) is a homodimeric enzyme found in the cytosol and mitochondrial matrix of the liver and other tissues. It catalyzes the glutathione-dependent isomerization of maleylacetoacetate to fumarylacetoacetate in the tyrosine catabolic pathway and can metabolize small halogenated carboxylic acids. GSTZ1a-1a crystals diffracted to a resolution of 3.1 Å and belonged to space group P1, with unit-cell parameters a = 42.0, b = 49.6, c = 54.6 Å, α = 82.9, β = 69.9, γ = 73.4°, with a calculated Matthews coefficient of 2.1 Å3 Da−1 assuming a dimer in the crystallographic asymmetric unit. Refinement of the structure is currently in progress.
Co-reporter:Dayne West;Melissa A. Pinard;Chingkuang Tu;David N. Silverman
Acta Crystallographica Section F 2014 Volume 70( Issue 10) pp:1324-1327
Publication Date(Web):
DOI:10.1107/S2053230X14018135
The binding of anions to carbonic anhydrase II (CA II) has been attributed to high affinity for the active-site zinc. An anion of interest is cyanate, for which contrasting binding modes have been reported in the literature. Previous spectroscopic data have shown cyanate behaving as an inhibitor, directly binding to the zinc, in contrast to previous crystallographic data that implied that cyanate acts as a substrate mimic that is not directly bound to the zinc but overlaps with the binding site of the substrate CO2. Wild-type and the V207I variant of CA II have been expressed and X-ray crystal structures of their cyanate complexes have been determined to 1.7 and 1.5 Å resolution, respectively. The rationale for the V207I CA II variant was its close proximity to the CO2-binding site. Both structures clearly show that the cyanate binds directly to the zinc. In addition, inhibition constants (∼40 µM) were measured using 18O-exchange mass spectrometry for wild-type and V207I CA II and were similar to those determined previously (Supuran et al., 1997). Hence, it is concluded that under the conditions of these experiments the binding of cyanate to CA II is directly to the zinc, displacing the zinc-bound solvent molecule, and not in a site that overlaps with the CO2 substrate-binding site.
Co-reporter:Kyle P. Heim;Joanna R. Long;Shweta Kailasan;Paula J. Crowley;L. Jeannine Brady
PNAS 2014 Volume 111 (Issue 44 ) pp:15746-15751
Publication Date(Web):2014-11-04
DOI:10.1073/pnas.1413018111
The cariogenic bacterium Streptococcus mutans uses adhesin P1 to adhere to tooth surfaces, extracellular matrix components, and other bacteria. A composite model of P1
based on partial crystal structures revealed an unusual complex architecture in which the protein forms an elongated hybrid
alpha/polyproline type II helical stalk by folding back on itself to display a globular head at the apex and a globular C-terminal
region at the base. The structure of P1’s N terminus and the nature of its critical interaction with the C-terminal region
remained unknown, however. We have cocrystallized a stable complex of recombinant N- and C-terminal fragments and here describe
a previously unidentified topological fold in which these widely discontinuous domains are intimately associated. The structure
reveals that the N terminus forms a stabilizing scaffold by wrapping behind the base of P1’s elongated stalk and physically
“locking” it into place. The structure is stabilized through a highly favorable ΔGsolvation on complex formation, along with extensive hydrogen bonding. We confirm the functional relevance of this intramolecular interaction
using differential scanning calorimetry and circular dichroism to show that disruption of the proper spacing of residues 989–1001
impedes folding and diminishes stability of the full-length molecule, including the stalk. Our findings clarify previously
unexplained functional and antigenic properties of P1.
Co-reporter:Rose Mikulski, Dayne West, Katherine H. Sippel, Balendu Sankara Avvaru, Mayank Aggarwal, Chingkuang Tu, Robert McKenna, and David N. Silverman
Biochemistry 2013 Volume 52(Issue 1) pp:
Publication Date(Web):December 6, 2012
DOI:10.1021/bi301099k
Variants of human carbonic anhydrase II (HCA II) with amino acid replacements at residues in contact with water molecules in the active-site cavity have provided insights into the proton transfer rates in this protein environment. X-ray crystallography and 18O exchange measured by membrane inlet mass spectrometry have been used to investigate structural and catalytic properties of variants of HCA II containing replacements of Tyr7 with Phe (Y7F) and Asn67 with Gln (N67Q). The rate constants for transfer of a proton from His64 to the zinc-bound hydroxide during catalysis were 4 and 9 μs–1 for Y7F and Y7F/N67Q, respectively, compared with a value of 0.8 μs–1 for wild-type HCA II. These higher values observed for Y7F and Y7F/N67Q HCA II could not be explained by differences in the values of the pKa of the proton donor (His64) and acceptor (zinc-bound hydroxide) or by the orientation of the side chain of the proton shuttle residue His64. They appeared to be associated with a reduced level of branching in the networks of hydrogen-bonded water molecules between proton shuttle residue His64 and the zinc-bound solvent molecule as observed in crystal structures at 1.5–1.6 Å resolution. Moreover, Y7F/N67Q HCA II is unique among the variants studied in having a direct, hydrogen-bonded chain of water molecules between the zinc-bound solvent and Nε of His64. This study provides the clearest example to date of the relevance of ordered water structure to rate constants for proton transfer in catalysis by carbonic anhydrase.
Co-reporter:Melissa A. Pinard, Christopher D. Boone, Brittany D. Rife, Claudiu T. Supuran, Robert McKenna
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 22) pp:7210-7215
Publication Date(Web):15 November 2013
DOI:10.1016/j.bmc.2013.08.033
Carbonic anhydrases (CAs, EC 4.2.1.1) are metalloenzymes that catalyze the reversible hydration of carbon dioxide and bicarbonate. Their pivotal role in metabolism, ubiquitous nature, and multiple isoforms (CA I–XIV) has made CAs an attractive drug target in clinical applications. The usefulness of CA inhibitors (CAIs) in the treatment of glaucoma and epilepsy are well documented. In addition several isoforms of CAs (namely, CA IX) also serve as biological markers for certain tumors, and therefore they have the potential for useful applications in the treatment of cancer. This is a structural study on the binding interactions of the widely used CA inhibitory drugs brinzolamide (marketed as Azopt®) and dorzolamide (marketed as Trusopt®) with CA II and a CA IX-mimic, which was created via site-directed mutagenesis of CA II cDNA such that the active site resembles that of CA IX. Also the inhibition of CA II and CA IX and molecular docking reveal brinzolamide to be a more potent inhibitor among the other catalytically active CA isoforms compared to dorzolamide. The structures show that the tail end of the sulfonamide inhibitor is critical in forming stabilizing interactions that influence tight binding; therefore, for future drug design it is the tail moiety that will ultimately determine isoform specificity.Overlay of BRZ (magenta) and DRZ (orange) in the active site of CA II. Hydrophobic region, red; hydrophilic region, blue.
Co-reporter:Özlen Güzel-Akdemir, Shyamasri Biswas, Katherine Lastra, Robert McKenna, Claudiu T. Supuran
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 21) pp:6674-6680
Publication Date(Web):1 November 2013
DOI:10.1016/j.bmc.2013.08.011
The crystal structure of 4-phenylacetamidomethyl-benzenesulfonamide (4ITP) bound to human carbonic anhydrase (hCA, EC 4.2.1.1) II is reported. 4ITP is a medium potency hCA I and II inhibitor (KIs of 54–75 nM), a strong mitochondrial CA VA/VB inhibitor (KIs of 8.3–8.6 nM) and a weak transmembrane CA inhibitor (KIs of 136–212 nM against hCA IX and XII). This elongated compound binds in an extended conformation to hCA II, with its tail lying towards the hydrophobic half of the active site whereas the sulfonamide moiety coordinates the zinc ion. The present structure was compared to that of structurally related aromatic sulfonamides, such as 4-phenylacetamido-benzene-sulfonamide (3OYS), 4-(2-mercaptophenylacetamido)-benzene-sulfonamide (2HD6) and 4-(3-nitrophenyl)-ureido-benzenesulfonamide (3N2P). Homology models of the hCA I, VA, VB, IX and XII structures were build which afforded an understanding of the amino acids involved in the binding of these compounds to these isoforms. The main conclusion of the study is that the orientation of the tail moiety and the presence of flexible linkers as well polar groups in it, strongly influence the potency and the selectivity of the sulfonamides for the inhibition of cytosolic, mitochondrial or transmembrane CA isoforms.
Co-reporter:Mayank Aggarwal, Bhargav Kondeti, Robert McKenna
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 6) pp:1526-1533
Publication Date(Web):15 March 2013
DOI:10.1016/j.bmc.2012.08.019
Carbonic anhydrases (CAs, EC 4.2.1.1) are a group of metalloenzymes that play important roles in carbon metabolism, pH regulation, CO2 fixation in plants, ion transport etc., and are found in all eukaryotic and many microbial organisms. This family of enzymes catalyzes the interconversion of CO2 and HCO3−. There are at least 16 different CA isoforms in the alpha structural class (α-CAs) that have been isolated in higher vertebrates, with CA isoform II (CA II) being ubiquitously abundant in all human cell types. CA inhibition has been exploited clinically for decades for various classes of diuretics and anti-glaucoma treatment. The characterization of the overexpression of CA isoform IX (CA IX) in certain tumors has raised interest in CA IX as a diagnostic marker and drug target for aggressive cancers and therefore the development of CA IX specific inhibitors. An important goal in the field of CA is to identify, rationalize, and design potential compounds that will preferentially inhibit CA IX over all other isoforms of CA. The variations in the active sites between isoforms of CA are subtle and this causes non-specific CA inhibition which leads to various side effects. In the case of CA IX inhibition, CA II along with other isoforms of CA provide off-target binding sites which is undesirable for cancer treatment. The focus of this article is on CA IX inhibition and two different structural approaches to CA isoform specific drug designing: tail approach and fragment addition approach.Figure optionsDownload full-size imageDownload as PowerPoint slide
Co-reporter:Shyamasri Biswas, Robert McKenna, Claudiu T. Supuran
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 20) pp:5646-5649
Publication Date(Web):15 October 2013
DOI:10.1016/j.bmcl.2013.08.019
The high resolution crystal structure of 5-(2-thienylacetamido)-1,3,4-thiadiazole-2-sulfonamide complexed to human (h) carbonic anhydrase (CA, EC 4.2.1.1) isoform hCA II is reported. The compound binds in a similar manner with acetazolamide when the sulfamoyl–thiadiazolyl–acetamido fragment of the two compounds is considered, but the thienyl tail was positioned in the subpocket 2, rarely observed by other investigated CA inhibitors. This positioning allows interaction with amino acid residues (such as Asn67, Ile91, Gln92 and Val121 which are variable in other isoforms of medicinal chemistry interest, such as hCA I, IX and XII. Indeed, the investigated sulfonamide was a medium potency hCA I and II inhibitor but was highly effective as a hCA IX and XII inhibitor. This different behavior with respect to acetazolamide (a promiscuous inhibitor of all these isoforms) has been explained by resolving the crystal structure, and may be used to design more isoform-selective compounds.
Co-reporter:Shyamasri Biswas, Fabrizio Carta, Andrea Scozzafava, Robert McKenna, Claudiu T. Supuran
Bioorganic & Medicinal Chemistry 2013 21(8) pp: 2314-2318
Publication Date(Web):
DOI:10.1016/j.bmc.2013.02.022
Co-reporter:S. Zoë Fisher ; Mayank Aggarwal ; Andrey Y. Kovalevsky ; David N. Silverman
Journal of the American Chemical Society 2012 Volume 134(Issue 36) pp:14726-14729
Publication Date(Web):August 28, 2012
DOI:10.1021/ja3068098
Carbonic anhydrases (CAs) catalyze the hydration of CO2 forming HCO3– and a proton, an important reaction for many physiological processes including respiration, fluid secretion, and pH regulation. As such, CA isoforms are prominent clinical targets for treating various diseases. The clinically used acetazolamide (AZM) is a sulfonamide that binds with high affinity to human CA isoform II (HCA II). There are several X-ray structures available of AZM bound to various CA isoforms, but these complexes do not show the charged state of AZM or the hydrogen atom positions of the protein and solvent. Neutron diffraction is a useful technique for directly observing H atoms and the mapping of H-bonding networks that can greatly contribute to rational drug design. To this end, the neutron structure of H/D exchanged HCA II crystals in complex with AZM was determined. The structure reveals the molecular details of AZM binding and the charged state of the bound drug. This represents the first determined neutron structure of a clinically used drug bound to its target.
Co-reporter:Fabrizio Carta, Mayank Aggarwal, Alfonso Maresca, Andrea Scozzafava, Robert McKenna and Claudiu T. Supuran
Chemical Communications 2012 vol. 48(Issue 13) pp:1868-1870
Publication Date(Web):14 Dec 2011
DOI:10.1039/C2CC16395K
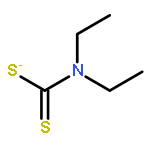
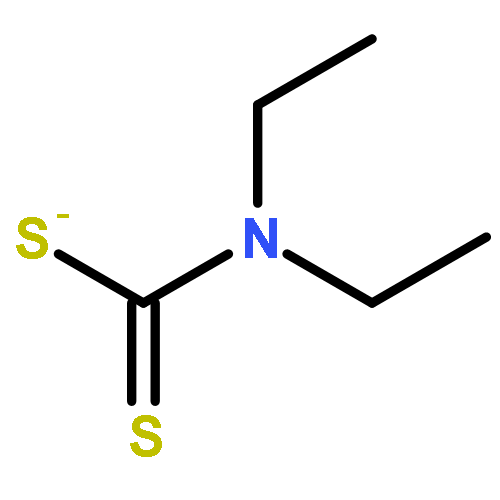
The zinc enzyme carbonic anhydrase (CA, EC 4.2.1.1) is inhibited by several classes of zinc-binders (sulfonamides, sulfamates, and sulfamides) as well as by compounds which do not interact with the metal ion (phenols, polyamines and coumarins). Here we report a new class of potent CA inhibitors which bind the zinc ion: the dithiocarbamates (DTCs). They coordinate to the zinc ion from the enzyme active site in monodentate manner and establish many favorable interactions with amino acid residues nearby. Several low nanomolar CA I, II and IX inhibitors were detected.
Co-reporter:Dayne West, Chae Un Kim, Chingkuang Tu, Arthur H. Robbins, Sol M. Gruner, David N. Silverman, and Robert McKenna
Biochemistry 2012 Volume 51(Issue 45) pp:
Publication Date(Web):October 25, 2012
DOI:10.1021/bi301155z
This work examines the effect of perturbing the position of bound CO2 in the active site of human carbonic anhydrase II (HCA II) on catalysis. Variants of HCA II in which Val143 was replaced with hydrophobic residues Ile, Leu, and Ala were examined. The efficiency of catalysis in the hydration of CO2 for these variants was characterized by 18O exchange mass spectrometry, and their structures were determined by X-ray crystallography at 1.7–1.5 Å resolution. The most hydrophobic substitutions, V143I and V143L, showed decreases in the level of catalysis, as much as 20-fold, while the replacement by the smaller V143A mutation showed an only moderate 2-fold decrease in activity. Structural data for all three variants show no significant change in the overall position of amino acid side chains in the active site compared with the wild type. However, V143A HCA II showed additional ordered water molecules in the active site compared to the number for the wild type. To further investigate the decrease in the catalytic efficiency of V143I HCA II, an X-ray crystallographic CO2 entrapment experiment was performed to 0.93 Å resolution. This structure revealed an unexpected shift in the CO2 substrate toward the zinc-bound solvent, placing it ∼0.3 Ǻ closer than previously observed in the wild type in conjunction with the observed dual occupancy of the product bicarbonate, presumably formed during the acquisition of data. These data suggest that the Ile substitution at position 143 reduced the catalytic efficiency, which is likely due to steric crowding resulting in destabilization of the transition state for conversion of CO2 into bicarbonate and a decreased product dissociation rate.
Co-reporter:C. Mark Maupin ; Norberto Castillo ; Srabani Taraphder ; Chingkuang Tu ; Robert McKenna ; David N. Silverman ;Gregory A. Voth
Journal of the American Chemical Society 2011 Volume 133(Issue 16) pp:6223-6234
Publication Date(Web):March 31, 2011
DOI:10.1021/ja1097594
In human carbonic anhydrase II (HCA II), the mutation of position 64 from histidine to alanine (H64A) disrupts the rate limiting proton transfer (PT) event, resulting in a reduction of the catalytic activity of the enzyme as compared to the wild-type. Potential of mean force (PMF) calculations utilizing the multistate empirical valence bond (MS-EVB) methodology for H64A HCA II yields a PT free energy barrier significantly higher than that found in the wild-type enzyme. This high barrier, determined in the absence of exogenous buffer and assuming no additional ionizable residues in the PT pathway, indicates the likelihood of alternate enzyme pathways that utilize either ionizable enzyme residues (self-rescue) and/or exogenous buffers (chemical rescue). It has been shown experimentally that the catalytic activity of H64A HCA II can be chemically rescued to near wild-type levels by the addition of the exogenous buffer 4-methylimidazole (4MI). Crystallographic studies have identified two 4MI binding sites, yet site-specific mutations intended to disrupt 4MI binding have demonstrated these sites to be nonproductive. In the present work, MS-EVB simulations show that binding of 4MI near Thr199 in the H64A HCA II mutant, a binding site determined by NMR spectroscopy, results in a viable chemical rescue pathway. Additional viable rescue pathways are also identified where 4MI acts as a proton transport intermediary from the active site to ionizable residues on the rim of the active site, revealing a probable mode of action for the chemical rescue pathway.
Co-reporter:Naama Hen ; Meir Bialer ; Boris Yagen ; Alfonso Maresca ; Mayank Aggarwal ; Arthur H. Robbins ; Robert McKenna ; Andrea Scozzafava ;Claudiu T. Supuran
Journal of Medicinal Chemistry 2011 Volume 54(Issue 11) pp:3977-3981
Publication Date(Web):April 20, 2011
DOI:10.1021/jm200209n
Aromatic amides comprising branched aliphatic carboxylic acids and 4-aminobenzenesulfonamide were evaluated for their inhibition of carbonic anhydrase (CA) isoforms. Of the most anticonvulsant-active compounds (2, 4, 13, 16, and 17), only 13, 16, and 17 were potent inhibitors of CAs VII and XIV. Compounds 9, 14, and 19 inhibited CA II, while 10 and 12 inhibited all isoforms. Structural studies suggest that differences in the active sites’ hydrophobicity modulate the affinity of the inhibitors.
Co-reporter:Fabrizio Carta, Vladimir Garaj, Alfonso Maresca, Jason Wagner, Balendu Sankara Avvaru, Arthur H. Robbins, Andrea Scozzafava, Robert McKenna, Claudiu T. Supuran
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 10) pp:3105-3119
Publication Date(Web):15 May 2011
DOI:10.1016/j.bmc.2011.04.005
Reaction of cyanuryl chloride with d,l-amino acids and amino alcohols afforded a new series of triazinyl-substituted benzenesulfonamides incorporating amino acyl/hydroxyalkyl-amino moieties. Inhibition studies of physiologically relevant human carbonic anhydrase (CA, EC 4.2.1.1) isoforms, such as CA I, II, IX, XII and XIV with these compounds are reported. They showed moderate-weak inhibition of the cytosolic, offtarget isozymes CA I and II, but many of them were low nanomolar inhibitors of the transmembrane, tumor-associated CA IX and XII (and also of CA XIV). The X-ray crystal structure of two of these compounds in adduct with CA II allowed us to understand the features associated with this strong inhibitory properties and possibly also their selectivity. Two of these compounds were also investigated for the inhibition of other human isoforms, that is, hCA IV, VA, VB, VI, VII and XIII, as well as inhibitors of the fungal pathogenic CAs Nce103 (Candida albicans) and Can2 (Cryptococcus neoformans), showing interesting activity. The 1,3,5-triazinyl-substituted benzenesulfonamides constitute thus a class of compounds with great potential for obtaining inhibitors targeting both α-class mammalian, tumor-associated, and β-class from pathogenic organisms CAs.
Co-reporter:Shyamasri Biswas, Mayank Aggarwal, Özlen Güzel, Andrea Scozzafava, Robert McKenna, Claudiu T. Supuran
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 12) pp:3732-3738
Publication Date(Web):15 June 2011
DOI:10.1016/j.bmc.2011.05.006
The X-ray crystal structures of the adducts of human carbonic anhydrase (hCA, EC 4.2.1.1) II complexed with two aromatic sulfonamides incorporating 2-thienylacetamido moieties are reported here. Although, the two inhibitors only differ by the presence of an additional 3-fluoro substituent on the 4-amino-benzenesulfonamide scaffold, their inhibition profiles against the cytosolic isoforms hCA I, II, III, VII and XIII are quite different. These differences were rationalized based on the obtained X-ray crystal structures, and their comparison with other sulfonamide CA inhibitors with clinical applications, such as acetazolamide, methazolamide and dichlorophenamide. The conformations of the 2-thienylacetamido tails in the hCA II adducts of the two sulfonamides were highly different, although the benzenesulfonamide parts were superimposable. Specific interactions between structurally different inhibitors and amino acid residues present only in some considered isoforms have thus been evidenced. These findings can explain the high affinity of the 2-thienylacetamido benzenesulfonamides for some pharmacologically relevant CAs (i.e., isoforms II and VII) being also useful to design high affinity, more selective sulfonamide inhibitors of various CAs.
Co-reporter:Fabio Pacchiano, Mayank Aggarwal, Balendu Sankara Avvaru, Arthur H. Robbins, Andrea Scozzafava, Robert McKenna and Claudiu T. Supuran
Chemical Communications 2010 vol. 46(Issue 44) pp:8371-8373
Publication Date(Web):05 Oct 2010
DOI:10.1039/C0CC02707C
4-Substituted-ureido benzenesulfonamides showing inhibitory activity against carbonic anhydrase (CA, EC 4.2.1.1) II between 3.3–226 nM were crystallized in complex with the enzyme. Hydrophobic interactions between the scaffold of the inhibitors in different hydrophobic pockets of the enzyme were observed, explaining the diverse inhibitory range of these derivatives.
Co-reporter:Jason Wagner, Balendu Sankara Avvaru, Arthur H. Robbins, Andrea Scozzafava, Claudiu T. Supuran, Robert McKenna
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 14) pp:4873-4878
Publication Date(Web):15 July 2010
DOI:10.1016/j.bmc.2010.06.028
We investigated a series of coumarinyl-substituted aromatic sulfonamides as inhibitors of four carbonic anhydrase (CA, EC 4.2.1.1) isoforms with medical applications, the cytosolic hCA I, and II, and the transmembrane, tumor-associated hCA IX and XII. Compounds incorporating 7-methoxy-coumarin-4-yl-acetamide-tails and benzenesulfonamide and benzene-1,3-disulfonamide scaffolds showed medium potency inhibition of hCA I (KIs of 73–131 nM), effective hCA II inhibition (KIs of 9.1–36 nM) and less effective hCA IX and XII inhibition (KIs of 55–128 nM). Only one compound, the derivatized 4-amino-6-trifluoromethyl-benzene-1,3-disulfonamide with the coumarinyl tail, showed effective inhibition of the transmembrane isoforms, with KIs of 5.9–14.2 nM, although it was less effective as hCA I and II inhibitor (KIs of 36–120 nM). An X-ray crystal structure of hCA II in complex with 4-(7-methoxy-coumarin-4-yl-acetamido)-benzenesulfonamide (KI of 9.1 nM against hCA II) showed the intact inhibitor coordinated to the zinc ion from the enzyme active site by the sulfonamide moiety, and participating in a edge-to-face stacking with Phe131, in addition to other hydrophobic and hydrophilic interactions with water molecules and amino acid residues from the active site. Thus, sulfonamides incorporating coumarin rings have a distinct inhibition mechanism compared to the coumarins, and may lead to compounds with interesting inhibition profiles against various α-CAs found in mammals or parasites, such as Plasmodium falciparum.
Co-reporter:John F. Domsic, Wilton Williams, Suzanne Z. Fisher, Chingkuang Tu, Mavis Agbandje-McKenna, David N. Silverman and Robert McKenna
Biochemistry 2010 Volume 49(Issue 30) pp:
Publication Date(Web):June 25, 2010
DOI:10.1021/bi1007645
The catalysis of CO2 hydration by human carbonic anhydrase II (HCA II) is limited in maximal velocity by proton transfer from a zinc-bound water molecule to the proton shuttle His64. This proton transfer occurs along a hydrogen-bonded water network, leading to the proton shuttle residue His64, which in turn transfers the proton to bulk solvent. The side chain of His64 occupies two conformations in wild-type HCA II, pointing inward toward the zinc or outward toward bulk solvent. Previously, several studies have examined the roles of residues of the active site cavity that interact with the solvent-mediated hydrogen-bonded network between His64 and the zinc-bound water. Here these studies are extended to examine the effects on proton transfer by mutation at Lys170 (to Ala, Asp, Glu, and His), a residue located near the side chain of His64 but over 15 Å away from the active site zinc. In all four variants, His64 is observed in the inward conformation associated with a decrease in the pKa of His64 by as much as 1.0 unit and an increase in the rate constant for proton transfer to as much as 4 μs−1, approximately 5-fold larger than wild-type HCA II. The results show a significant extension of the effective active site of HCA II from the zinc-bound water at the base of the conical cavity in the enzyme to Lys170 near the rim of the cavity. These data emphasize that the active site of HCA II is extended to include residues that, at first glance, appear to be too far from the zinc to exert any catalytic effects.
Co-reporter:Balendu Sankara Avvaru, Chae Un Kim, Katherine H. Sippel, Sol M. Gruner, Mavis Agbandje-McKenna, David N. Silverman and Robert McKenna
Biochemistry 2010 Volume 49(Issue 2) pp:
Publication Date(Web):December 9, 2009
DOI:10.1021/bi902007b
The crystal structure of human carbonic anhydrase II (HCA II) obtained at 0.9 Å resolution reveals that a water molecule, termed deep water, Dw, and bound in a hydrophobic pocket of the active site forms a short, strong hydrogen bond with the zinc-bound solvent molecule, a conclusion based on the observed oxygen−oxygen distance of 2.45 Å. This water structure has similarities with hydrated hydroxide found in crystals of certain inorganic complexes. The energy required to displace Dw contributes in significant part to the weak binding of CO2 in the enzyme−substrate complex, a weak binding that enhances kcat for the conversion of CO2 into bicarbonate. In addition, this short, strong hydrogen bond is expected to contribute to the low pKa of the zinc-bound water and to promote proton transfer in catalysis.
Co-reporter:Balendu Sankara Avvaru, Jason M. Wagner, Alfonso Maresca, Andrea Scozzafava, Arthur H. Robbins, Claudiu T. Supuran, Robert McKenna
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 15) pp:4376-4381
Publication Date(Web):1 August 2010
DOI:10.1016/j.bmcl.2010.06.082
We investigated the inhibitory activity of several 1,3,4-thiadiazole-sulfonamides against all catalytically active CA (EC 4.2.1.1), CA I–XV. The tail derivatizing the 5-position in the 1,3,4-thiadiazole-2-sulfonamide scaffold was observed to be critical as an inhibitory determinant of these compounds. The high resolution X-ray crystal structure of hCA II in complex with 5-(1-adamantylcarboxamido)-1,3,4-thiadiazole-2-sulfonamide, showed the adamantyl moiety of the inhibitor residing in a less utilized binding pocket than that of most hydrophobic inhibitors, lined by the amino acid residues Ile91, Val121 and Phe131. This binding site may explain the diverse inhibition profiles of 5-carboxamide- and sufonamide-derivatized 1,3,4-thiadiazole-2-sulfonamides and offers a hot spot for designing isoform selective inhibitors, considering that residues 91 and 131 are highly variable among the 13 catalytically active isoforms.
Co-reporter:Katherine H. Sippel, Caroli Genis, Lakshmanan Govindasamy, Mavis Agbandje-McKenna, James J. Kiddle, Brian C. Tripp, and Robert McKenna
The Journal of Physical Chemistry Letters 2010 Volume 1(Issue 19) pp:2898-2902
Publication Date(Web):September 16, 2010
DOI:10.1021/jz100954h
Thioxolone acts as a prodrug in the presence of carbonic anhydrase II (CA II), whereby the molecule is cleaved by thioester hydrolysis to the carbonic anhydrase inhibitor, 4-mercaptobenzene-1,3-diol (TH0). Thioxolone was soaked in crystals of the proton transfer mutant H64A of CA II in an effort to capture a reaction intermediate via X-ray crystallography. Structure determination of the 1.2 Å resolution data revealed that TH0 had been modified to a 4,4′-disulfanediyldibenzene-1,3-diol, a product of crystallization conditions, and a zinc ligated 2,4-dihydroxybenzenesulfenic acid, most likely induced by radiation damage. Neither ligand was conceivably a result of an enzymatic mechanism.Keywords (keywords): carbonic anhydrase; free radical damage; sulfenic acid; synchrotron radiation; thioxolone;
Co-reporter:Caroli Genis, Katherine H. Sippel, Nicolette Case, Wengang Cao, Balendu Sankara Avvaru, Lawrence J. Tartaglia, Lakshmanan Govindasamy, Chingkuang Tu, Mavis Agbandje-McKenna, David N. Silverman, Charles J. Rosser and Robert McKenna
Biochemistry 2009 Volume 48(Issue 6) pp:
Publication Date(Web):January 26, 2009
DOI:10.1021/bi802035f
Recently, a convincing body of evidence has accumulated suggesting that the overexpression of carbonic anhydrase isozyme IX (CA IX) in some cancers contributes to the acidification of the extracellular matrix, which in turn promotes the growth and metastasis of the tumor. These observations have made CA IX an attractive drug target for the selective treatment of certain cancers. Currently, there is no available X-ray crystal structure of CA IX, and this lack of availability has hampered the rational design of selective CA IX inhibitors. In light of these observations and on the basis of structural alignment homology, using the crystal structure of carbonic anhydrase II (CA II) and the sequence of CA IX, a double mutant of CA II with Ala65 replaced by Ser and Asn67 replaced by Gln has been constructed to resemble the active site of CA IX. This CA IX mimic has been characterized kinetically using 18O-exchange and structurally using X-ray crystallography, alone and in complex with five CA sulfonamide-based inhibitors (acetazolamide, benzolamide, chlorzolamide, ethoxzolamide, and methazolamide), and compared to CA II. This structural information has been evaluated by both inhibition studies and in vitro cytotoxicity assays and shows a correlated structure−activity relationship. Kinetic and structural studies of CA II and CA IX mimic reveal chlorzolamide to be a more potent inhibitor of CA IX, inducing an active-site conformational change upon binding. Additionally, chlorzolamide appears to be cytotoxic to prostate cancer cells. This preliminary study demonstrates that the CA IX mimic may provide a useful model to design more isozyme-specific CA IX inhibitors, which may lead to development of new therapeutic treatments of some cancers.
Co-reporter:Balendu Sankara Avvaru, Scott A. Busby, Michael J. Chalmers, Patrick R. Griffin, Balasubramanian Venkatakrishnan, Mavis Agbandje-McKenna, David N. Silverman and Robert McKenna
Biochemistry 2009 Volume 48(Issue 31) pp:
Publication Date(Web):July 7, 2009
DOI:10.1021/bi9007512
Human carbonic anhydrase II (HCA II) is a monomeric zinc-containing metalloenzyme that catalyzes the hydration of CO2 to form bicarbonate and a proton. The properties of the zinc have been extensively elucidated in catalysis but less well studied as a contributor to structure and stability. Apo-HCA II (without zinc) was prepared and compared to holo-HCA II: in crystallographic structural features, in backbone amide H/D exchange, and in thermal stability. The removal of zinc from the active site has no effect on either the topological fold of the enzyme or the ordered water network in the active site. However, the removal of the zinc alters the collective electrostatics of the apo-HCA II that result in the following differences from that of the holoenzyme: (1) the main thermal unfolding transition of the apo-HCA II is lowered by 8 °C, (2) the relative increase in thermal mobility of atoms of the apo-HCA II was not observed in the vicinity of the active site but manifested on the surface of the enzyme, and (3) the side chain of His 64, the proton shuttle residue that sits on the rim of the active site, is oriented outward and is associated with additional ordered “external” waters, as opposed to a near equal inward and outward orientation in the holo-HCA II.
Co-reporter:C. Mark Maupin, Jiayin Zheng, Chingkuang Tu, Robert McKenna, David N. Silverman and Gregory A. Voth
Biochemistry 2009 Volume 48(Issue 33) pp:
Publication Date(Web):July 27, 2009
DOI:10.1021/bi901037u
The rate-limiting proton transfer (PT) event in the site-specific mutant N67L of human carbonic anhydrase II (HCA II) has been examined by kinetic, X-ray, and simulation approaches. The X-ray crystallography studies, which were previously reported, and molecular dynamics (MD) simulations indicate that the proton shuttling residue, His64, predominantly resides in the outward orientation with a significant disruption of the ordered water in the active site for the dehydration pathway. While disorder is seen in the active-site water, water cluster analysis indicates that the N67L mutant may form water clusters similar to those seen in the wild-type (WT). For the hydration pathway of the enzyme, the active site water cluster analysis reveals an inability of the N67L mutant to stabilize water clusters when His64 is in the inward orientation, thereby favoring PT when His64 is in the outward orientation. The preference of the N67L mutant to carry out the PT when His64 is in the outward orientation for both the hydration and dehydration pathway is reasoned to be the main cause of the observed reduction in the overall rate. To probe the mechanism of PT, solvent H/D kinetic isotope effects (KIEs) were experimentally studied with catalysis measured by the exchange of 18O between CO2 and water. The values obtained from the KIEs were determined as a function of the deuterium content of solvent, using the proton inventory method. No differences were detected in the overarching mechanism of PT between WT and N67L HCA II, despite changes in the active-site water structure and/or the orientation of His64.
Co-reporter:Albert A. Barrese, III, Caroli Genis, S. Zoe Fisher, Jared N. Orwenyo, Mudalige Thilak Kumara, Subodh K. Dutta, Eric Phillips, James J. Kiddle, Chingkuang Tu, David N. Silverman, Lakshmanan Govindasamy, Mavis Agbandje-McKenna, Robert McKenna and Brian C. Tripp
Biochemistry 2008 Volume 47(Issue 10) pp:
Publication Date(Web):February 12, 2008
DOI:10.1021/bi702385k
This paper examines the functional mechanism of thioxolone, a compound recently identified as a weak inhibitor of human carbonic anhydrase II by Iyer et al. (2006) J. Biomol. Screening 11, 782−791. Thioxolone lacks sulfonamide, sulfamate, or hydroxamate functional groups that are typically found in therapeutic carbonic anhydrase (CA) inhibitors, such as acetazolamide. Analytical chemistry and biochemical methods were used to investigate the fate of thioxolone upon binding to CA II, including Michaelis–Menten kinetics of 4-nitrophenyl acetate esterase cleavage, liquid chromatography–mass spectrometry (LC-MS), oxygen-18 isotope exchange studies, and X-ray crystallography. Thioxolone is proposed to be a prodrug inhibitor that is cleaved via a CA II zinc-hydroxide mechanism known to catalyze the hydrolysis of esters. When thioxolone binds in the active site of CA II, it is cleaved and forms 4-mercaptobenzene-1,3-diol via the intermediate S-(2,4-thiophenyl)hydrogen thiocarbonate. The esterase cleavage product binds to the zinc active site via the thiol group and is therefore the active CA inhibitor, while the intermediate is located at the rim of the active-site cavity. The time-dependence of this inhibition reaction was investigated in detail. Because this type of prodrug inhibitor mechanism depends on cleavage of ester bonds, this class of inhibitors may have advantages over sulfonamides in determining isozyme specificity. A preliminary structure–activity relationship study with a series of structural analogues of thioxolone yielded similar estimates of inhibition constants for most compounds, although two compounds with bromine groups at the C1 carbon of thioxolone were not inhibitory, suggesting a possible steric effect.
Co-reporter:Patrick S. Quint, John F. Domsic, Diane E. Cabelli, Robert McKenna and David N. Silverman
Biochemistry 2008 Volume 47(Issue 16) pp:
Publication Date(Web):March 29, 2008
DOI:10.1021/bi7024518
The function in the structure, stability, and catalysis of the interfaces between subunits in manganese superoxide dismutase (MnSOD) is currently under scrutiny. Glu162 in homotetrameric human MnSOD spans a dimeric interface and forms a hydrogen bond with His163 of an adjacent subunit which is a direct ligand of the manganese. We have examined the properties of two site-specific mutants of human MnSOD in which Glu162 is replaced with Asp (E162D) and Ala (E162A). The X-ray crystal structures of E162D and E162A MnSOD reveal no significant structural changes compared with the wild type other than the removal of the hydrogen bond interaction with His163 in E162A MnSOD. In the case of E162D MnSOD, an intervening solvent molecule fills the void created by the mutation to conserve the hydrogen bond interaction between His163 and residue 162. These mutants retain their tetrameric structure and their specificity for manganese over iron. Each has catalytic activity in the disproportionation of superoxide that is typically 5–25% of that of the wild-type enzyme and a level of product inhibition greater by approximately 2-fold. Differential scanning calorimetry indicates that the hydrogen bond between Glu162 and His163 contributes to the stability of MnSOD, with the major unfolding transition occurring at 81 °C for E162A compared to 90 °C for wild-type MnSOD. These results suggest that Glu162 at the tetrameric interface in human MnSOD supports stability and efficient catalysis and has a significant role in regulating product inhibition.
Co-reporter:Jiayin Zheng, Balendu Sankara Avvaru, Chingkuang Tu, Robert McKenna and David N. Silverman
Biochemistry 2008 Volume 47(Issue 46) pp:
Publication Date(Web):October 22, 2008
DOI:10.1021/bi801473w
Catalysis by the zinc metalloenzyme human carbonic anhydrase II (HCA II) is limited in maximal velocity by proton transfer between His64 and the zinc-bound solvent molecule. Asn62 extends into the active site cavity of HCA II adjacent to His64 and has been shown to be one of several hydrophilic residues participating in a hydrogen-bonded solvent network within the active site. We compared several site-specific mutants of HCA II with replacements at position 62 (Ala, Val, Leu, Thr, and Asp). The efficiency of catalysis in the hydration of CO2 for the resulting mutants has been characterized by 18O exchange, and the structures of the mutants have been determined by X-ray crystallography to 1.5−1.7 Å resolution. Each of these mutants maintained the ordered water structure observed by X-ray crystallography in the active site cavity of wild-type HCA II; hence, this water structure was not a variable in comparing with wild type the activities of mutants at residue 62. Crystal structures of wild-type and N62T HCA II showed both an inward and outward orientation of the side chain of His64; however, other mutants in this study showed predominantly inward (N62A, N62V, N62L) or predominantly outward (N62D) orientations of His64. A significant role of Asn62 in HCA II is to permit two conformations of the side chain of His64, the inward and outward, that contributes to maximal efficiency of proton transfer between the active site and solution. The site-specific mutant N62D had a mainly outward orientation of His64, yet the difference in pKa between the proton donor His64 and zinc-bound hydroxide was near zero, as in wild-type HCA II. The rate of proton transfer in catalysis by N62D HCA II was 5% that of wild type, showing that His64 mainly in the outward orientation is associated with inefficient proton transfer compared with His64 in wild type which shows both inward and outward orientations. These results emphasize the roles of the residues of the hydrophilic side of the active site cavity in maintaining efficient catalysis by carbonic anhydrase.
Co-reporter:David N. Silverman and Robert McKenna
Accounts of Chemical Research 2007 Volume 40(Issue 8) pp:669
Publication Date(Web):June 6, 2007
DOI:10.1021/ar7000588
Considerable attention has been focused on proton transfer through intervening water molecules in complex macromolecules of biological interest, such as bacteriorhodopsin, cytochrome c oxidase, and many others. Proton transfer in catalysis by carbonic anhydrase provides a useful model for the study of the properties of such proton translocations. High-resolution X-ray crystallography in combination with measurements of catalysis have revealed new details of this process. A prominent proton shuttle residue His64 shows evidence of structural mobility, which appears to enhance proton transfer between the active site and bulk solvent. Moreover, the properties of the imidazole side chain of His64, including its conformations and pKa, are finely tuned by surrounding residues of the active-site cavity. The structure of a network of ordered solvent molecules located between His64 and the active site are also sensitive to surrounding residues. These features combine to provide efficient proton-transfer rates as great as 106 s−1 necessary to sustain rapid catalysis.
Co-reporter:S. Zoë Fisher, Iyerus Tariku, Nicolette M. Case, Chingkuang Tu, Teri Seron, David N. Silverman, Paul J. Linser, Robert McKenna
Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics 2006 Volume 1764(Issue 8) pp:1413-1419
Publication Date(Web):August 2006
DOI:10.1016/j.bbapap.2006.06.013
Carbonic anhydrases (CAs) are zinc-containing metalloenzymes that catalyze the interconversion of carbon dioxide and bicarbonate. The α-class CAs are found predominantly in vertebrates, but they are also expressed in insects like mosquitoes. Recently, an α-CA from the midgut of Aedes aegypti larvae (AaCA1) was identified, cloned, and subsequently shown to share high sequence homologous to human CA I (HCA I). This paper presents the bacterial expression, purification, and kinetic characterization of the soluble CA domain of AaCA1. The data show AaCA1 is a highly active CA that displays inhibition by methazolamide and ethoxzolamide with nM affinity. Additionally, a homology model of AaCA1, based on the crystal structure of HCA I, is presented and the overall structure, active site, and surface charge properties are compared to those of HCA I and II. Measurements of catalysis show that AaCA1 is more like HCA II in terms of proton transfer, but more similar to HCA I in terms of conversion of carbon dioxide to bicarbonate, and these differences are rationalized in terms of structure. These results also indicate that amino acid differences in the active site of AaCA1 compared to human CAs could be used to design specific CA inhibitors for the management of mosquito populations.
Co-reporter:Farzaneh Tondnevis, Thomas M. Weiss, Tsutomu Matsui, Linda B. Bloom, Robert McKenna
Journal of Structural Biology (June 2016) Volume 194(Issue 3) pp:272-281
Publication Date(Web):1 June 2016
DOI:10.1016/j.jsb.2016.03.003
Sliding clamps are opened and loaded onto primer template junctions by clamp loaders, and once loaded on DNA, confer processivity to replicative polymerases. Previously determined crystal structures of eukaryotic and T4 clamp loader-clamp complexes have captured the sliding clamps in either closed or only partially open interface conformations. In these solution structure studies, we have captured for the first time the clamp loader-sliding clamp complex from Escherichia coli using size exclusion chromatography coupled to small angle X-ray scattering (SEC-SAXS). The data suggests the sliding clamp is in an open conformation which is wide enough to permit duplex DNA binding. The data also provides information about spatial arrangement of the sliding clamp with respect to the clamp loader subunits and is compared to complex crystal structures determined from other organisms.
Co-reporter:Rose Mikulski, Balendu Sankara Avvaru, Chingkuang Tu, Nicolette Case, Robert McKenna, David N. Silverman
Archives of Biochemistry and Biophysics (15 February 2011) Volume 506(Issue 2) pp:181-187
Publication Date(Web):15 February 2011
DOI:10.1016/j.abb.2010.12.004
Co-reporter:Rose Mikulski, John F. Domsic, George Ling, Chingkuang Tu, Arthur H. Robbins, David N. Silverman, Robert McKenna
Archives of Biochemistry and Biophysics (15 December 2011) Volume 516(Issue 2) pp:97-102
Publication Date(Web):15 December 2011
DOI:10.1016/j.abb.2011.09.011
Co-reporter:Balendu Sankara Avvaru, Daniel J. Arenas, Chingkuang Tu, D.B. Tanner, Robert McKenna, David N. Silverman
Archives of Biochemistry and Biophysics (1 October 2010) Volume 502(Issue 1) pp:53-59
Publication Date(Web):1 October 2010
DOI:10.1016/j.abb.2010.07.010
Co-reporter:Christopher D. Boone, Sonika Gill, Chingkuang Tu, David N. Silverman, Robert McKenna
Archives of Biochemistry and Biophysics (1 November 2013) Volume 539(Issue 1) pp:31-37
Publication Date(Web):1 November 2013
DOI:10.1016/j.abb.2013.09.001
Co-reporter:Fabio Pacchiano, Mayank Aggarwal, Balendu Sankara Avvaru, Arthur H. Robbins, Andrea Scozzafava, Robert McKenna and Claudiu T. Supuran
Chemical Communications 2010 - vol. 46(Issue 44) pp:NaN8373-8373
Publication Date(Web):2010/10/05
DOI:10.1039/C0CC02707C
4-Substituted-ureido benzenesulfonamides showing inhibitory activity against carbonic anhydrase (CA, EC 4.2.1.1) II between 3.3–226 nM were crystallized in complex with the enzyme. Hydrophobic interactions between the scaffold of the inhibitors in different hydrophobic pockets of the enzyme were observed, explaining the diverse inhibitory range of these derivatives.
Co-reporter:Fabrizio Carta, Mayank Aggarwal, Alfonso Maresca, Andrea Scozzafava, Robert McKenna and Claudiu T. Supuran
Chemical Communications 2012 - vol. 48(Issue 13) pp:NaN1870-1870
Publication Date(Web):2011/12/14
DOI:10.1039/C2CC16395K
The zinc enzyme carbonic anhydrase (CA, EC 4.2.1.1) is inhibited by several classes of zinc-binders (sulfonamides, sulfamates, and sulfamides) as well as by compounds which do not interact with the metal ion (phenols, polyamines and coumarins). Here we report a new class of potent CA inhibitors which bind the zinc ion: the dithiocarbamates (DTCs). They coordinate to the zinc ion from the enzyme active site in monodentate manner and establish many favorable interactions with amino acid residues nearby. Several low nanomolar CA I, II and IX inhibitors were detected.







![Benzenesulfonamide, 4-[[[(pentafluorophenyl)amino]carbonyl]amino]-](http://img.cochemist.com/ccimg/706000/705945-33-1.png)
![Benzenesulfonamide, 4-[[[(pentafluorophenyl)amino]carbonyl]amino]-](http://img.cochemist.com/ccimg/706000/705945-33-1_b.png)
![Benzenesulfonamide, 4-[[[(4-fluorophenyl)amino]carbonyl]amino]-](http://img.cochemist.com/ccimg/178700/178606-66-1.png)
![Benzenesulfonamide, 4-[[[(4-fluorophenyl)amino]carbonyl]amino]-](http://img.cochemist.com/ccimg/178700/178606-66-1_b.png)










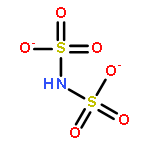








![[(sulfonatoperoxy)sulfonyl]oxidanide](http://img.cochemist.com/ccimg/15100/15092-81-6.png)
![[(sulfonatoperoxy)sulfonyl]oxidanide](http://img.cochemist.com/ccimg/15100/15092-81-6_b.png)