Co-reporter:Yoshiki Shibuya, Hung V.-T. Nguyen, and Jeremiah A. Johnson
ACS Macro Letters September 19, 2017 Volume 6(Issue 9) pp:963-963
Publication Date(Web):August 21, 2017
DOI:10.1021/acsmacrolett.7b00529
A convergent brush-first ring-opening metathesis polymerization (ROMP) approach for the synthesis of mikto-brush-arm star polymers (MBASPs) via cross-linking of dissimilar bottlebrush polymers is reported. Living bottlebrush polymers prepared via ROMP of norbornene-terminated poly(ethylene glycol) (PEG) or polystyrene (PS) macromonomers (MMs) were mixed together in a desired ratio and exposed to a bis-norbornene cross-linker to yield MBASPs with narrow size distributions. Dynamic light scattering (DLS) and transmission electron microscopy (TEM) revealed that the solution morphologies of MBASPs depended on the feed ratio of the PEG and PS bottlebrush polymers at the cross-linking stage. This work provides a robust and modular strategy for the synthesis of a new type of miktoarm star polymer wherein the star arms are bottlebrush polymers.
Co-reporter:Hung V.-T. Nguyen, Qixian Chen, Joseph T. Paletta, Peter Harvey, Yivan Jiang, Hui Zhang, Michael D. Boska, M. Francesca Ottaviani, Alan Jasanoff, Andrzej Rajca, and Jeremiah A. Johnson
ACS Central Science July 26, 2017 Volume 3(Issue 7) pp:800-800
Publication Date(Web):July 12, 2017
DOI:10.1021/acscentsci.7b00253
Metal-free magnetic resonance imaging (MRI) agents could overcome the established toxicity associated with metal-based agents in some patient populations and enable new modes of functional MRI in vivo. Herein, we report nitroxide-functionalized brush-arm star polymer organic radical contrast agents (BASP-ORCAs) that overcome the low contrast and poor in vivo stability associated with nitroxide-based MRI contrast agents. As a consequence of their unique nanoarchitectures, BASP-ORCAs possess per-nitroxide transverse relaxivities up to ∼44-fold greater than common nitroxides, exceptional stability in highly reducing environments, and low toxicity. These features combine to provide for accumulation of a sufficient concentration of BASP-ORCA in murine subcutaneous tumors up to 20 h following systemic administration such that MRI contrast on par with metal-based agents is observed. BASP-ORCAs are, to our knowledge, the first nitroxide MRI contrast agents capable of tumor imaging over long time periods using clinical high-field 1H MRI techniques.
Co-reporter:Awaneesh Singh;Alex M. Jordan;LaShanda T. J. Korley;Santidan Biswas;Anna C. Balazs;Yuwei Gu;Mao Chen;Mingjiang Zhong
ACS Central Science February 22, 2017 Volume 3(Issue 2) pp:124-134
Publication Date(Web):January 13, 2017
DOI:10.1021/acscentsci.6b00335
Light-initiated additive manufacturing techniques typically rely on layer-by-layer addition or continuous extraction of polymers formed via nonliving, free radical polymerization methods that render the final materials “dead” toward further monomer insertion; the polymer chains within the materials cannot be reactivated to induce chain extension. An alternative “living additive manufacturing” strategy would involve the use of photocontrolled living radical polymerization to spatiotemporally insert monomers into dormant “parent” materials to generate more complex and diversely functionalized “daughter” materials. Here, we demonstrate a proof-of-concept study of living additive manufacturing using end-linked polymer gels embedded with trithiocarbonate iniferters that can be activated by photoinduced single-electron transfer from an organic photoredox catalyst in solution. This system enables the synthesis of a wide range of chemically and mechanically differentiated daughter gels from a single type of parent gel via light-controlled modification of the parent’s average composition, strand length, and/or cross-linking density. Daughter gels that are softer than their parent, stiffer than their parent, larger but with the same modulus as their parent, thermally responsive, polarity responsive, healable, and weldable are all realized.
Co-reporter:Mao Chen, Shihong Deng, Yuwei Gu, Jun Lin, Michelle J. MacLeod, and Jeremiah A. Johnson
Journal of the American Chemical Society February 15, 2017 Volume 139(Issue 6) pp:2257-2257
Publication Date(Web):February 2, 2017
DOI:10.1021/jacs.6b10345
Strategies for switching polymerizations between “ON” and “OFF” states offer new possibilities for materials design and fabrication. While switching of controlled radical polymerization has been achieve using light, applied voltage, allosteric effects, chemical reagents, pH, and mechanical force, it is still challenging to introduce multiple external switches using the same catalyst to achieve logic gating of controlled polymerization reactions. Herein, we report an easy-to-synthesize thermally responsive organo-/hydro-gel that features covalently bound 10-phenylphenothiazine (PTH). With this “Gel-PTH”, we demonstrate switching of controlled radical polymerization reactions using temperature “LOW”/“HIGH”, light “ON”/“OFF”, and catalyst presence “IN”/“OUT”. Various iniferters/initiators and a wide range of monomers including acrylates, methacrylates, acrylamides, vinyl esters, and vinyl amides were polymerized by RAFT/iniferter and ATRP methods using Gel-PTH and a readily available compact fluorescent light (CFL) source. In all cases, polymer molar masses increased linearly with conversion, and narrow molar mass distributions were obtained. To further highlight the utility of Gel-PTH, we achieved “AND” gating of controlled radical polymerization wherein various combinations of three stimuli were required to induce polymer chain growth. Finally, block copolymer synthesis and catalyst recycling were demonstrated. Logic-controlled polymerization with Gel-PTH offers a straightforward approach to achieve multiplexed external switching of polymer chain growth using a single catalyst without the need for addition of exogenous reagents.
Co-reporter:M. J. MacLeod;J. A. Johnson
Polymer Chemistry (2010-Present) 2017 vol. 8(Issue 31) pp:4488-4493
Publication Date(Web):2017/08/08
DOI:10.1039/C7PY00922D
We report the synthesis of uniform oligomeric polyMOF ligands with alkyne end groups via an iterative exponential growth (IEG) strategy. These ligands were used to prepare a diblock copolymer via copper-catalyzed azide–alkyne cycloaddition “click” chemistry with azide-terminated polystyrene (PS). In the presence of Zn ions, this novel block copolymer forms a “block co-polyMOF” (BCPMOF) comprised of crystalline polyMOF domains embedded in an amorphous PS matrix. BCPMOFs represent a new type of hybrid polyMOF–polymer material.
Co-reporter:Shuting Feng;Mao Chen;Livia Giordano;Mingjun Huang;Wenxu Zhang;Chibueze V. Amanchukwu;Robinson Anandakathir;Yang Shao-Horn
Journal of Materials Chemistry A 2017 vol. 5(Issue 45) pp:23987-23998
Publication Date(Web):2017/11/21
DOI:10.1039/C7TA08321A
Electrolyte instability is one of the greatest impediments that must be overcome for the practical development of rechargeable aprotic Li–air batteries. In this work, we establish a comprehensive framework for evaluation of the stability of potential organic electrolytes for aprotic Li–air batteries that is based on four key descriptors: Bond dissociation energy, deprotonation free energy (i.e., Acidity), Nucleophilic substitution free energy, and Electrochemical oxidation/reduction. These parameters were calculated for several classes of organic compounds. The chemical stability of the molecules was studied experimentally under conditions designed to mimic the aprotic Li–air battery environment (heating in the presence of excess KO2 and Li2O2). In general, the calculated and experimental data agreed well for alkanes, alkenes, ethers, aromatics, carbonates, and S-containing and N-containing compounds. Using this dataset, we identified functional groups and other structural features of organic molecules that may be suitable for aprotic Li–air battery electrolyte design.
Co-reporter:Mao Chen, Mingjiang Zhong, and Jeremiah A. Johnson
Chemical Reviews 2016 Volume 116(Issue 17) pp:10167-10211
Publication Date(Web):March 15, 2016
DOI:10.1021/acs.chemrev.5b00671
The use of light to mediate controlled radical polymerization has emerged as a powerful strategy for rational polymer synthesis and advanced materials fabrication. This review provides a comprehensive survey of photocontrolled, living radical polymerizations (photo-CRPs). From the perspective of mechanism, all known photo-CRPs are divided into either (1) intramolecular photochemical processes or (2) photoredox processes. Within these mechanistic regimes, a large number of methods are summarized and further classified into subcategories based on the specific reagents, catalysts, etc., involved. To provide a clear understanding of each subcategory, reaction mechanisms are discussed. In addition, applications of photo-CRP reported so far, which include surface fabrication, particle preparation, photoresponsive gel design, and continuous flow technology, are summarized. We hope this review will not only provide informative knowledge to researchers in this field but also stimulate new ideas and applications to further advance photocontrolled reactions.
Co-reporter:Aleksandr V. Zhukhovitskiy; Michael G. Mavros; K. T. Queeney; Tony Wu; Troy Van Voorhis
Journal of the American Chemical Society 2016 Volume 138(Issue 27) pp:8639-8652
Publication Date(Web):July 1, 2016
DOI:10.1021/jacs.6b04962
Surface passivation has enabled the development of silicon-based solar cells and microelectronics. However, a number of emerging applications require a paradigm shift from passivation to functionalization, wherein surface functionality is installed proximal to the silicon surface. To address this need, we report here the use of persistent aminocarbenes to functionalize hydrogen-terminated silicon surfaces via Si–H insertion reactions. Through the use of model compounds (H–Si(TMS)3 and H–Si(OTMS)3), nanoparticles (H–SiNPs), and planar Si(111) wafers (H–Si(111)), we demonstrate that among different classes of persistent carbenes, the more electrophilic and nucleophilic ones, in particular, a cyclic (alkyl)(amino)carbene (CAAC) and an acyclic diaminocarbene (ADAC), are able to undergo insertion into Si–H bonds at the silicon surface, forming persistent C–Si linkages and simultaneously installing amine or aminal functionality in proximity to the surface. The CAAC (6) is particularly notable for its clean insertion reactivity under mild conditions that produces monolayers with 21 ± 3% coverage of Si(111) atop sites, commensurate with the expected maximum of ∼20%. Atomic force and transmission electron microscopy, nuclear magnetic resonance, X-ray photoelectron, and infrared spectroscopy, and time-of-flight secondary ion mass spectrometry provided evidence for the surface Si–H insertion process. Furthermore, computational studies shed light on the reaction energetics and indicated that CAAC 6 should be particularly effective at binding to silicon dihydride, trihydride, and coupled monohyride motifs, as well as oxidized surface sites. Our results pave the way for the further development of persistent carbenes as universal ligands for silicon and potentially other nonmetallic substrates.
Co-reporter:Yivan Jiang; Matthew R. Golder; Hung V.-T. Nguyen; Yufeng Wang; Mingjiang Zhong; Jonathan C. Barnes; Deborah J. C. Ehrlich
Journal of the American Chemical Society 2016 Volume 138(Issue 30) pp:9369-9372
Publication Date(Web):July 12, 2016
DOI:10.1021/jacs.6b04964
Studies on the phase segregation of unimolecular block copolymers (BCPs) are limited by a lack of reliable, versatile methods for the synthesis of such polymers on the preparative scale. Herein, we describe an advancement of Iterative Exponential Growth (IEG) wherein chiral allyl-based IEG oligomers are subjected to thiol–ene reactions and converted into unimolecular BCPs. With this strategy we have synthesized uniform BCPs with molar masses up to 12.1 kDa on ∼1 g scale. BCPs composed of decane-based side chains and either triethyleneglycol- or thioglycerol-based side chains phase-segregate into hexagonal cylinder morphologies. The assembly is not driven by side-chain crystallization, but is instead the result of amorphous BCP assembly.
Co-reporter:Jonathan C. Barnes, Peter M. Bruno, Hung V.-T. Nguyen, Longyan Liao, Jenny Liu, Michael T. Hemann, and Jeremiah A. Johnson
Journal of the American Chemical Society 2016 Volume 138(Issue 38) pp:12494-12501
Publication Date(Web):September 14, 2016
DOI:10.1021/jacs.6b06321
Single-nanoparticle (NP) combination chemotherapeutics are quickly emerging as attractive alternatives to traditional chemotherapy due to their ability to increase drug solubility, reduce off-target toxicity, enhance blood circulation lifetime, and increase the amount of drug delivered to tumors. In the case of NP-bound drugs, that is, NP-prodrugs, the current standard of practice is to assume that the subcellular mechanism of action for each drug released from the NP mirrors that of the unbound, free-drug. Here, we use an RNAi signature assay for the first time to examine the mechanism of action of multidrug-conjugated NP prodrugs relative to their small molecule prodrugs and native drug mechanisms of action. Additionally, the effective additive contribution of three different drugs in a single-NP platform is characterized. The results indicate that some platinum(IV) cisplatin prodrugs, although cytotoxic, may not have the expected mechanism of action for cisplatin. This insight was utilized to develop a novel platinum(IV) oxaliplatin prodrug and incorporate it into a three-drug-conjugated NP, where each drug’s mechanism of action is preserved, to treat tumor-bearing mice with otherwise lethal levels of chemotherapy.
Co-reporter:Ken Kawamoto, Mingjiang Zhong, Karim R. Gadelrab, Li-Chen Cheng, Caroline A. Ross, Alfredo Alexander-Katz, and Jeremiah A. Johnson
Journal of the American Chemical Society 2016 Volume 138(Issue 36) pp:11501-11504
Publication Date(Web):September 1, 2016
DOI:10.1021/jacs.6b07670
We report the synthesis of Janus bottlebrush block copolymers by graft-through polymerization of branched diblock macromonomers. Self-assembly of the bottlebrushes was characterized by small-angle X-ray scattering, atomic force microscopy, and scanning electron microscopy. Phase separation and packing models of the bottlebrushes were computed, and their self-assembly behavior was corroborated experimentally in bulk and in thin films. Lamellar, hexagonal cylinder, and gyroid phases were observed and modeled. The A-branch-B Janus bottlebrush structure provides several unique advantages in the context of bottlebrush polymer assembly, including access to the first examples of gyroid phases.
Co-reporter:Yufeng Wang, Mingjiang Zhong, Jiwon V. Park, Aleksandr V. Zhukhovitskiy, Weichao Shi, and Jeremiah A. Johnson
Journal of the American Chemical Society 2016 Volume 138(Issue 33) pp:10708-10715
Publication Date(Web):July 27, 2016
DOI:10.1021/jacs.6b06712
We report a stepwise assembly strategy for the integration of metal–organic cages (MOCs) into block copolymers (BCPs). This approach creates “block co-polyMOC” (BCPMOC) materials whose microscopic structures and mechanical properties are readily tunable by adjusting the size and geometry of the MOCs and the composition of the BCPs. In the first assembly step, BCPs functionalized with a pyridyl ligand on the chain end form star-shaped polymers triggered by metal-coordination-induced MOC assembly. The type of MOC junction employed precisely determines the number of arms for the star polymer. In the second step, microphase separation of the BCP is induced, physically cross-linking the star polymers and producing the desired BCPMOC networks in the bulk or gel state. We demonstrate that large spherical M12L24 MOCs, small paddlewheel M2L4 MOCs, or a mixture of both can be incorporated into BCPMOCs to provide materials with tailored branch functionality, phase separation, microdomain spacing, and mechanical properties. Given the synthetic and functional diversity of MOCs and BCPs, our method should enable access to BCPMOCs for a wide range of applications.
Co-reporter:Aleksandr V. Zhukhovitskiy, Julia Zhao, Mingjiang Zhong, Eric G. Keeler, Eric A. Alt, Paul Teichen, Robert G. Griffin, Michael J. A. Hore, Adam P. Willard, and Jeremiah A. Johnson
Macromolecules 2016 Volume 49(Issue 18) pp:6896-6902
Publication Date(Web):September 6, 2016
DOI:10.1021/acs.macromol.6b01607
Polymer gels are often very soft due to their low branch functionality (f) and the inevitable presence of defects (e.g., primary loops or dangling chains). Polymer metal–organic cage (polyMOC) gels are a relatively new class of supramolecular gels with precisely defined junction structures made possible by subcomponent assembly of nanoscale MOCs connected by polymer chains. Herein, we report that variation of the molecular weight and architecture of the polymer component of polyMOCs provides an entry into gels with ultra-high f. For example, materials with f ∼ 9–12, i.e., ∼ 9–12 polymer chains connect each MOC within the gel network, are realized. As a consequence of their increased f, these gels display exemplary mechanical properties at low concentrations (down to 240 μM) of metal ions and only 5.4–5.9 wt % of polymer. Furthermore, X-ray and neutron scattering pointed to an additional level of structural hierarchy that arises from the assembly of M12L24 MOCs into clusters. The relationships between polymer and polyMOC network structure revealed here will facilitate the design of high-performance polyMOCs.
Co-reporter:Aleksandr V. Zhukhovitskiy, Michelle J. MacLeod, and Jeremiah A. Johnson
Chemical Reviews 2015 Volume 115(Issue 20) pp:11503
Publication Date(Web):September 22, 2015
DOI:10.1021/acs.chemrev.5b00220
Co-reporter:Michelle J. MacLeod
Journal of the American Chemical Society 2015 Volume 137(Issue 25) pp:7974-7977
Publication Date(Web):June 17, 2015
DOI:10.1021/jacs.5b02452
N-Heterocyclic carbenes (NHCs) have emerged as versatile ligands for surface functionalization. Their ease of synthesis and ability to form strong bonds with a range of substrates provide a unique complement to traditional surface modification methods. Gold nanoparticles (NPs) are a particularly useful class of materials whose applications intimately depend on surface functionalization. Here we report the development of PEGylated-NHC ligands for Au-NP surfaces and the first example of NHC-functionalized NPs that are compatible with biologically relevant conditions. Our PEGylated-NHC-Au-NPs are stable toward aggregation in aqueous solutions in the pH range of 3–14, in <250 mM electrolyte solutions, at high and low temperatures (95 and −78 °C), in cell culture media, and in aqueous H2O2 solutions. This work demonstrates for the first time that NHCs can serve as anchors for water-soluble Au-NPs and opens the door to potential biomedical applications of NHC surface anchors.
Co-reporter:Mao Chen and Jeremiah A. Johnson
Chemical Communications 2015 vol. 51(Issue 31) pp:6742-6745
Publication Date(Web):17 Mar 2015
DOI:10.1039/C5CC01562F
Herein, we report simple flow reactor designs that enable photo-controlled living radical polymerization (photo-CRP) from trithiocarbonates (TTCs) with significant enhancements in scalability and reaction rates compared to the analogous batch reactions. We also demonstrate the “on/off” photo-switchability of this reaction under flow conditions.
Co-reporter:Mao Chen, Michelle J. MacLeod, and Jeremiah A. Johnson
ACS Macro Letters 2015 Volume 4(Issue 5) pp:566
Publication Date(Web):April 30, 2015
DOI:10.1021/acsmacrolett.5b00241
Living radical polymerization of acrylates and acrylamides from trithiocarbonate iniferters using a compact fluorescent lamp (CFL) bulb and 10-phenylphenothiazine as an organic photoredox catalyst is reported. With this system, chain growth can be efficiently switched between “on” and “off” in response to visible light. Polymer molar masses increase linearly with conversion, and narrow molar mass distributions are obtained. The excellent fidelity of the trithiocarbonate-iniferter enables the preparation of triblock copolymers from macro-iniferters under the same visible-light mediated protocol, using UV light without a photoredox catalyst or under traditional thermally induced RAFT conditions. We expect that the simplicity and efficiency of this metal-free, visible-light-mediated polymerization will enable the synthesis and modification of a range of materials under mild conditions.
Co-reporter:Ken Kawamoto, Scott C. Grindy, Jenny Liu, Niels Holten-Andersen, and Jeremiah A. Johnson
ACS Macro Letters 2015 Volume 4(Issue 4) pp:458
Publication Date(Web):April 9, 2015
DOI:10.1021/acsmacrolett.5b00221
The inverse-electron demand Diels–Alder cycloaddition of tetrazines and olefins has emerged as a powerful coupling reaction for the formation of polymer gels with diverse applications. Tetrazines are also excellent ligands for metal atoms. For example, 3,6-bis(2-pyridyl)-1,2,4,5-tetrazines (bptz) have been used to generate discrete supramolecular Mxbptzy metal clusters and extended 2D grid structures. We reasoned that both the Diels–Alder and the metal-coordination modes of reactivity of bptz derivatives could be leveraged in the context of hydrogel design to yield novel hybrid materials. Here we report on the formation of supramolecular hydrogels via substoichiometric Diels–Alder functionalization of bptz ligands bound to the ends of poly(ethylene glycol) (PEG) chains followed by metal-coordination-induced gelation in the presence of Ni2+ and Fe2+ salts. Our results show that simple bptz-based polymers are versatile precursors to a diverse range of novel functional materials.
Co-reporter:Frank A. Leibfarth;Timothy F. Jamison
PNAS 2015 Volume 112 (Issue 34 ) pp:10617-10622
Publication Date(Web):2015-08-25
DOI:10.1073/pnas.1508599112
We report a semiautomated synthesis of sequence and architecturally defined, unimolecular macromolecules through a marriage
of multistep flow synthesis and iterative exponential growth (Flow-IEG). The Flow-IEG system performs three reactions and
an in-line purification in a total residence time of under 10 min, effectively doubling the molecular weight of an oligomeric
species in an uninterrupted reaction sequence. Further iterations using the Flow-IEG system enable an exponential increase
in molecular weight. Incorporating a variety of monomer structures and branching units provides control over polymer sequence
and architecture. The synthesis of a uniform macromolecule with a molecular weight of 4,023 g/mol is demonstrated. The user-friendly
nature, scalability, and modularity of Flow-IEG provide a general strategy for the automated synthesis of sequence-defined,
unimolecular macromolecules. Flow-IEG is thus an enabling tool for theory validation, structure–property studies, and advanced
applications in biotechnology and materials science.
Co-reporter:Ken Kawamoto, Mingjiang Zhong, Rui Wang, Bradley D. Olsen, and Jeremiah A. Johnson
Macromolecules 2015 Volume 48(Issue 24) pp:8980-8988
Publication Date(Web):December 1, 2015
DOI:10.1021/acs.macromol.5b02243
Through the use of macromolecular design and efficient chemical reactions it is now possible to control the composition of polymer networks and gels with excellent precision. In contrast, topological defects are still impossible to avoid and are generally difficult to quantify. For example, primary loops that form when a bifunctional monomer (A2) reacts twice with the same f functional (f > 2) monomer (Bf) during formation of an end-linked A2 + Bf network represent a pervasive defect that has a detrimental effect on mechanical integrity. Methods for the quantitative analysis of primary loops in such materials have recently emerged; however, these methods have only been applied to the simplest network structure: A2 + B3. Herein, we report strategies for counting primary loops in tetrafunctional (A2 + B4) networks and networks with mixed tri- and tetrafunctional (A2 + B3/B4) junctions. We apply these strategies to the quantitative analysis of primary loops in a series of end-linked poly(ethylene glycol) hydrogels synthesized via copper-catalyzed azide–alkyne cycloaddition “click” chemistry. Our results show that A2 + B4 networks are particularly susceptible to cyclic defects compared to A2 + B3 networks and that higher-order cyclic species must play a significant role in the gel point of the former materials. Our experimental results were compared to rate theory and Monte Carlo simulations. This work reveals new structural insights into a widely studied family of materials and sets the stage for the development of strategies to tune network defects in such gels.
Co-reporter:Aleksr V. Zhukhovitskiy;Julie Geng ;Dr. Jeremiah A. Johnson
Chemistry - A European Journal 2015 Volume 21( Issue 15) pp:5685-5688
Publication Date(Web):
DOI:10.1002/chem.201500052
Abstract
We report the discovery that 1,3-bis(aryl)imidazolidin-2-ylidenes, one of the most widely studied classes of N-heterocyclic carbenes (NHCs), undergo quantitative conversion to zwitterionic “NHC-CDI” amidinates upon heating to ≈100 °C in solution. The mechanism of this novel NHC decomposition process is studied in detail. These studies enabled the rational synthesis of a new class of bench stable amidinates from a panel of NHCs and carbodiimides. We expect these constructs to find utility in a variety of applications.
Co-reporter:Aleksr V. Zhukhovitskiy;Julie Geng ;Dr. Jeremiah A. Johnson
Chemistry - A European Journal 2015 Volume 21( Issue 15) pp:
Publication Date(Web):
DOI:10.1002/chem.201500674
Abstract
Invited for the cover of this issue is the group of Jeremiah A. Johnson at the Massachusetts Institute of Technology. The image depicts the conversion of 1,3-bis(aryl)imidazolidin-2-ylidenes to zwitterionic “NHC-CDI” amidinates (NHC-CDI = N-heterocyclic carbene–carbodiimide). Read the full text of the article at 10.1002/chem.201500052.
Co-reporter:Aleksr V. Zhukhovitskiy;Julie Geng ;Dr. Jeremiah A. Johnson
Chemistry - A European Journal 2015 Volume 21( Issue 15) pp:
Publication Date(Web):
DOI:10.1002/chem.201590057
Co-reporter:Longyan Liao ; Jenny Liu ; Erik C. Dreaden ; Stephen W. Morton ; Kevin E. Shopsowitz ; Paula T. Hammond
Journal of the American Chemical Society 2014 Volume 136(Issue 16) pp:5896-5899
Publication Date(Web):April 11, 2014
DOI:10.1021/ja502011g
The synthesis of polymer therapeutics capable of controlled loading and synchronized release of multiple therapeutic agents remains a formidable challenge in drug delivery and synthetic polymer chemistry. Herein, we report the synthesis of polymer nanoparticles (NPs) that carry precise molar ratios of doxorubicin, camptothecin, and cisplatin. To our knowledge, this work provides the first example of orthogonally triggered release of three drugs from single NPs. The highly convergent synthetic approach opens the door to new NP-based combination therapies for cancer.
Co-reporter:Huaxing Zhou ; Eva-Maria Schön ; Muzhou Wang ; Matthew J. Glassman ; Jenny Liu ; Mingjiang Zhong ; David Díaz Díaz ; Bradley D. Olsen
Journal of the American Chemical Society 2014 Volume 136(Issue 26) pp:9464-9470
Publication Date(Web):May 29, 2014
DOI:10.1021/ja5042385
Molecular defects critically impact the properties of materials. Here we introduce a paradigm called “isotopic labeling disassembly spectrometry” (ILDaS) that facilitates unprecedented precise experimental correlations between elastically inactive network defects (dangling chains and primary loops) and network formation kinetics and precursor structure. ILDaS is inspired by classical crossover experiments, which are often used to interrogate whether a reaction mechanism proceeds via an inter- or intramolecular pathway. We show that if networks are designed from labeled bifunctional monomers that transfer their labels to multifunctional junctions upon network formation, then the extent of junction labeling correlates directly with the number of dangling chains and cyclic imperfections within the network. We demonstrate two complementary ILDaS approaches that enable defect measurements with short analysis times, low cost, and synthetic versatility applicable to a broad range of network materials including polydisperse polymer precursors. The results will spur new experimental and theoretical investigations into the interplay between polymer network structure and properties.
Co-reporter:Angela X. Gao, Longyan Liao, and Jeremiah A. Johnson
ACS Macro Letters 2014 Volume 3(Issue 9) pp:854
Publication Date(Web):August 13, 2014
DOI:10.1021/mz5004097
A panel of acid-labile bis-norbornene cross-linkers was synthesized and evaluated for the formation of acid-degradable brush-arm star polymers (BASPs) via the brush-first ring-opening metathesis polymerization (ROMP) method. An acetal-based cross-linker was identified that, when employed in conjunction with a poly(ethylene glycol) (PEG) macromonomer, provided highly controlled BASP formation reactions. A combination of this new cross-linker with a novel doxorubicin (DOX)-branch-PEG macromonomer provided BASPs that simultaneously degrade and release cytotoxic DOX in vitro.
Co-reporter:Alan O. Burts;Angela X. Gao
Macromolecular Rapid Communications 2014 Volume 35( Issue 2) pp:168-173
Publication Date(Web):
DOI:10.1002/marc.201300618
Co-reporter:Alan O. Burts;Longyan Liao;Ying Y. Lu;David A. Tirrell
Photochemistry and Photobiology 2014 Volume 90( Issue 2) pp:380-385
Publication Date(Web):
DOI:10.1111/php.12182
Abstract
New strategies for the synthesis of multifunctional particles that respond to external stimuli and release biologically relevant agents will enable the discovery of new formulations for drug delivery. In this article, we combine two powerful methods: brush-first ring-opening metathesis polymerization and copper-catalyzed azide–alkyne cycloaddition click chemistry, for the synthesis of a novel class of brush-arm star polymers (BASPs) that simultaneously degrade and release the anticancer drug doxorubicin (DOX) in response to 365 nm light. In vitro cell viability studies were performed to study the toxicity of azide- and DOX-loaded BASPs. The former were completely nontoxic. The latter showed minimal toxicity in the absence of light; UV-triggered DOX release led to IC50 values that were similar to that of free DOX.
Co-reporter:Aleksandr V. Zhukhovitskiy ; Michael G. Mavros ; Troy Van Voorhis
Journal of the American Chemical Society 2013 Volume 135(Issue 20) pp:7418-7421
Publication Date(Web):May 13, 2013
DOI:10.1021/ja401965d
New strategies to access functional monolayers could augment current surface modification methods. Here we present addressable N-heterocyclic carbene (ANHC) anchors for gold surfaces. A suite of experimental and theoretical methods was used to characterize ANHC monolayers. We demonstrate grafting of highly fluorinated polymers from surface-bound ANHCs. This work establishes ANHCs as viable anchors for gold surfaces.
Co-reporter:Dr. Huaxing Zhou ;Dr. Jeremiah A. Johnson
Angewandte Chemie International Edition 2013 Volume 52( Issue 8) pp:2235-2238
Publication Date(Web):
DOI:10.1002/anie.201207966
Co-reporter:Dr. Huaxing Zhou ;Dr. Jeremiah A. Johnson
Angewandte Chemie 2013 Volume 125( Issue 8) pp:2291-2294
Publication Date(Web):
DOI:10.1002/ange.201207966
Co-reporter:Bradley D. Olsen
PNAS 2013 Volume 110 (Issue 22 ) pp:E1973
Publication Date(Web):2013-05-28
DOI:10.1073/pnas.1304496110
Co-reporter:Jenny Liu ; Alan O. Burts ; Yongjun Li ; Aleksandr V. Zhukhovitskiy ; M. Francesca Ottaviani ; Nicholas J. Turro
Journal of the American Chemical Society 2012 Volume 134(Issue 39) pp:16337-16344
Publication Date(Web):September 6, 2012
DOI:10.1021/ja3067176
We describe the parallel, one-pot synthesis of core-photocleavable, poly(norbornene)-co-poly(ethylene glycol) (PEG) brush-arm star polymers (BASPs) via a route that combines the “graft-through” and “arm-first” methodologies for brush polymer and star polymer synthesis, respectively. In this method, ring-opening metathesis polymerization of a norbornene–PEG macromonomer generates small living brush initiators. Transfer of various amounts of this brush initiator to vials containing a photocleavable bis-norbornene cross-linker yielded a series of water-soluble BASPs with low polydispersities and molecular weights that increased geometrically as a function of the amount of bis-norbornene added. The BASP cores were cleaved upon exposure to UV light; the extent of photo-disassembly depended on the amount of cross-linker. EPR spectroscopy of nitroxide-labeled BASPs was used to probe differences between the BASP core and surface environments. We expect that BASPs will find applications as easy-to-synthesize, stimuli-responsive core–shell nanostructures.
Co-reporter:Huaxing Zhou;Jiyeon Woo;Bradley D. Olsen;Muzhou Wang;Alexandra M. Cok
PNAS 2012 Volume 109 (Issue 47 ) pp:19119-19124
Publication Date(Web):2012-11-20
DOI:10.1073/pnas.1213169109
Much of our fundamental knowledge related to polymer networks is built on an assumption of ideal end-linked network structure.
Real networks invariably possess topological imperfections that negatively affect mechanical properties; modifications of
classical network theories have been developed to account for these defects. Despite decades of effort, there are no known
experimental protocols for precise quantification of even the simplest topological network imperfections: primary loops. Here
we present a simple conceptual framework that enables primary loop quantification in polymeric materials. We apply this framework
to measure the fraction of primary loop junctions in trifunctional PEG-based hydrogels. We anticipate that the concepts described
here will open new avenues of theoretical and experimental research related to polymer network structure.
Co-reporter:Weibin Li, Hoyong Chung, Chris Daeffler, Jeremiah A. Johnson, and Robert H. Grubbs
Macromolecules 2012 Volume 45(Issue 24) pp:9595-9603
Publication Date(Web):December 4, 2012
DOI:10.1021/ma301666x
To address the practical issues of polymer molecular weight determination, the first accurate polymer weight-average molecular weight determination method in diverse living/controlled polymerization via DOSY (diffusion-ordered NMR spectroscopy) is reported. Based on the linear correlation between the logarithm of diffusion coefficient (log D) and the molecular weights (log Mw), external calibration curves were created to give predictions of molecular weights of narrowly dispersed polymers. This method was successfully applied to atom transfer radical polymerization (ATRP), reversible addition–fragmentation chain transfer (RAFT), and ring-opening metathesis polymerization (ROMP), with weight-average molecular weights given by this method closely correlated to those obtained from GPC measurement.
Co-reporter:Yan Xia ; Yongjun Li ; Alan O. Burts ; M. Francesca Ottaviani ; David A. Tirrell ; Jeremiah A. Johnson ; Nicholas J. Turro ;Robert H. Grubbs
Journal of the American Chemical Society 2011 Volume 133(Issue 49) pp:19953-19959
Publication Date(Web):October 24, 2011
DOI:10.1021/ja2085349
Spin-labeled polylactide brush polymers were synthesized via ring-opening metathesis polymerization (ROMP), and nitroxide radicals were incorporated at three different locations of brush polymers: the end and the middle of the backbone, and the end of the side chains (periphery). Electron paramagnetic resonance (EPR) was used to quantitatively probe the macromolecular structure of brush polymers in dilute solutions. The peripheral spin-labels showed significantly higher mobility than the backbone labels, and in dimethylsulfoxide (DMSO), the backbone end labels were shown to be more mobile than the middle labels. Reduction of the nitroxide labels by a polymeric reductant revealed location-dependent reactivity of the nitroxide labels: peripheral nitroxides were much more reactive than the backbone nitroxides. In contrast, almost no difference was observed when a small molecule reductant was used. These results reveal that the dense side chains of brush polymers significantly reduce the interaction of the backbone region with external macromolecules, but allow free diffusion of small molecules.
Co-reporter:Mao Chen and Jeremiah A. Johnson
Chemical Communications 2015 - vol. 51(Issue 31) pp:NaN6745-6745
Publication Date(Web):2015/03/17
DOI:10.1039/C5CC01562F
Herein, we report simple flow reactor designs that enable photo-controlled living radical polymerization (photo-CRP) from trithiocarbonates (TTCs) with significant enhancements in scalability and reaction rates compared to the analogous batch reactions. We also demonstrate the “on/off” photo-switchability of this reaction under flow conditions.
.jpg)






































![1H-1,2,3-Triazole-1-ethanol, 4-[[(2R)-3-[4-[[(2R)-2-oxiranylmethoxy]methyl]-1H-1,2,3-triazol-1-yl]-2-(phenylmethoxy)propoxy]methyl]-α-[[[1-[(2R)-2-(phenylmethoxy)-3-(2-propyn-1-yloxy)propyl]-1H-1,2,3-triazol-4-yl]methoxy]methyl]-, 1-acetate, (αR)-](/data/chemimg/3721800/1809614-24-1.png)
![1H-1,2,3-Triazole-1-ethanol, 4-[[(2R)-3-[4-[[(2R)-2-oxiranylmethoxy]methyl]-1H-1,2,3-triazol-1-yl]-2-(phenylmethoxy)propoxy]methyl]-α-[[[1-[(2R)-2-(phenylmethoxy)-3-(2-propyn-1-yloxy)propyl]-1H-1,2,3-triazol-4-yl]methoxy]methyl]-, 1-acetate, (αR)-](/data/chemimg/3721800/1809614-24-1_b.png)
![1H-1,2,3-Triazole-1-ethanol, 4-[[(2R)-3-[4-[[(2R)-2-(acetyloxy)-3-azidopropoxy]methyl]-1H-1,2,3-triazol-1-yl]-2-(phenylmethoxy)propoxy]methyl]-α-[[[1-[(2R)-3-[[3-[(1,1-dimethylethyl)dimethylsilyl]-2-propyn-1-yl]oxy]-2-(phenylmethoxy)propyl]-1H-1,2,3-triazol-4-yl]methoxy]methyl]-, 1-acetate, (αR)-](/data/chemimg/3721800/1809614-23-0.png)
![1H-1,2,3-Triazole-1-ethanol, 4-[[(2R)-3-[4-[[(2R)-2-(acetyloxy)-3-azidopropoxy]methyl]-1H-1,2,3-triazol-1-yl]-2-(phenylmethoxy)propoxy]methyl]-α-[[[1-[(2R)-3-[[3-[(1,1-dimethylethyl)dimethylsilyl]-2-propyn-1-yl]oxy]-2-(phenylmethoxy)propyl]-1H-1,2,3-triazol-4-yl]methoxy]methyl]-, 1-acetate, (αR)-](/data/chemimg/3721800/1809614-23-0_b.png)
![1H-1,2,3-Triazole-1-ethanol, α-[[[1-[(2R)-3-[[3-[(1,1-dimethylethyl)dimethylsilyl]-2-propyn-1-yl]oxy]-2-(phenylmethoxy)propyl]-1H-1,2,3-triazol-4-yl]methoxy]methyl]-4-[[(2R)-3-[4-[[(2R)-2-oxiranylmethoxy]methyl]-1H-1,2,3-triazol-1-yl]-2-(phenylmethoxy)propoxy]methyl]-, 1-acetate, (αR)-](/data/chemimg/3721800/1809614-22-9.png)
![1H-1,2,3-Triazole-1-ethanol, α-[[[1-[(2R)-3-[[3-[(1,1-dimethylethyl)dimethylsilyl]-2-propyn-1-yl]oxy]-2-(phenylmethoxy)propyl]-1H-1,2,3-triazol-4-yl]methoxy]methyl]-4-[[(2R)-3-[4-[[(2R)-2-oxiranylmethoxy]methyl]-1H-1,2,3-triazol-1-yl]-2-(phenylmethoxy)propoxy]methyl]-, 1-acetate, (αR)-](/data/chemimg/3721800/1809614-22-9_b.png)
![1H-1,2,3-Triazole, 4-[[(2R)-2-oxiranylmethoxy]methyl]-1-[(2R)-2-(phenylmethoxy)-3-(2-propyn-1-yloxy)propyl]-](/data/chemimg/3721700/1809614-21-8.png)
![1H-1,2,3-Triazole, 4-[[(2R)-2-oxiranylmethoxy]methyl]-1-[(2R)-2-(phenylmethoxy)-3-(2-propyn-1-yloxy)propyl]-](/data/chemimg/3721700/1809614-21-8_b.png)
![2-Propanol, 1-azido-3-[[1-[(2R)-3-[[3-[(1,1-dimethylethyl)dimethylsilyl]-2-propyn-1-yl]oxy]-2-(phenylmethoxy)propyl]-1H-1,2,3-triazol-4-yl]methoxy]-, 2-acetate, (2R)-](/data/chemimg/3721700/1809614-20-7.png)
![2-Propanol, 1-azido-3-[[1-[(2R)-3-[[3-[(1,1-dimethylethyl)dimethylsilyl]-2-propyn-1-yl]oxy]-2-(phenylmethoxy)propyl]-1H-1,2,3-triazol-4-yl]methoxy]-, 2-acetate, (2R)-](/data/chemimg/3721700/1809614-20-7_b.png)
![1H-1,2,3-Triazole, 1-[(2R)-3-[[3-[(1,1-dimethylethyl)dimethylsilyl]-2-propyn-1-yl]oxy]-2-(phenylmethoxy)propyl]-4-[[(2R)-2-oxiranylmethoxy]methyl]-](/data/chemimg/3721700/1809614-19-4.png)
![1H-1,2,3-Triazole, 1-[(2R)-3-[[3-[(1,1-dimethylethyl)dimethylsilyl]-2-propyn-1-yl]oxy]-2-(phenylmethoxy)propyl]-4-[[(2R)-2-oxiranylmethoxy]methyl]-](/data/chemimg/3721700/1809614-19-4_b.png)
![1H-1,2,3-Triazole-1-ethanol, 4-[[(2R)-2-(acetyloxy)-3-[4-[[(2R)-2-(acetyloxy)-3-[4-[[(2R)-2-(acetyloxy)-3-[4-[[(2R)-2-oxiranylmethoxy]methyl]-1H-1,2,3-triazol-1-yl]propoxy]methyl]-1H-1,2,3-triazol-1-yl]propoxy]methyl]-1H-1,2,3-triazol-1-yl]propoxy]methyl]-α-[[[1-[(2R)-2-(acetyloxy)-3-[[1-[(2R)-2-(acetyloxy)-3-[[1-[(2R)-2-(acetyloxy)-3-(2-propyn-1-yloxy)propyl]-1H-1,2,3-triazol-4-yl]methoxy]propyl]-1H-1,2,3-triazol-4-yl]methoxy]propyl]-1H-1,2,3-triazol-4-yl]methoxy]methyl]-, 1-acetate, (αR)-](/data/chemimg/3721700/1809614-15-0.png)
![1H-1,2,3-Triazole-1-ethanol, 4-[[(2R)-2-(acetyloxy)-3-[4-[[(2R)-2-(acetyloxy)-3-[4-[[(2R)-2-(acetyloxy)-3-[4-[[(2R)-2-oxiranylmethoxy]methyl]-1H-1,2,3-triazol-1-yl]propoxy]methyl]-1H-1,2,3-triazol-1-yl]propoxy]methyl]-1H-1,2,3-triazol-1-yl]propoxy]methyl]-α-[[[1-[(2R)-2-(acetyloxy)-3-[[1-[(2R)-2-(acetyloxy)-3-[[1-[(2R)-2-(acetyloxy)-3-(2-propyn-1-yloxy)propyl]-1H-1,2,3-triazol-4-yl]methoxy]propyl]-1H-1,2,3-triazol-4-yl]methoxy]propyl]-1H-1,2,3-triazol-4-yl]methoxy]methyl]-, 1-acetate, (αR)-](/data/chemimg/3721700/1809614-15-0_b.png)
![1H-1,2,3-Triazole-1-ethanol, 4-[[(2R)-2-(acetyloxy)-3-[4-[[(2R)-2-oxiranylmethoxy]methyl]-1H-1,2,3-triazol-1-yl]propoxy]methyl]-α-[[[1-[(2R)-2-(acetyloxy)-3-(2-propyn-1-yloxy)propyl]-1H-1,2,3-triazol-4-yl]methoxy]methyl]-, 1-acetate, (αR)-](/data/chemimg/3721700/1809614-12-7.png)
![1H-1,2,3-Triazole-1-ethanol, 4-[[(2R)-2-(acetyloxy)-3-[4-[[(2R)-2-oxiranylmethoxy]methyl]-1H-1,2,3-triazol-1-yl]propoxy]methyl]-α-[[[1-[(2R)-2-(acetyloxy)-3-(2-propyn-1-yloxy)propyl]-1H-1,2,3-triazol-4-yl]methoxy]methyl]-, 1-acetate, (αR)-](/data/chemimg/3721700/1809614-12-7_b.png)
![1H-1,2,3-Triazole-1-ethanol, 4-[[(2R)-2-(acetyloxy)-3-[4-[[(2R)-2-(acetyloxy)-3-azidopropoxy]methyl]-1H-1,2,3-triazol-1-yl]propoxy]methyl]-α-[[[1-[(2R)-2-(acetyloxy)-3-[[3-[(1,1-dimethylethyl)dimethylsilyl]-2-propyn-1-yl]oxy]propyl]-1H-1,2,3-triazol-4-yl]methoxy]methyl]-, 1-acetate, (αR)-](/data/chemimg/3721700/1809614-11-6.png)
![1H-1,2,3-Triazole-1-ethanol, 4-[[(2R)-2-(acetyloxy)-3-[4-[[(2R)-2-(acetyloxy)-3-azidopropoxy]methyl]-1H-1,2,3-triazol-1-yl]propoxy]methyl]-α-[[[1-[(2R)-2-(acetyloxy)-3-[[3-[(1,1-dimethylethyl)dimethylsilyl]-2-propyn-1-yl]oxy]propyl]-1H-1,2,3-triazol-4-yl]methoxy]methyl]-, 1-acetate, (αR)-](/data/chemimg/3721700/1809614-11-6_b.png)
![1H-1,2,3-Triazole-1-ethanol, α-[[[1-[(2R)-2-(acetyloxy)-3-[[3-[(1,1-dimethylethyl)dimethylsilyl]-2-propyn-1-yl]oxy]propyl]-1H-1,2,3-triazol-4-yl]methoxy]methyl]-4-[[(2R)-2-(acetyloxy)-3-[4-[[(2R)-2-oxiranylmethoxy]methyl]-1H-1,2,3-triazol-1-yl]propoxy]methyl]-, 1-acetate, (αR)-](/data/chemimg/3721700/1809614-10-5.png)
![1H-1,2,3-Triazole-1-ethanol, α-[[[1-[(2R)-2-(acetyloxy)-3-[[3-[(1,1-dimethylethyl)dimethylsilyl]-2-propyn-1-yl]oxy]propyl]-1H-1,2,3-triazol-4-yl]methoxy]methyl]-4-[[(2R)-2-(acetyloxy)-3-[4-[[(2R)-2-oxiranylmethoxy]methyl]-1H-1,2,3-triazol-1-yl]propoxy]methyl]-, 1-acetate, (αR)-](/data/chemimg/3721700/1809614-10-5_b.png)
![1H-1,2,3-Triazole-1-ethanol, 4-[[(2R)-2-oxiranylmethoxy]methyl]-α-[(2-propyn-1-yloxy)methyl]-, 1-acetate, (αR)-](/data/chemimg/3721700/1809614-09-2.png)
![1H-1,2,3-Triazole-1-ethanol, 4-[[(2R)-2-oxiranylmethoxy]methyl]-α-[(2-propyn-1-yloxy)methyl]-, 1-acetate, (αR)-](/data/chemimg/3721700/1809614-09-2_b.png)
![1H-1,2,3-Triazole-1-ethanol, 4-[[(2R)-2-(acetyloxy)-3-azidopropoxy]methyl]-α-[[[3-[(1,1-dimethylethyl)dimethylsilyl]-2-propyn-1-yl]oxy]methyl]-, 1-acetate, (αR)-](/data/chemimg/3721700/1809614-08-1.png)
![1H-1,2,3-Triazole-1-ethanol, 4-[[(2R)-2-(acetyloxy)-3-azidopropoxy]methyl]-α-[[[3-[(1,1-dimethylethyl)dimethylsilyl]-2-propyn-1-yl]oxy]methyl]-, 1-acetate, (αR)-](/data/chemimg/3721700/1809614-08-1_b.png)
![1H-1,2,3-Triazole-1-ethanol, α-[[[3-[(1,1-dimethylethyl)dimethylsilyl]-2-propyn-1-yl]oxy]methyl]-4-[[(2R)-2-oxiranylmethoxy]methyl]-, 1-acetate, (αR)-](/data/chemimg/3721700/1809614-07-0.png)
![1H-1,2,3-Triazole-1-ethanol, α-[[[3-[(1,1-dimethylethyl)dimethylsilyl]-2-propyn-1-yl]oxy]methyl]-4-[[(2R)-2-oxiranylmethoxy]methyl]-, 1-acetate, (αR)-](/data/chemimg/3721700/1809614-07-0_b.png)
![1H-1,2,3-Triazole-1-ethanol, 4-[[(2R)-2-(acetyloxy)-3-[4-[[(2S)-2-(acetyloxy)-3-[4-[[(2R)-2-(acetyloxy)-3-[4-[[(2S)-2-oxiranylmethoxy]methyl]-1H-1,2,3-triazol-1-yl]propoxy]methyl]-1H-1,2,3-triazol-1-yl]propoxy]methyl]-1H-1,2,3-triazol-1-yl]propoxy]methyl]-α-[[[1-[(2R)-2-(acetyloxy)-3-[[1-[(2S)-2-(acetyloxy)-3-[[1-[(2R)-2-(acetyloxy)-3-(2-propyn-1-yloxy)propyl]-1H-1,2,3-triazol-4-yl]methoxy]propyl]-1H-1,2,3-triazol-4-yl]methoxy]propyl]-1H-1,2,3-triazol-4-yl]methoxy]methyl]-, 1-acetate, (αS)-](/data/chemimg/3721700/1809614-01-4.png)
![1H-1,2,3-Triazole-1-ethanol, 4-[[(2R)-2-(acetyloxy)-3-[4-[[(2S)-2-(acetyloxy)-3-[4-[[(2R)-2-(acetyloxy)-3-[4-[[(2S)-2-oxiranylmethoxy]methyl]-1H-1,2,3-triazol-1-yl]propoxy]methyl]-1H-1,2,3-triazol-1-yl]propoxy]methyl]-1H-1,2,3-triazol-1-yl]propoxy]methyl]-α-[[[1-[(2R)-2-(acetyloxy)-3-[[1-[(2S)-2-(acetyloxy)-3-[[1-[(2R)-2-(acetyloxy)-3-(2-propyn-1-yloxy)propyl]-1H-1,2,3-triazol-4-yl]methoxy]propyl]-1H-1,2,3-triazol-4-yl]methoxy]propyl]-1H-1,2,3-triazol-4-yl]methoxy]methyl]-, 1-acetate, (αS)-](/data/chemimg/3721700/1809614-01-4_b.png)
![1H-1,2,3-Triazole-1-ethanol, 4-[[(2R)-2-(acetyloxy)-3-[4-[[(2S)-2-oxiranylmethoxy]methyl]-1H-1,2,3-triazol-1-yl]propoxy]methyl]-α-[[[1-[(2R)-2-(acetyloxy)-3-(2-propyn-1-yloxy)propyl]-1H-1,2,3-triazol-4-yl]methoxy]methyl]-, 1-acetate, (αS)-](/data/chemimg/3721700/1809613-98-6.png)
![1H-1,2,3-Triazole-1-ethanol, 4-[[(2R)-2-(acetyloxy)-3-[4-[[(2S)-2-oxiranylmethoxy]methyl]-1H-1,2,3-triazol-1-yl]propoxy]methyl]-α-[[[1-[(2R)-2-(acetyloxy)-3-(2-propyn-1-yloxy)propyl]-1H-1,2,3-triazol-4-yl]methoxy]methyl]-, 1-acetate, (αS)-](/data/chemimg/3721700/1809613-98-6_b.png)
![1H-1,2,3-Triazole-1-ethanol, 4-[[(2R)-2-(acetyloxy)-3-[4-[[(2S)-2-(acetyloxy)-3-azidopropoxy]methyl]-1H-1,2,3-triazol-1-yl]propoxy]methyl]-α-[[[1-[(2R)-2-(acetyloxy)-3-[[3-[(1,1-dimethylethyl)dimethylsilyl]-2-propyn-1-yl]oxy]propyl]-1H-1,2,3-triazol-4-yl]methoxy]methyl]-, 1-acetate, (αS)-](/data/chemimg/3721700/1809613-97-5.png)
![1H-1,2,3-Triazole-1-ethanol, 4-[[(2R)-2-(acetyloxy)-3-[4-[[(2S)-2-(acetyloxy)-3-azidopropoxy]methyl]-1H-1,2,3-triazol-1-yl]propoxy]methyl]-α-[[[1-[(2R)-2-(acetyloxy)-3-[[3-[(1,1-dimethylethyl)dimethylsilyl]-2-propyn-1-yl]oxy]propyl]-1H-1,2,3-triazol-4-yl]methoxy]methyl]-, 1-acetate, (αS)-](/data/chemimg/3721700/1809613-97-5_b.png)
![1H-1,2,3-Triazole-1-ethanol, α-[[[1-[(2R)-2-(acetyloxy)-3-[[3-[(1,1-dimethylethyl)dimethylsilyl]-2-propyn-1-yl]oxy]propyl]-1H-1,2,3-triazol-4-yl]methoxy]methyl]-4-[[(2R)-2-(acetyloxy)-3-[4-[[(2S)-2-oxiranylmethoxy]methyl]-1H-1,2,3-triazol-1-yl]propoxy]methyl]-, 1-acetate, (αS)-](/data/chemimg/3721700/1809613-96-4.png)
![1H-1,2,3-Triazole-1-ethanol, α-[[[1-[(2R)-2-(acetyloxy)-3-[[3-[(1,1-dimethylethyl)dimethylsilyl]-2-propyn-1-yl]oxy]propyl]-1H-1,2,3-triazol-4-yl]methoxy]methyl]-4-[[(2R)-2-(acetyloxy)-3-[4-[[(2S)-2-oxiranylmethoxy]methyl]-1H-1,2,3-triazol-1-yl]propoxy]methyl]-, 1-acetate, (αS)-](/data/chemimg/3721700/1809613-96-4_b.png)
![1H-1,2,3-Triazole-1-ethanol, 4-[[(2S)-2-oxiranylmethoxy]methyl]-α-[(2-propyn-1-yloxy)methyl]-, 1-acetate, (αR)-](/data/chemimg/3721700/1809613-95-3.png)
![1H-1,2,3-Triazole-1-ethanol, 4-[[(2S)-2-oxiranylmethoxy]methyl]-α-[(2-propyn-1-yloxy)methyl]-, 1-acetate, (αR)-](/data/chemimg/3721700/1809613-95-3_b.png)
![1H-1,2,3-Triazole-1-ethanol, 4-[[(2S)-2-(acetyloxy)-3-azidopropoxy]methyl]-α-[[[3-[(1,1-dimethylethyl)dimethylsilyl]-2-propyn-1-yl]oxy]methyl]-, 1-acetate, (αR)-](/data/chemimg/3721700/1809613-94-2.png)
![1H-1,2,3-Triazole-1-ethanol, 4-[[(2S)-2-(acetyloxy)-3-azidopropoxy]methyl]-α-[[[3-[(1,1-dimethylethyl)dimethylsilyl]-2-propyn-1-yl]oxy]methyl]-, 1-acetate, (αR)-](/data/chemimg/3721700/1809613-94-2_b.png)
![1H-1,2,3-Triazole-1-ethanol, α-[[[3-[(1,1-dimethylethyl)dimethylsilyl]-2-propyn-1-yl]oxy]methyl]-4-[[(2S)-2-oxiranylmethoxy]methyl]-, 1-acetate, (αR)-](/data/chemimg/3721700/1809613-93-1.png)
![1H-1,2,3-Triazole-1-ethanol, α-[[[3-[(1,1-dimethylethyl)dimethylsilyl]-2-propyn-1-yl]oxy]methyl]-4-[[(2S)-2-oxiranylmethoxy]methyl]-, 1-acetate, (αR)-](/data/chemimg/3721700/1809613-93-1_b.png)


![5,12-Naphthacenedione, 7,8,9,10-tetrahydro-6,8,11-trihydroxy-8-(2-hydroxyacetyl)-1-methoxy-10-[[2,3,6-trideoxy-3-[[[[2-nitro-3-[(2-propyn-1-ylamino)carbonyl]phenyl]methoxy]carbonyl]amino]-α-L-lyxo-hexopyranosyl]oxy]-, (8S,10S)-](/data/chemimg/161300/1260094-12-9.png)
![5,12-Naphthacenedione, 7,8,9,10-tetrahydro-6,8,11-trihydroxy-8-(2-hydroxyacetyl)-1-methoxy-10-[[2,3,6-trideoxy-3-[[[[2-nitro-3-[(2-propyn-1-ylamino)carbonyl]phenyl]methoxy]carbonyl]amino]-α-L-lyxo-hexopyranosyl]oxy]-, (8S,10S)-](/data/chemimg/161300/1260094-12-9_b.png)








![Acetic acid, 2-[2-[2-(2-azidoethoxy)ethoxy]ethoxy]-](/data/chemimg/1152400/172531-37-2.png)
![Acetic acid, 2-[2-[2-(2-azidoethoxy)ethoxy]ethoxy]-](/data/chemimg/1152400/172531-37-2_b.png)
![1H-Isoindole-1,3(2H)-dione, 2-[2-[2-(2-methoxyethoxy)ethoxy]ethyl]-](http://img.cochemist.com/ccimg/167300/167221-50-3.png)
![1H-Isoindole-1,3(2H)-dione, 2-[2-[2-(2-methoxyethoxy)ethoxy]ethyl]-](http://img.cochemist.com/ccimg/167300/167221-50-3_b.png)


![1-Propyne, 3,3'-[[2,2-bis[(2-propyn-1-yloxy)methyl]-1,3-propanediyl]bis(oxy)]bis-](/data/chemimg/2234900/127751-08-0.png)
![1-Propyne, 3,3'-[[2,2-bis[(2-propyn-1-yloxy)methyl]-1,3-propanediyl]bis(oxy)]bis-](/data/chemimg/2234900/127751-08-0_b.png)


![Ethanol, 2-[2-[2-(2-azidoethoxy)ethoxy]ethoxy]-](/data/chemimg/976600/86770-67-4.png)
![Ethanol, 2-[2-[2-(2-azidoethoxy)ethoxy]ethoxy]-](/data/chemimg/976600/86770-67-4_b.png)



![METHANESULFONIC ACID;2-[2-(2-METHOXYETHOXY)ETHOXY]ETHANOL](http://img.cochemist.com/ccimg/74700/74654-05-0.png)
![METHANESULFONIC ACID;2-[2-(2-METHOXYETHOXY)ETHOXY]ETHANOL](http://img.cochemist.com/ccimg/74700/74654-05-0_b.png)




![L-[1-13C]GLUCOSE](http://img.cochemist.com/ccimg/56300/56253-90-8.png)
![L-[1-13C]GLUCOSE](http://img.cochemist.com/ccimg/56300/56253-90-8_b.png)








![3-Pyridinamine, 6-[6-(2-pyridinyl)-1,2,4,5-tetrazin-3-yl]-](/data/chemimg/3748300/1055983-02-2.png)
![3-Pyridinamine, 6-[6-(2-pyridinyl)-1,2,4,5-tetrazin-3-yl]-](/data/chemimg/3748300/1055983-02-2_b.png)
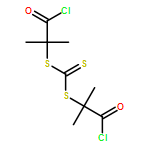

![Propanoic acid, 2,2'-[carbonothioylbis(thio)]bis[2-methyl-](/data/chemimg/104200/355120-40-0.png)
![Propanoic acid, 2,2'-[carbonothioylbis(thio)]bis[2-methyl-](/data/chemimg/104200/355120-40-0_b.png)


![1-azido-11-[(tert-butyldimethylsilyl)oxy]-3,6,9-trioxaundecane](http://img.cochemist.com/ccimg/134200/134179-42-3.png)
![1-azido-11-[(tert-butyldimethylsilyl)oxy]-3,6,9-trioxaundecane](http://img.cochemist.com/ccimg/134200/134179-42-3_b.png)






![Pentanoic acid, 5-oxo-5-[[6-[6-(2-pyridinyl)-1,2,4,5-tetrazin-3-yl]-3-pyridinyl]amino]-](/data/chemimg/3767600/1233234-76-8.png)
![Pentanoic acid, 5-oxo-5-[[6-[6-(2-pyridinyl)-1,2,4,5-tetrazin-3-yl]-3-pyridinyl]amino]-](/data/chemimg/3767600/1233234-76-8_b.png)