Co-reporter:Daniel H. Ess ; Robert J. Nielsen ; William A. Goddard III ;Roy A. Periana
Journal of the American Chemical Society 2009 Volume 131(Issue 33) pp:11686-11688
Publication Date(Web):August 4, 2009
DOI:10.1021/ja902748c
Absolutely localized molecular orbital energy decomposition analysis of C−H activation transition states (TSs), including Pt, Au, Ir, Ru, W, Sc, and Re metal centers, shows an electrophilic, ambiphilic, and nucleophilic charge transfer (CT) continuum irrespective of the bonding paradigm (oxidative addition, σ-bond metathesis, oxidative hydrogen migration, 1,2-substitution). Pt(II) insertion and Au(III) substitution TSs are highly electrophilic and dominated by C−H bond to metal/ligand orbital stabilization, while Ir−X and Ru−X (X = R, NH2, OR, or BOR2) substitution TSs are ambiphilic in nature. In this ambiphilic activation regime, an increase in one direction of CT typically leads to a decrease in the reverse direction. Comparison of Tp(CO)Ru−OH and Tp(CO)Ru−NH2 complexes showed no evidence for the classic dπ−pπ repulsion model. Complexes such as and Cp(CO)2W−B(OR)2, (PNP)Ir(I), Cp2ScMe, and (acac-κO,κO)2Re(III)−OH were found to mediate nucleophilic C−H activation, where the CT is dominated by the metal/ligand orbital to C−H antibonding orbital interaction. This CT continuum ultimately affects the metal−alkyl intermediate polarization and possible functionalization reactions. This analysis will impact the design of new activation reactions and stimulate the discovery of more nucleophilic activation complexes.
Co-reporter:Daniel H. Ess, Jeremy Kister, Ming Chen and William R. Roush
Organic Letters 2009 Volume 11(Issue 23) pp:5538-5541
Publication Date(Web):November 9, 2009
DOI:10.1021/ol902364d
Density functional theory was used to locate transition states for hydroboration reactions of allenes with 9-borabicyclo[3.3.1]nonane and 10-R-9-borabicyclo[3.3.2]decane, as well as transition states for [1,3]-boratropic shift and aldehyde addition reactions of the derived allylboranes. The origin of kinetic versus thermodynamic control in the allene hydroboration step is described.
Co-reporter:Daniel H. Ess, Steven M. Bischof, Jonas Oxgaard, Roy A. Periana and William A. Goddard III.
Organometallics 2008 Volume 27(Issue 24) pp:6440-6445
Publication Date(Web):November 13, 2008
DOI:10.1021/om8006568
Chelate-assisted and internal electrophilic substitution type transition states were studied using a DFT-based energy decomposition method. Interaction energies for benzene and methane C−H bond activation by (acac-O,O)2Ir(X) complexes (X = CH3COO and OH) were evaluated using the absolutely localized molecular orbital energy decomposition analysis (ALMO-EDA). A ratio of ∼1.5:1 for forward to reverse charge-transfer between (acac-O,O)2Ir(X) and benzene or methane transition state fragments confirms “ambiphilic” bonding, the result of an interplay between the electrophilic iridium center and the internal base component. This analysis also revealed that polarization effects account for a significant amount of transition state stabilization. The energy penalty to deform reactants into their transition state geometry, distortion energy, was also used to understand the large activation energy difference between six-membered and four-membered acetate-assisted transition states and help explain why these complexes do not activate the methane C−H bond.

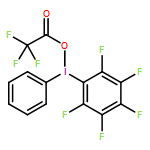
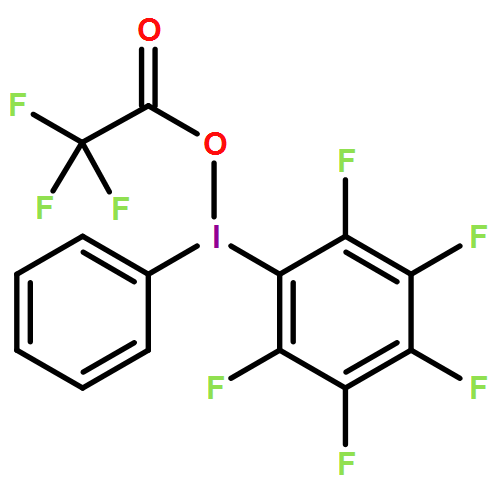


![[Bis(trifluoroacetoxy)iodo]pentafluorobenzene](/data/chemimg/515400/14353-88-9.png)
![[Bis(trifluoroacetoxy)iodo]pentafluorobenzene](/data/chemimg/515400/14353-88-9_b.png)