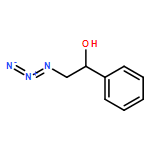
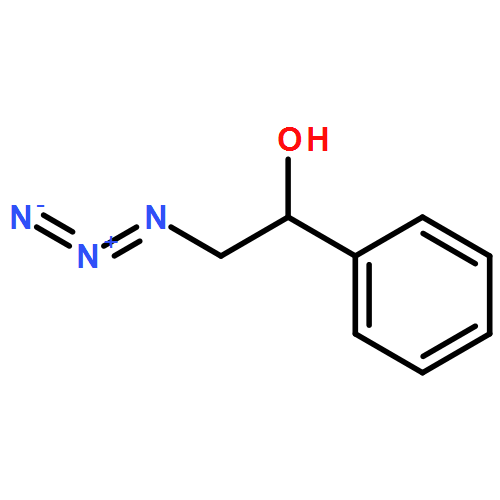
Co-reporter:Anthony P. Silvestri;Philip A. Cistrone; Dr. Philip E. Dawson
Angewandte Chemie International Edition 2017 Volume 56(Issue 35) pp:10438-10442
Publication Date(Web):2017/08/21
DOI:10.1002/anie.201705065
AbstractCopper-mediated coupling between alkynes to generate a structurally rigid, linear 1,3-diyne linkage has been known for over a century. However, the mechanistic requirement to simultaneously maintain CuI and an oxidant has limited its practical utility, especially for complex functional molecules in aqueous solution. We find that addition of a specific bpy-diol ligand protects unprotected peptides from CuII-mediated oxidative damage through the formation of an insoluble CuII gel which solves the critical challenge of applying Glaser coupling to substrates that are degraded by CuII. The generality of this method is illustrated through the conjugation of a series of polar and nonpolar labels onto a fully unprotected GLP-1R agonist through a linear 7 Å diynyl linker.
Co-reporter:Anthony P. Silvestri;Philip A. Cistrone; Dr. Philip E. Dawson
Angewandte Chemie 2017 Volume 129(Issue 35) pp:10574-10578
Publication Date(Web):2017/08/21
DOI:10.1002/ange.201705065
AbstractCopper-mediated coupling between alkynes to generate a structurally rigid, linear 1,3-diyne linkage has been known for over a century. However, the mechanistic requirement to simultaneously maintain CuI and an oxidant has limited its practical utility, especially for complex functional molecules in aqueous solution. We find that addition of a specific bpy-diol ligand protects unprotected peptides from CuII-mediated oxidative damage through the formation of an insoluble CuII gel which solves the critical challenge of applying Glaser coupling to substrates that are degraded by CuII. The generality of this method is illustrated through the conjugation of a series of polar and nonpolar labels onto a fully unprotected GLP-1R agonist through a linear 7 Å diynyl linker.
Co-reporter:Anthony P. Silvestri
Organic & Biomolecular Chemistry 2017 vol. 15(Issue 24) pp:5131-5134
Publication Date(Web):2017/06/21
DOI:10.1039/C7OB01094J
Amphiphilic fluorocarbons have unique properties that facilitate their self assembly and adhesion to both inorganic and biological substrates. Incorporation of these moieties into valuable constructs typically require complex synthetic routes that have limited their use. Here, the base-catalyzed diastereoselective synthesis of 6-methyl-2,4,6-tris(trifluoromethyl)tetrahydro-2H-pyran-2,4-diol is reported. Trimerization of trifluoroacetone in the presence of 5 mol% KHMDS delivers one of four diastereomers selectively in 81% yield with no column chromatography. Temperature screening revealed the reversibility of this trimerization and the funneling of material into the most thermodynamically stable oxane. Subsequent functionalization with boronic acids is reported.
Co-reporter:Joachim Weidmann; Elena Dimitrijević; Jörg D. Hoheisel
Organic Letters 2016 Volume 18(Issue 2) pp:164-167
Publication Date(Web):December 24, 2015
DOI:10.1021/acs.orglett.5b03111
A protection strategy is described for the efficient synthesis of peptide o-aminoanilides using in situ neutralization protocols for Boc-SPPS. On-resin protection of Boc-protected aminoacyl o-aminoanilides is achieved with 2-chlorobenzyl chloroformate. Activation through a peptidyl-benzotriazole intermediate allows for facile conversion to peptide-thioesters for use in native chemical ligation. In addition to providing a robust alternative to established thioester resins, as a latent thioester, the peptide o-aminoanilide has broad utility in convergent ligation strategies.
Co-reporter:Philip A. Cistrone and Philip E. Dawson
ACS Combinatorial Science 2016 Volume 18(Issue 3) pp:139
Publication Date(Web):February 25, 2016
DOI:10.1021/acscombsci.5b00195
Performing sequential reactions for the orthogonal derivatization of peptides in solution often requires intermediate handling and purification steps. To solve these problems, we have exploited the distinct adsorption kinetics of peptides toward particulate reversed-phase (RP) C18 silica material, enabling consecutive reactions to be performed without intermediate elution. To illustrate this approach, sequential CuAAC/click reactions were used to modify an analog of the bicyclic peptide sunflower trypsin inhibitor 1 (SFTI-1), a potent scaffold for trypsin and chymotrypsin-like enzyme inhibitors. The SFTI-1 scaffold was synthesized containing both β-azido alanine and propargyl glycine residues. Despite the mutual reactivity of these groups, site isolation on RP silica enabled consecutive click reactions and associated washing steps to be performed while the peptide remained immobilized. Importantly, this approach eliminated side products that could form between two peptides or within a single peptide. These studies suggest a broad utility for RP silica in solving both peptide handling problems and in improving synthetic workflows.Keywords: CuAAC/click chemistry; peptides; reversed-phase silica
Co-reporter:Erika J. Olson, Bernhard C. Lechtenberg, Chunxia Zhao, Elena Rubio de la Torre, Ilaria Lamberto, Stefan J. Riedl, Philip E. Dawson, and Elena B. Pasquale
ACS Medicinal Chemistry Letters 2016 Volume 7(Issue 9) pp:841
Publication Date(Web):June 25, 2016
DOI:10.1021/acsmedchemlett.6b00132
EphA4 is a receptor tyrosine kinase with a critical role in repulsive axon guidance and synaptic function. However, aberrant EphA4 activity can inhibit neural repair after injury and exacerbate neurodegenerative diseases such as amyotrophic lateral sclerosis (ALS) and Alzheimer’s. We previously identified the cyclic peptide APY-d2 (APYCVYRβASWSC-nh2, containing a disulfide bond) as a potent and selective EphA4 antagonist. However, APY-d2 lacks sufficient plasma stability to be useful for EphA4 inhibition in vivo through peripheral administration. Using structure–activity relationship studies, we show that protecting the peptide N-terminus from proteolytic degradation dramatically increases the persistence of the active peptide in plasma and that a positively charged peptide N-terminus is essential for high EphA4 binding affinity. Among several improved APY-d2 derivatives, the cyclic peptides APY-d3 (βAPYCVYRβASWSC-nh2) and APY-d4 (βAPYCVYRβAEWEC-nh2) combine high stability in plasma and cerebrospinal fluid with slightly enhanced potency. These properties make them valuable research tools and leads toward development of therapeutics for neurological diseases.Keywords: ALS; Alzheimer’s disease; aminopeptidase; Peptide inhibitor; protease resistance; SAR;
Co-reporter:Valle Palomo, Sebastián A. Díaz, Michael H. Stewart, Kimihiro Susumu, Igor L. Medintz, and Philip E. Dawson
ACS Nano 2016 Volume 10(Issue 6) pp:6090
Publication Date(Web):May 20, 2016
DOI:10.1021/acsnano.6b01682
Fluorescence-based assays for hydrolases that cleave within the substrate (endopeptidases) are common, while developing substrates for proteases that selectively cleave from peptide termini (exopeptidases) is more challenging, since the termini are specifically recognized by the enzyme and cannot be modified to facilitate a Förster resonance energy transfer (FRET)-based approach. The development of a robust system that enables the quenching of fluorescent particles by simple amino acid side chains would find broad utility for peptide sensors and would be advantageous for exopeptidases. Here we describe a quantum dot (QD)-based electron transfer (ET) sensor that is able to allow direct, quantitative monitoring of both exopeptidase and endopeptidase activity. The incorporation of 3,4-dihydroxyphenylalanine (DOPA) into the sequence of a peptide allows for the quenching of QD photoluminescence through an ET mechanism. DOPA is a nonproteinogenic amino acid that can replace a phenylalanine or tyrosine residue in a peptide sequence without severely altering structural properties, allowing for its introduction at multiple positions within a biologically active peptide substrate. Consequently, the quenching system presented here is ideally suited for incorporation into diverse peptide substrates for enzyme recognition, digestion, and activity sensing. Our findings suggest a broad utility of a small ET-capable amino acid side chain in detecting enzyme activity through ET-mediated QD luminescence quenching.Keywords: DOPA; electron transfer; exopeptidase; nanoparticle; quantum dot; sensor
Co-reporter:Juan B. Blanco-Canosa; Brunello Nardone; Fernando Albericio
Journal of the American Chemical Society 2015 Volume 137(Issue 22) pp:7197-7209
Publication Date(Web):May 15, 2015
DOI:10.1021/jacs.5b03504
The broad utility of native chemical ligation (NCL) in protein synthesis has fostered a search for methods that enable the efficient synthesis of C-terminal peptide-thioesters, key intermediates in NCL. We have developed an N-acylurea (Nbz) approach for the synthesis of thioester peptide precursors that efficiently undergo thiol exchange yielding thioester peptides and subsequently NCL reaction. However, the synthesis of some glycine-rich sequences revealed limitations, such as diacylated products that can not be converted into N-acylurea peptides. Here, we introduce a new N-acylurea linker bearing an o-amino(methyl)aniline (MeDbz) moiety that enables in a more robust peptide chain assembly. The generality of the approach is illustrated by the synthesis of a pentaglycine sequence under different coupling conditions including microwave heating at coupling temperatures up to 90 C, affording the unique and desired N-acyl-N′-methylacylurea (MeNbz) product. Further extension of the method allowed the synthesis of all 20 natural amino acids and their NCL reactions. The kinetic analysis of the ligations using model peptides shows the MeNbz peptide rapidly converts to arylthioesters that are efficient at NCL. Finally, we show that the new MeDbz linker can be applied to the synthesis of cysteine-rich proteins such the cyclotides Kalata B1 and MCoTI-II through a one cyclization/folding step in the ligation/folding buffer.
Co-reporter:Dr. Naila Assem;David J. Ferreira;Dr. Dennis W. Wolan;Dr. Philip E. Dawson
Angewandte Chemie International Edition 2015 Volume 54( Issue 30) pp:8665-8668
Publication Date(Web):
DOI:10.1002/anie.201502607
Abstract
Macrocyclization is a broadly applied approach for overcoming the intrinsically disordered nature of linear peptides. Herein, it is shown that dichloroacetone (DCA) enhances helical secondary structures when introduced between peptide nucleophiles, such as thiols, to yield an acetone-linked bridge (ACE). Aside from stabilizing helical structures, the ketone moiety embedded in the linker can be modified with diverse molecular tags by oxime ligation. Insights into the structure of the tether were obtained through co-crystallization of a constrained S-peptide in complex with RNAse S. The scope of the acetone-linked peptides was further explored through the generation of N-terminus to side chain macrocycles and a new approach for generating fused macrocycles (bicycles). Together, these studies suggest that acetone linking is generally applicable to peptide macrocycles with a specific utility in the synthesis of stabilized helices that incorporate functional tags.
Co-reporter:Dr. Naila Assem;David J. Ferreira;Dr. Dennis W. Wolan;Dr. Philip E. Dawson
Angewandte Chemie 2015 Volume 127( Issue 30) pp:8789-8792
Publication Date(Web):
DOI:10.1002/ange.201502607
Abstract
Macrocyclization is a broadly applied approach for overcoming the intrinsically disordered nature of linear peptides. Herein, it is shown that dichloroacetone (DCA) enhances helical secondary structures when introduced between peptide nucleophiles, such as thiols, to yield an acetone-linked bridge (ACE). Aside from stabilizing helical structures, the ketone moiety embedded in the linker can be modified with diverse molecular tags by oxime ligation. Insights into the structure of the tether were obtained through co-crystallization of a constrained S-peptide in complex with RNAse S. The scope of the acetone-linked peptides was further explored through the generation of N-terminus to side chain macrocycles and a new approach for generating fused macrocycles (bicycles). Together, these studies suggest that acetone linking is generally applicable to peptide macrocycles with a specific utility in the synthesis of stabilized helices that incorporate functional tags.
Co-reporter:Kelly Boeneman, James B. Delehanty, Juan B. Blanco-Canosa, Kimihiro Susumu, Michael H. Stewart, Eunkeu Oh, Alan L. Huston, Glyn Dawson, Sampat Ingale, Ryan Walters, Miriam Domowicz, Jeffrey R. Deschamps, W. Russ Algar, Stassi DiMaggio, Janet Manono, Christopher M. Spillmann, Darren Thompson, Travis L. Jennings, Philip E. Dawson, and Igor L. Medintz
ACS Nano 2013 Volume 7(Issue 5) pp:3778
Publication Date(Web):May 28, 2013
DOI:10.1021/nn400702r
Cell penetrating peptides facilitate efficient intracellular uptake of diverse materials ranging from small contrast agents to larger proteins and nanoparticles. However, a significant impediment remains in the subsequent compartmentalization/endosomal sequestration of most of these cargoes. Previous functional screening suggested that a modular peptide originally designed to deliver palmitoyl-protein thioesterase inhibitors to neurons could mediate endosomal escape in cultured cells. Here, we detail properties relevant to this peptide’s ability to mediate cytosolic delivery of quantum dots (QDs) to a wide range of cell-types, brain tissue culture and a developing chick embryo in a remarkably nontoxic manner. The peptide further facilitated efficient endosomal escape of large proteins, dendrimers and other nanoparticle materials. We undertook an iterative structure–activity relationship analysis of the peptide by discretely modifying key components including length, charge, fatty acid content and their order using a comparative, semiquantitative assay. This approach allowed us to define the key motifs required for endosomal escape, to select more efficient escape sequences, along with unexpectedly identifying a sequence modified by one methylene group that specifically targeted QDs to cellular membranes. We interpret our results within a model of peptide function and highlight implications for in vivo labeling and nanoparticle-mediated drug delivery by using different peptides to co-deliver cargoes to cells and engage in multifunctional labeling.Keywords: cargo; cell penetrating peptide; cellular labeling; cytosol; dendrimer; endosomal escape; fusogenic; membrane; nanoparticle; peptide; protein; quantum dot
Co-reporter:Sampat Ingale and Philip E. Dawson
Organic Letters 2011 Volume 13(Issue 11) pp:2822-2825
Publication Date(Web):May 9, 2011
DOI:10.1021/ol200775h
Triazole tethers have been explored for stabilization of secondary structures in peptides. Despite the utility of this approach, cyclization efficiency in complex peptides remains a significant challenge. A robust, on-resin protocol for side chain to side chain macrocyclization by CuAAC is described. This protocol was applied to the synthesis of a series of 21 amino acid helical peptides presenting a binding dipeptide motif from the membrane proximal external region (MPER) of HIV-1 gp41.
Co-reporter:Philip E. Dawson
Israel Journal of Chemistry 2011 Volume 51( Issue 8-9) pp:862-867
Publication Date(Web):
DOI:10.1002/ijch.201100128
Abstract
Of the many approaches proposed to generalize the native chemical ligation approach for protein synthesis, the simple procedure of global desulfurization of peptide thiols has become the most widely adopted. In this review, the development of the native ligation–desulfurization strategy is described, focusing on the conversion of Cys to Ala following ligation at N-terminal Cys residues. Subsequent variations on this theme have broadened the scope to other natural amino acids including Phe, Leu, Val, and Lys, and even non-native peptide linkages such as isopeptide bonds on lysine side chains. Using insights from both selenocysteine–peptide side reactions and radical initiated desulfurization procedures, a new method for the selective deselenization of peptides containing both selenocysteine and cysteine residues has been developed. Together, these approaches represent a robust and flexible methodology for the synthesis of complex polypeptides without the use of protecting groups.
Co-reporter:Florence M. Brunel, John D. Lewis, Giuseppe Destito, Nicole F. Steinmetz, Marianne Manchester, Heidi Stuhlmann and Philip E. Dawson
Nano Letters 2010 Volume 10(Issue 3) pp:1093-1097
Publication Date(Web):February 17, 2010
DOI:10.1021/nl1002526
Multivalent nanoparticle platforms are attractive for biomedical applications because of their improved target specificity, sensitivity, and solubility. However, their controlled assembly remains a considerable challenge. An efficient hydrazone ligation chemistry was applied to the assembly of Cowpea mosaic virus (CPMV) nanoparticles with individually tunable levels of a VEGFR-1 ligand and a fluorescent PEGylated peptide. The nanoparticles recognized VEGFR-1 on endothelial cell lines and VEGFR1-expressing tumor xenografts in mice, validating targeted CPMV as a nanoparticle platform in vivo.
Co-reporter:Juan B. Blanco-Canosa ; Igor L. Medintz ; Dorothy Farrell ; Hedi Mattoussi
Journal of the American Chemical Society 2010 Volume 132(Issue 29) pp:10027-10033
Publication Date(Web):July 2, 2010
DOI:10.1021/ja910988d
Water solubilized nanoparticles such as CdSe−ZnS core−shell nanocrystals (quantum dots, QDs) have great potential in bioimaging and sensing applications due to their excellent photophysical properties. However, the efficient modification of QDs with complex biomolecules represents a significant challenge. Here, we describe a straightforward arylhydrazone approach for the chemoselective covalent modification of QDs that is compatible with neutral pH and micromolar concentrations of the peptide target. The kinetics of covalent modification can be monitored spectroscopically at 354 nm in the presence of the QD and average peptide/QD ratios from 2:1 to 11:1 were achieved with excellent control over the desired valency. These results suggest that aniline catalyzed hydrazone ligation has the potencial to provide a general method for the controlled assembly of a variety of nanoparticle-biomolecule hybrids.
Co-reporter:Talia Shekhter, Norman Metanis, Philip E. Dawson and Ehud Keinan
Molecular BioSystems 2010 vol. 6(Issue 1) pp:241-248
Publication Date(Web):22 Sep 2009
DOI:10.1039/B912753D
The glutaredoxin (Grx) family of oxidoreductases has a conserved residue at position 8 that varies between Arginine in Grx1 and Lysine in Grx3. It has been proposed that this Arg/Lys change is the main cause for the 35 mV difference in redox potential between the two enzymes. To gain insights into the catalytic machinery of Grx3 and directly evaluate the role of residue 8 in the catalysis of thiol–disulfide exchange by this enzyme, we synthesized the “wild type” enzyme (sGrx3), and four analogues substituting the lysine at position 8 with arginine, ornithine (Orn), citrulline (Cit) and norvaline (Nva). The redox potential and equilibration kinetics with thioredoxin (Trx1) were determined for each enzyme by fluorescence intensity. While minor effects on redox potential were observed, we found that residue 8 had a more marked effect on the catalytic efficiency of this enzyme. Surprisingly, truncation of the functional group resulted in a more efficient enzyme, Lys8Nva, exhibiting rate constants that are an order of magnitude higher than sGrx3 for both forward and reverse reactions. These observations pose the question why would a residue that reduces the rate of enzyme turnover be evolutionarily conserved? The significant changes in the kinetic parameters suggest that this position plays an important role in the thiol–disulfide exchange reaction by affecting the nucleophilicthiolate through electrostatic or hydrogen bonding interactions. Since the reduced Grx has an exposed thiol that could easily be alkylated, either Arg or Lys could act as a gatekeeper that deters unwanted electrophiles from attacking the active site thiolate.
Co-reporter:Anouk Dirksen Dr.;Subramanian Yegneswaran Dr.;PhilipE. Dawson Dr.
Angewandte Chemie 2010 Volume 122( Issue 11) pp:2067-2071
Publication Date(Web):
DOI:10.1002/ange.200906756
Co-reporter:Anouk Dirksen Dr.;Subramanian Yegneswaran Dr.;PhilipE. Dawson Dr.
Angewandte Chemie International Edition 2010 Volume 49( Issue 11) pp:2023-2027
Publication Date(Web):
DOI:10.1002/anie.200906756
Co-reporter:Dr. Norman Metanis; Ehud Keinan; Philip E. Dawson
Angewandte Chemie International Edition 2010 Volume 49( Issue 39) pp:7049-7053
Publication Date(Web):
DOI:10.1002/anie.201001900
Co-reporter:Anouk Dirksen, Philip E Dawson
Current Opinion in Chemical Biology 2008 Volume 12(Issue 6) pp:760-766
Publication Date(Web):December 2008
DOI:10.1016/j.cbpa.2008.10.009
Chemoselective ligation methods have increased the efficiency of bioconjugation, enabling complex macromolecules to be assembled. In particular, these methods have been utilized for the ligation and modification of peptides and proteins. The chemical synthesis of proteins from unprotected peptide fragments has enabled the introduction of unnatural amino acids, site-specific isotopic labeling, and the site-specific attachment of affinity tags or labels for imaging. A greater insight into current ligation methods has led to higher reaction rates, higher reaction yields, and greater biocompatibility, thereby increasing the impact of chemoselective ligation reactions in chemical biology.
Co-reporter:Anouk Dirksen and Philip E. Dawson
Bioconjugate Chemistry 2008 Volume 19(Issue 12) pp:2543
Publication Date(Web):November 17, 2008
DOI:10.1021/bc800310p
A high-yielding and rapid chemoselective ligation approach is presented that uses aniline catalysis to activate aromatic aldehydes toward two amine nucleophiles, namely, 6-hydrazinopyridyl and aminooxyacetyl groups. The rates of these ligations are resolved for model reactions with unprotected peptides. The resulting hydrazone and oxime conjugates are attained under ambient conditions with rate constants of 101−103 M−1 s−1. These rate constants exceed those of current chemoselective ligation chemistries and enable efficient labeling of peptides and proteins at low μM concentrations, at neutral pH, without using a large excess of one of the components. The utility of the approach is demonstrated by the p-fluorobenzylation of human serum albumin and by the fluorescent labeling of an unprotected peptide with Alexa Fluor 488.
Co-reporter:JuanB. Blanco-Canosa Dr. ;PhilipE. Dawson Dr.
Angewandte Chemie International Edition 2008 Volume 47( Issue 36) pp:6851-6855
Publication Date(Web):
DOI:10.1002/anie.200705471
Co-reporter:JuanB. Blanco-Canosa Dr. ;PhilipE. Dawson Dr.
Angewandte Chemie 2008 Volume 120( Issue 36) pp:6957-6961
Publication Date(Web):
DOI:10.1002/ange.200705471
Co-reporter:Anouk Dirksen Dr.;Tilman M. Hackeng Dr. Dr.
Angewandte Chemie 2006 Volume 118(Issue 45) pp:
Publication Date(Web):19 OCT 2006
DOI:10.1002/ange.200602877
Anilin agiert in Form des orange gezeichneten Intermediats als nucleophiler Katalysator in der Oxim-Ligation in wässriger Lösung. Die Oxim-Ligation zweier ungeschützter Peptidfragmente wurde bei pH 4.5 und pH 7 beschleunigt, was die Anwendungsbreite dieser Reaktion auf Bedingungen ausdehnt, die für die Biokonjugation relevant sind (siehe Schema).
Co-reporter:Anouk Dirksen Dr.;Tilman M. Hackeng Dr. Dr.
Angewandte Chemie International Edition 2006 Volume 45(Issue 45) pp:
Publication Date(Web):19 OCT 2006
DOI:10.1002/anie.200602877
Aniline acts as a nucleophilic catalyst of oxime ligation in aqueous solution through formation of the orange-colored intermediate. The method is demonstrated by acceleration of the oxime ligation of peptides at pH 4.5 and pH 7, which extends the scope of this reaction to conditions relevant for the bioconjugation of macromolecules (see scheme).
Co-reporter:Florence M. Brunel and Philip E. Dawson
Chemical Communications 2005 (Issue 20) pp:2552-2554
Publication Date(Web):11 Mar 2005
DOI:10.1039/B419015G
We present a straightforward and high yielding method to synthesize constrained helical peptides via thioether ligation; this method represents an attractive alternative to the formation of lactam bridge constraints to induce helicity in peptides.
Co-reporter:Amy C. Beltran Dr. Dr.;Joel M. Gottesfeld Dr.
ChemBioChem 2005 Volume 6(Issue 1) pp:
Publication Date(Web):9 DEC 2004
DOI:10.1002/cbic.200400184
The basic helix-loop-helix (bHLH) domain defines a class of transcription factors that are essential for the regulation of many genes involved in cell differentiation and development. To determine the role of the DNA sequence in driving dimerization specificity of bHLH transcription factors, we analyzed the DNA sequence in and around a consensus hexanucleotide binding site (E-box). The bHLH domains of two transcription factors, E12 and TAL1, were chemically synthesized. The minimal DNA binding domain for both the E12 homodimer and the E12–TAL1 heterodimer was determined, thereby extending the E-box by two base pairs. Additional studies indicate that the presence of a thymine in the first flanking position 5′ to the E-box prevents DNA binding of both dimer complexes. The presence of a thymine or cytosine in a flanking position two bases 5′ to the E-box decreases the affinity for the E12 homodimer twofold but completely inactivates DNA binding for the E12–TAL1 heterodimer. Access to synthetic DNA and protein enabled the analysis of specific interactions between a conserved arginine residue in the basic helix of each bHLH domain and adenine in a flanking position two bases 5′ to the E-box. Our results indicate a key role of the DNA sequence in driving dimerization specificity among bHLH transcription factors.
Co-reporter:Ashraf Brik;Lawrence J. D'Souza;Ehud Keinan;Flavio Grynszpan
ChemBioChem 2002 Volume 3(Issue 9) pp:
Publication Date(Web):28 AUG 2002
DOI:10.1002/1439-7633(20020902)3:9<799::AID-CBIC799>3.0.CO;2-7
The cover picture shows a schematic representation of the mechanistic and catalytic diversity exhibited by mutants of 4-oxalocrotonate tautomerase (4-OT). A designed single amino acid substitution can alter the catalytic activity and mechanism of this enzyme. While the wild-type 4-OT catalyzes only the tautomerization of oxalocrotonate through a general acid/base machanism, the Pro1Ala mutant catalyzes two reactions—the original tautomerization reaction through an acid/base mechanism and the decarboxylation of oxaloacetate by a nucleophilic mechanism. This bifunctional mutant suggests that a new synthetic family of nucleophilic catalysts could be generated on the basis of the 4-OT scaffold through selection methods and rational protein engineering. For more details, see the article by Brik et al. on p. 845 ff.
Co-reporter:Ashraf Brik;Lawrence J. D'Souza;Ehud Keinan;Flavio Grynszpan
ChemBioChem 2002 Volume 3(Issue 9) pp:
Publication Date(Web):28 AUG 2002
DOI:10.1002/1439-7633(20020902)3:9<845::AID-CBIC845>3.0.CO;2-2
A designed single amino acid substitution can alter the catalytic activity and mechanism of 4-oxalocrotonate tautomerase (4-OT). While the wild-type enzyme catalyzes only the tautomerization of oxalocrotonate, the Pro1Ala mutant (P1A) catalyzes two reactions—the original tautomerization reaction and the decarboxylation of oxaloacetate. Although the N-terminal amine group of P1A is involved in both reactions, our results support a nucleophilic mechanism for the decarboxylase activity, in contrast to the general acid/base mechanism that has been previously established for the tautomerase activity. These findings demonstrate that a single catalytic group in a 4-OT mutant can catalyze two reactions by two different mechanisms.
Co-reporter:Chiara Marinzi, Steven J. Bark, John Offer, Philip E. Dawson
Bioorganic & Medicinal Chemistry 2001 Volume 9(Issue 9) pp:2323-2328
Publication Date(Web):September 2001
DOI:10.1016/S0968-0896(01)00136-5
Highly chemoselective amide forming ligation reactions have facilitated the synthetic access to proteins and other amide-linked bioconjugates. In order to generalize this approach, a Nα-2-phenyl ethanethiol scaffold has been developed to promote S to N acyl transfer in a manner analogous to native chemical ligation with N-terminal cysteine residues. Analysis of scaffold-mediated ligation reactions in aqueous solution indicate that the ligation rate at Xaa-Gly junctions is sufficient for the synthesis of large polypeptides. In addition, it was found that the ligation rate is independent of the stereocenter in the scaffold and S- to N-acyl transfer is rate limiting. These studies indicate that the Nα-2-phenyl ethanethiol scaffold is a good candidate for the development of a ligation chemistry for the formation of Xaa-Gly peptides and other unhindered amides.The use of a Nα-2-phenyl ethanethiol scaffold for amide forming ligations between unprotected peptides is reported.
Co-reporter:Liang Z. Yan
Angewandte Chemie International Edition 2001 Volume 40(Issue 19) pp:
Publication Date(Web):2 OCT 2001
DOI:10.1002/1521-3773(20011001)40:19<3625::AID-ANIE3625>3.0.CO;2-Q
In contrast to DNA, proteins with topologically linked backbone structures have not yet been observed in Nature. We have chemically synthesized a p53 protein catenane (see schematic representation) consisting of interlocking cyclic polypeptides to demonstrate that such structures are synthetically attainable and can significantly stabilize the folded conformation of a protein.
Co-reporter:Liang Z. Yan
Angewandte Chemie 2001 Volume 113(Issue 19) pp:
Publication Date(Web):2 OCT 2001
DOI:10.1002/1521-3757(20011001)113:19<3737::AID-ANGE3737>3.0.CO;2-W
Anders als DNA wurden Proteine mit topologisch verbundenen Rückgratstrukturen bisher nicht in der Natur beobachtet. Um zu zeigen, dass solche Strukturen präparativ zugänglich sind und die gefaltete Konformation eines Proteins stabilisieren können, wurde ein aus ineinandergreifenden cyclischen Polypeptiden aufgebautes p53-Proteincatenan (siehe schematische Darstellung) chemisch synthetisiert.
Co-reporter:Anthony P. Silvestri and Philip E. Dawson
Organic & Biomolecular Chemistry 2017 - vol. 15(Issue 24) pp:NaN5134-5134
Publication Date(Web):2017/06/05
DOI:10.1039/C7OB01094J
Amphiphilic fluorocarbons have unique properties that facilitate their self assembly and adhesion to both inorganic and biological substrates. Incorporation of these moieties into valuable constructs typically require complex synthetic routes that have limited their use. Here, the base-catalyzed diastereoselective synthesis of 6-methyl-2,4,6-tris(trifluoromethyl)tetrahydro-2H-pyran-2,4-diol is reported. Trimerization of trifluoroacetone in the presence of 5 mol% KHMDS delivers one of four diastereomers selectively in 81% yield with no column chromatography. Temperature screening revealed the reversibility of this trimerization and the funneling of material into the most thermodynamically stable oxane. Subsequent functionalization with boronic acids is reported.
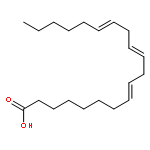
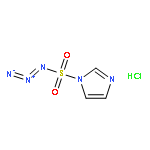
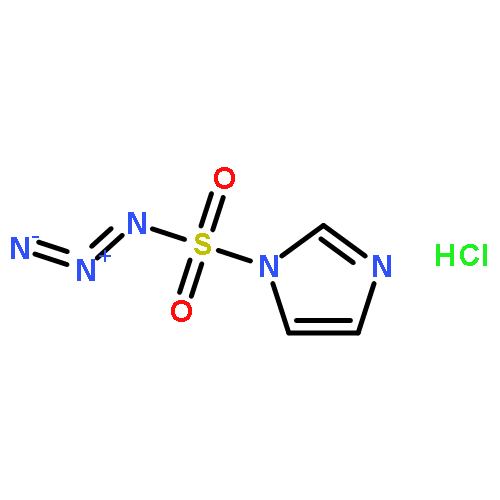
.jpg)
![1H-Thieno[3,4-d]imidazole-4-pentanamide, hexahydro-2-oxo-N-3,6,9,12-tetraoxapentadec-14-yn-1-yl-, (3aS,4S,6aR)-](/data/chemimg/186200/1262681-31-1.png)
![1H-Thieno[3,4-d]imidazole-4-pentanamide, hexahydro-2-oxo-N-3,6,9,12-tetraoxapentadec-14-yn-1-yl-, (3aS,4S,6aR)-](/data/chemimg/186200/1262681-31-1_b.png)














![(S)-2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-3-(2,2-dimethylbenzo[d][1,3]dioxol-5-yl)propanoic acid](http://img.cochemist.com/ccimg/852300/852288-18-7.png)
![(S)-2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-3-(2,2-dimethylbenzo[d][1,3]dioxol-5-yl)propanoic acid](http://img.cochemist.com/ccimg/852300/852288-18-7_b.png)











![Carbamic acid, [(1S)-1-formyl-3-methylbutyl]-, 9H-fluoren-9-ylmethylester](http://img.cochemist.com/ccimg/146900/146803-42-1.png)
![Carbamic acid, [(1S)-1-formyl-3-methylbutyl]-, 9H-fluoren-9-ylmethylester](http://img.cochemist.com/ccimg/146900/146803-42-1_b.png)




![Butanoic acid, 4-azido-2-[[(1,1-dimethylethoxy)carbonyl]amino]-, (S)-](http://img.cochemist.com/ccimg/120100/120042-08-2.png)
![Butanoic acid, 4-azido-2-[[(1,1-dimethylethoxy)carbonyl]amino]-, (S)-](http://img.cochemist.com/ccimg/120100/120042-08-2_b.png)