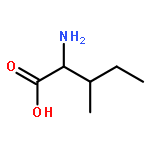
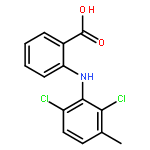
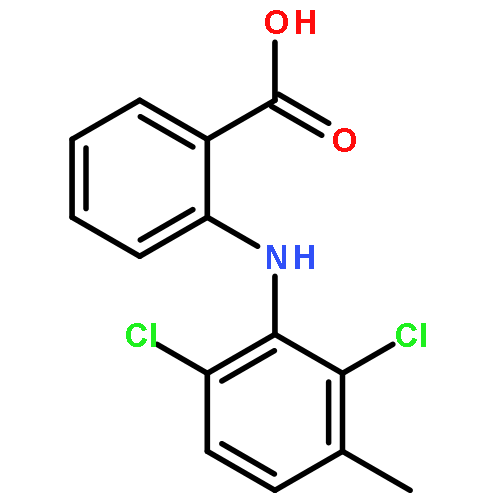
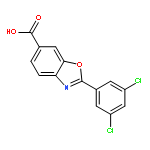
Co-reporter:Xinyi Li;Joel N Buxbaum
Molecular Neurodegeneration 2011 Volume 6( Issue 1) pp:
Publication Date(Web):2011 December
DOI:10.1186/1750-1326-6-79
Since the mid-1990's a trickle of publications from scattered independent laboratories have presented data suggesting that the systemic amyloid precursor transthyretin (TTR) could interact with the amyloidogenic β-amyloid (Aβ) peptide of Alzheimer's disease (AD). The notion that one amyloid precursor could actually inhibit amyloid fibril formation by another seemed quite far-fetched. Further it seemed clear that within the CNS, TTR was only produced in choroid plexus epithelial cells, not in neurons. The most enthusiastic of the authors proclaimed that TTR sequestered Aβ in vivo resulting in a lowered TTR level in the cerebrospinal fluid (CSF) of AD patients and that the relationship was salutary. More circumspect investigators merely showed in vitro interaction between the two molecules. A single in vivo study in Caenorhabditis elegans suggested that wild type human TTR could suppress the abnormalities seen when Aβ was expressed in the muscle cells of the worm. Subsequent studies in human Aβ transgenic mice, including those from our laboratory, also suggested that the interaction reduced the Aβ deposition phenotype. We have reviewed the literature analyzing the relationship including recent data examining potential mechanisms that could explain the effect. We have proposed a model which is consistent with most of the published data and current notions of AD pathogenesis and can serve as a hypothesis which can be tested.
Co-reporter:Joel N. Buxbaum;Natàlia Reixach
Cellular and Molecular Life Sciences 2009 Volume 66( Issue 19) pp:3095-3101
Publication Date(Web):2009 October
DOI:10.1007/s00018-009-0109-0
Transthyretin (TTR) (formerly, thyroxine binding prealbumin) is an evolutionarily conserved serum and cerebrospinal fluid protein that transports holo-retinol-binding protein and thyroxine. Its serum concentration has been widely used to assess clinical nutritional status. It is also well known that wild-type transthyretin and approximately 100 different mutants give rise to a variety of forms of systemic amyloid deposition. It has been suspected and recently established that TTR can suppress the Alzheimer’s disease phenotype in transgenic animal models of cerebral Aβ deposition. Thus, while TTR is a systemic amyloid precursor, in the brain it seems to have an anti-amyloidogenic effect. TTR is found in other organs as a result of local synthesis or transport, suggesting that it may have other, as yet undiscovered, functions. It is possible that its capacity to bind many classes of compounds allows it to serve as an endogenous detoxifier of molecules with potential pathologic effects.
Co-reporter:Natàlia Reixach;Zhengyi Ye;Joel N. Buxbaum;Linsey Friske;Coree Levy;Pritam Das;Todd Golde;Amanda R. Roberts;Eliezer Masliah;Tamas Bartfai
PNAS 2008 Volume 105 (Issue 7 ) pp:2681-2686
Publication Date(Web):2008-02-19
DOI:10.1073/pnas.0712197105
Cells that have evolved to produce large quantities of secreted proteins to serve the integrated functions of complex multicellular
organisms are equipped to compensate for protein misfolding. Hepatocytes and plasma cells have well developed chaperone and
proteasome systems to ensure that secreted proteins transit the cell efficiently. The number of neurodegenerative disorders
associated with protein misfolding suggests that neurons are particularly sensitive to the pathogenic effects of aggregates
of misfolded molecules because those systems are less well developed in this lineage. Aggregates of the amyloidogenic (Aβ1–42) peptide play a major role in the pathogenesis of Alzheimer's disease (AD), although the precise mechanism is unclear. In
genetic studies examining protein–protein interactions that could constitute native mechanisms of neuroprotection in vivo, overexpression of a WT human transthyretin (TTR) transgene was ameliorative in the APP23 transgenic murine model of human
AD. Targeted silencing of the endogenous TTR gene accelerated the development of the neuropathologic phenotype. Intraneuronal
TTR was seen in the brains of normal humans and mice and in AD patients and APP23 mice. The APP23 brains showed colocalization
of extracellular TTR with Aβ in plaques. Using surface plasmon resonance we obtained in vitro evidence of direct protein–protein interaction between TTR and Aβ aggregates. These findings suggest that TTR is protective
because of its capacity to bind toxic or pretoxic Aβ aggregates in both the intracellular and extracellular environment in
a chaperone-like manner. The interaction may represent a unique normal host defense mechanism, enhancement of which could
be therapeutically useful.
Co-reporter:Miguel Luz Soares, Teresa Coelho, Alda Sousa, Gösta Holmgren, Maria João Saraiva, Daniel L Kastner and Joel N Buxbaum
European Journal of Human Genetics 2004 12(3) pp:225-237
Publication Date(Web):November 19, 2003
DOI:10.1038/sj.ejhg.5201095
Familial amyloid polyneuropathy (FAP) is a lethal autosomal dominant disorder in which fibrils derived from mutant forms of transthyretin (TTR), the normal plasma carrier of thyroxine (T4) and retinol-binding protein, are deposited in tissues. Over 80 TTR sequence variants are associated with FAP, but the amino-acid substitutions alone do not completely explain the variability in disease penetrance, pathology and clinical course. To analyze the factors possibly contributing to this phenotypic variability, we characterized the variations within the wild-type and mutant (Val30Met) TTR genes and their flanking sequences by performing extended microsatellite haplotype analyses, sequencing and single-nucleotide polymorphism haplotyping of genomic DNA from Portuguese and Swedish carriers of V30M. We identified 10 new polymorphisms in the TTR untranslated regions, eight resulting from single-base substitutions and two arising from insertion/deletions in dinucleotide repeat sequences. The data suggest that the onset of symptoms of FAP V30M may be modulated by an interval downstream of TTR on the accompanying noncarrier chromosome (defined by microsatellites D18S457 and D18S456), but not by the immediately 5'- and 3'-flanking sequences of TTR. During the course of these studies, we also encountered the first instance in which the previously described intragenic haplotype III may be associated with V30M FAP in the Portuguese population.
Co-reporter:Natàlia Reixach;Songpon Deechongkit;Xin Jiang;Jeffery W. Kelly;Joel N. Buxbaum;
Proceedings of the National Academy of Sciences 2004 101(9) pp:2817-2822
Publication Date(Web):February 23, 2004
DOI:10.1073/pnas.0400062101
The transthyretin (TTR) amyloidoses are human diseases in which the misfolded TTR protein aggregates in tissues with subsequent
visceral, peripheral, and autonomic nerve dysfunction. Recent reports have stressed the importance of oligomeric intermediates
as major cytotoxic species in various forms of amyloidogenesis. We have examined the cytotoxic effects of several quaternary
structural states of wild-type and variant TTR proteins on cells of neural lineage. TTR amyloid fibrils and soluble aggregates
>100 kDa were not toxic. Incubation of TTR under the conditions of the cell assay and analysis by size-exclusion chromatography
and SDS/PAGE reveal that monomeric TTR or relatively small, rapidly formed aggregates of a maximum size of six subunits were
the major cytotoxic species. Small molecules that stabilize the native tetrameric state were shown to prevent toxicity. The
studies are consistent with a model in which the misfolded TTR monomer rapidly aggregates to form transient low molecular
mass assemblies (<100 kDa) that are highly cytotoxic in tissue culture.
Co-reporter:Joel N. Buxbaum
FEBS Letters (20 August 2009) Volume 583(Issue 16) pp:2663-2673
Publication Date(Web):20 August 2009
DOI:10.1016/j.febslet.2009.07.031
The amyloidoses are the prototype gain of toxic function protein misfolding diseases. As such, several naturally occurring animal models and their inducible variants provided some of the first insights into these disorders of protein aggregation. With greater analytic knowledge and the increasing flexibility of transgenic and gene knockout technology, new models have been generated allowing the interrogation of phenomena that have not been approachable in more reductionist systems, i.e. behavioral readouts in the neurodegenerative diseases, interactions among organ systems in the transthyretin amyloidoses and taking pre-clinical therapeutic trials beyond cell culture. The current review describes the features of both transgenic and non-transgenic models and discusses issues that appear to be unresolved even when viewed in their organismal context.
.jpg)