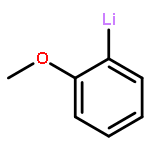
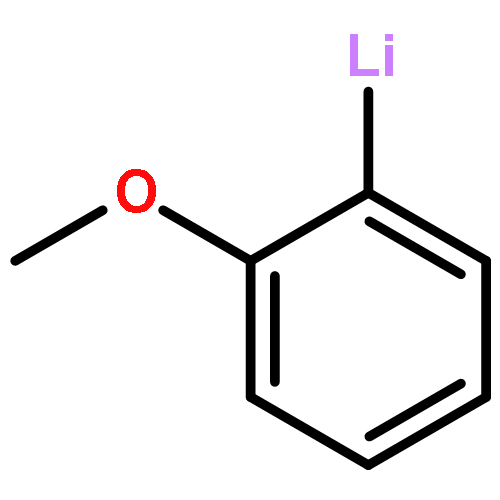
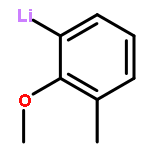
Co-reporter:Keith Izod, James M. Watson, Salima M. El-Hamruni, Ross W. Harrington, and Paul G. Waddell
Organometallics June 12, 2017 Volume 36(Issue 11) pp:2218-2218
Publication Date(Web):May 16, 2017
DOI:10.1021/acs.organomet.7b00262
The reaction between {(Me3Si)2CH}PH(C6H4-2-OMe) (4) and 1 equiv of BH3·SMe2 yields the phosphine–borane {(Me3Si)2CH}PH(BH3)(C6H4-2-OMe) (5). Subsequent reaction between 5 and 1 equiv of n-BuLi in THF gives the phosphido–borane complex [{(Me3Si)2CH}P(BH3)(C6H4-2-OMe)]Li(THF) (6a), which was isolated as a colorless microcrystalline solid. Treatment of 5 with 1 equiv of PhCH2M yields the corresponding complexes [{(Me3Si)2CH}P(BH3)(C6H4-2-OMe)]ML (ML = Na(THF) (6b), K(pmdeta) (6c); pmdeta = N,N,N′,N″,N″-pentamethyldiethylenetriamine), after crystallization in the presence of the corresponding coligand. While compounds 6b,c are stable toward heat, compound 6a decomposes on heating to 50 °C in toluene to give the cluster [[{(Me3Si)2CH}PH(C6H4-2-O)]Li]6 (7) and the tertiary phosphine–borane {(Me3Si)2CH}P(BH3)(Me)(C6H4-2-OMe) (8). Related C–O cleavage reactions are observed when MgI2 is treated with 2 equiv of 6a and when CaI2 is treated with 2 equiv of [{(Me3Si)2CH}P(BH3)(C6H4-2-OMe)]K in THF, giving [{(Me3Si)2CH}P(BH3)(C6H4-2-O)Mg(THF)2]2 (9) and [{(Me3Si)2CH}P(BH3)(C6H4-2-O)Ca(THF)]4 (10), respectively, along with 1 equiv of 8 in each case. In contrast, the reaction between SrI2 and 2 equiv of [{(Me3Si)2CH}P(BH3)(C6H4-2-OMe)]K in THF yields [{(Me3Si)2CH}P(BH3)(C6H4-2-OMe)Sr(THF)4] (11).
Co-reporter:Dr. Keith Izod;Peter Evans;Dr. Paul G. Waddell
Angewandte Chemie 2017 Volume 129(Issue 20) pp:5685-5689
Publication Date(Web):2017/05/08
DOI:10.1002/ange.201701867
AbstractThere is growing interest in compounds containing functionalized E=E multiple bonds (E=Si, Ge, Sn, Pb) because of their potential to exhibit novel physical and chemical properties. However, compounds containing multiple functionalizations are rare, with scarcity increasing with increasing degree of substitution. The first ditetrelene R2E=ER2 in which the E=E bond is substituted by four heteroatoms (other than Si) is described. The tetraphosphadisilene {(Mes)2P}2Si=Si{P(Mes)2}2 (7) is readily isolated from the reaction between SiBr4 and [(Mes)2P]Li, the latter of which acts as a sacrificial reducing agent. The structure of 7 is presented, while the bonding in, and stability of 7 were probed using DFT calculations.
Co-reporter:Dr. Keith Izod;Peter Evans;Dr. Paul G. Waddell
Angewandte Chemie International Edition 2017 Volume 56(Issue 20) pp:5593-5597
Publication Date(Web):2017/05/08
DOI:10.1002/anie.201701867
AbstractThere is growing interest in compounds containing functionalized E=E multiple bonds (E=Si, Ge, Sn, Pb) because of their potential to exhibit novel physical and chemical properties. However, compounds containing multiple functionalizations are rare, with scarcity increasing with increasing degree of substitution. The first ditetrelene R2E=ER2 in which the E=E bond is substituted by four heteroatoms (other than Si) is described. The tetraphosphadisilene {(Mes)2P}2Si=Si{P(Mes)2}2 (7) is readily isolated from the reaction between SiBr4 and [(Mes)2P]Li, the latter of which acts as a sacrificial reducing agent. The structure of 7 is presented, while the bonding in, and stability of 7 were probed using DFT calculations.
Co-reporter:Keith Izod;Peter Evans;Paul G. Waddell
Dalton Transactions 2017 vol. 46(Issue 40) pp:13824-13834
Publication Date(Web):2017/10/17
DOI:10.1039/C7DT02238G
The reaction between (Dipp)2PH and one equivalent of n-BuLi, PhCH2Na or PhCH2K in THF gives the complexes [(Dipp)2P]Li(THF)3 (2a), {[(Dipp)2P]Na(THF)2}2 (3a) and [(Dipp)2P]K(THF)4 (4a), respectively [Dipp = 2,6-iPr2C6H3]. Exposure of these compounds to vacuum yields the alternative solvates [(Dipp)2P]Li(THF)2 (2b), [(Dipp)2P]Na(THF)1.5 (3b), and [(Dipp)2P]K (4b), respectively; the alternative adduct [(Dipp)2P]Na(PMDETA) (3c) was prepared by treatment of 3a with PMDETA. Treatment of (Dipp)(Mes)PH or (Mes)2PH with one equivalent of n-BuLi in THF gives the complexes [(Dipp)(Mes)P]Li(THF)3 (7a) and [(Mes)2P]2Li2(THF)2(OEt2) (8a) after crystallisation from diethyl ether [Mes = 2,4,6-Me3C6H2]; crystallisation of 8a from hexane gives the alternative adduct [(Mes)2P]Li(THF)3 (8b). Exposure of 7a, 8a and 8b to vacuum leads to loss of coordinated solvent, yielding the solvates [(Dipp)(Mes)P]Li(THF)2 (7b) and [(Mes)2P]Li(THF) (8c). The solid-state structures of complexes 2a, 3a, 3c, 4a, 7a, 8a, and 8b have been determined by X-ray crystallography. Variable-temperature 31P{1H} and 7Li NMR spectroscopy indicates that 2b, 3b and 7b are subject to a monomer–dimer equilibrium in solution, where the monomeric forms are favoured at low temperature. In contrast, variable-temperature 31P{1H} and 7Li NMR spectroscopy suggests that 8c is subject to a dynamic equilibrium between a dimer and a cyclic trimer in solution, where the trimer is favoured at low temperatures.
Co-reporter:Keith Izod, Peter Evans, Paul G. Waddell, and Michael R. Probert
Inorganic Chemistry 2016 Volume 55(Issue 20) pp:10510-10522
Publication Date(Web):September 30, 2016
DOI:10.1021/acs.inorgchem.6b01566
A rare P–E π interaction between the lone pair of a planar P center and the vacant p orbital at the Ge or Sn center provides efficient stabilization for P-substituted tetrylenes (R2P)2E (E = Ge, Sn) and enables isolation of the first example of a compound with a crystallographically authenticated P═Sn bond. Subtle changes in the electronic properties of the bulky aryl substituents in these compounds change the preference for planar versus pyramidal P centers in the solid state; however, variable-temperature NMR spectroscopy indicates that in solution these species are subject to a dynamic equilibrium, which interconverts the planar and pyramidal P centers. Consistent with this, density functional theory studies suggest that there is only a small energy difference between the planar and pyramidal forms of these compounds and reveal a small singlet–triplet energy separation, suggesting potentially interesting reactivities.
Co-reporter:Keith Izod, Casey M. Dixon, Ross W. Harrington and Michael R. Probert
Chemical Communications 2015 vol. 51(Issue 4) pp:679-681
Publication Date(Web):21 Nov 2014
DOI:10.1039/C4CC08740B
The reaction between the phosphine–borane-stabilised dicarbanion complex [1,2-C6H4{CHP(BH3)Cy2}2][Li(THF)n]2 and Cp2Sn gives the unusual stannyl–stannylene [[1,2-C6H4{CHP(BH3)Cy2}2]Sn]2·1½PhMe, in which one dicarbanion ligand chelates a tin centre, while the other bridges a tin–tin bond. The stannylene centre is stabilised by an agostic-type B–H⋯Sn interaction.
Co-reporter:Keith Izod, Corinne Wills, Salima El-Hamruni, Ross W. Harrington, Paul G. Waddell, and Michael R. Probert
Organometallics 2015 Volume 34(Issue 11) pp:2406-2414
Publication Date(Web):February 27, 2015
DOI:10.1021/om501183p
The reaction between (4-tBuC6H4CH2)2Ca and one equivalent of PhP(BH3)(CH2SiMe3)2 (1) in diethyl ether gives the dimeric complex [[PhP(BH3){CH(SiMe3)}2]Ca(OEt2)]2 (2) in good yield. Similar reactions between 1 and one equivalent of either (PhCH2)2Sr(THF) or (PhCH2)2Ba yield the corresponding dimers [[PhP(BH3){CH(SiMe3)}2]Sr(THF)1.75(OEt2)0.25]2 (3) and [[PhP(BH3){CH(SiMe3)}2]Ba(OEt2)1.75(THF)0.25]2 (4), respectively. Unexpectedly, an attempt to prepare 3 from a one-pot reaction between SrI2, 1, and two equivalents of PhCH2K gave the complex [[PhP(BH3){CH(SiMe3)}2]2Sr3K2(OEt2)(THF)2]2·Et2O (5) in low yield. While superficially similar, compounds 2, 3, and 4 crystallize with distinct structures, which differ either in the chirality of the carbanion centers or in the nature of the bridging group. Compounds 2, 3, and 4 decompose slowly in THF solution to give ethylene, Ae(OEt)2 (or Ae(OCH═CH2)2), and the monocarbanion derivatives [PhP(BH3){CH(SiMe3)}{CH2SiMe3}]2Ae(THF)n, according to NMR spectroscopy [Ae = Ca, Sr, Ba].
Co-reporter:Keith Izod and Paul G. Waddell
Organometallics 2015 Volume 34(Issue 12) pp:2726-2730
Publication Date(Web):February 24, 2015
DOI:10.1021/om5010868
The reactions between two equivalents of PhCH2K and AeI2 (Ae = Ca, Sr, Ba) in THF give the alkaline earth metal benzyls [(PhCH2)2Ca(THF)4] (3), [(PhCH2)4Sr2(THF)3] (4), and [(PhCH2)6Ba3(THF)4] (5), as orange, crystalline solids in good yield. While 3 crystallizes as a molecular species, compound 4 crystallizes as a ribbon polymer and 5 crystallizes as a 2-dimensional sheet in which the metal ions are coordinated by either benzyl and THF molecules or benzyl ligands alone.
Co-reporter:Dr. Keith Izod;Daniel G. Rayner;Dr. Salima M. El-Hamruni;Dr. Ross W. Harrington;Dr. Ulrich Baisch
Angewandte Chemie International Edition 2014 Volume 53( Issue 14) pp:3636-3640
Publication Date(Web):
DOI:10.1002/anie.201308002
Abstract
N-Heterocyclic carbenes and their heavier homologues are, in part, stabilized by delocalization of the N lone pairs into the vacant p-orbital at carbon (or a heavier Group 14 element center). These interactions are usually absent in the corresponding P-substituted species, owing to the large barrier to planarization of phosphorus. However, judicious selection of the substituents at phosphorus has enabled the synthesis of a diphosphagermylene, [(Dipp)2P]2Ge, in which one of the P centers is planar (Dipp=2,6-diisopropylphenyl). The planar nature of this P center and the correspondingly short PGe distance suggest a significant degree of PGe multiple bond character that is due to delocalization of the phosphorus lone pair into the vacant p-orbital at germanium. DFT calculations support this proposition and NBO and AIM analyses are consistent with a GeP bond order greater than unity.
Co-reporter:Dr. Keith Izod;Daniel G. Rayner;Dr. Salima M. El-Hamruni;Dr. Ross W. Harrington;Dr. Ulrich Baisch
Angewandte Chemie 2014 Volume 126( Issue 14) pp:3710-3714
Publication Date(Web):
DOI:10.1002/ange.201308002
Abstract
N-Heterocyclic carbenes and their heavier homologues are, in part, stabilized by delocalization of the N lone pairs into the vacant p-orbital at carbon (or a heavier Group 14 element center). These interactions are usually absent in the corresponding P-substituted species, owing to the large barrier to planarization of phosphorus. However, judicious selection of the substituents at phosphorus has enabled the synthesis of a diphosphagermylene, [(Dipp)2P]2Ge, in which one of the P centers is planar (Dipp=2,6-diisopropylphenyl). The planar nature of this P center and the correspondingly short PGe distance suggest a significant degree of PGe multiple bond character that is due to delocalization of the phosphorus lone pair into the vacant p-orbital at germanium. DFT calculations support this proposition and NBO and AIM analyses are consistent with a GeP bond order greater than unity.
Co-reporter:Keith Izod, Corinne Wills, Emma Anderson, Ross W. Harrington, and Michael R. Probert
Organometallics 2014 Volume 33(Issue 19) pp:5283-5294
Publication Date(Web):September 5, 2014
DOI:10.1021/om5005995
The reaction between iPr2PCl and Ph2P(BH3)CH2Li gives the mixed phosphine/phosphine-borane Ph2P(BH3)CH2PiPr2 (1a) in good yield. Thermolysis of 1a leads to borane migration and the formation of Ph2PCH2P(BH3)iPr2 (2a) along with small amounts of Ph2P(BH3)CH2P(BH3)iPr2 (3a) and Ph2PCH2PiPr2 (4a). Compound 3a may be synthesized directly from the reaction of 1a with BH3·SMe2, while 4a can be prepared cleanly by heating 1a in methanol under reflux. Kinetic studies on the conversion of 1a to 2a reveal the reaction to be apparently first order in 1a, suggesting a dissociative process, and yield the activation parameters ΔH⧧ = 63 ± 8 kJ mol–1, ΔS⧧ = −145 ± 24 J K–1 mol–1, and ΔG⧧ = 106 ± 8 kJ mol–1, the negative entropy of activation conversely suggesting an associative process. DFT studies suggest that concerted migration of borane within a molecule of 1a is disfavored, but that both the dissociative and associative mechanisms for borane migration operate simultaneously. Metalation of 1a–4a with nBuLi in the presence of tmeda gives the complexes [{Ph2P(BH3)}CHPiPr2]Li(tmeda) (1b), [Ph2PCH{P(BH3)iPr2}]Li(tmeda) (2b), [{Ph2P(BH3)}CH{P(BH3)iPr2}]Li(tmeda) (3b), and [Ph2PCHPiPr2]Li(tmeda) (4b), respectively, which adopt similar structures in the solid state. Analysis of the crystal structures suggests that the phosphine-borane groups stabilize the adjacent charge to a greater extent than the phosphine groups. This is supported by DFT calculations, which show that the greatest delocalization of negative charge from the carbanion is into the P–C(Ph) or P–C(Pr) σ*-orbitals of the phosphine-borane substituents.
Co-reporter:Keith Izod, Casey M. Dixon, Ross W. Harrington and Michael R. Probert
Chemical Communications 2015 - vol. 51(Issue 4) pp:NaN681-681
Publication Date(Web):2014/11/21
DOI:10.1039/C4CC08740B
The reaction between the phosphine–borane-stabilised dicarbanion complex [1,2-C6H4{CHP(BH3)Cy2}2][Li(THF)n]2 and Cp2Sn gives the unusual stannyl–stannylene [[1,2-C6H4{CHP(BH3)Cy2}2]Sn]2·1½PhMe, in which one dicarbanion ligand chelates a tin centre, while the other bridges a tin–tin bond. The stannylene centre is stabilised by an agostic-type B–H⋯Sn interaction.


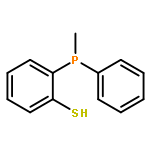
![Phosphine, [2-(methylthio)phenyl]phenyl-](http://img.cochemist.com/ccimg/93400/93383-29-0.png)
![Phosphine, [2-(methylthio)phenyl]phenyl-](http://img.cochemist.com/ccimg/93400/93383-29-0_b.png)


![Phosphine sulfide, [1-(dimethylphosphinothioyl)ethenyl]diphenyl-](http://img.cochemist.com/ccimg/686400/686353-37-7.png)
![Phosphine sulfide, [1-(dimethylphosphinothioyl)ethenyl]diphenyl-](http://img.cochemist.com/ccimg/686400/686353-37-7_b.png)
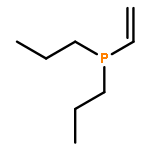
![Phosphine, [bis(trimethylsilyl)methyl]dipropyl-](http://img.cochemist.com/ccimg/686400/686353-34-4.png)
![Phosphine, [bis(trimethylsilyl)methyl]dipropyl-](http://img.cochemist.com/ccimg/686400/686353-34-4_b.png)




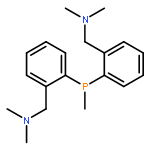
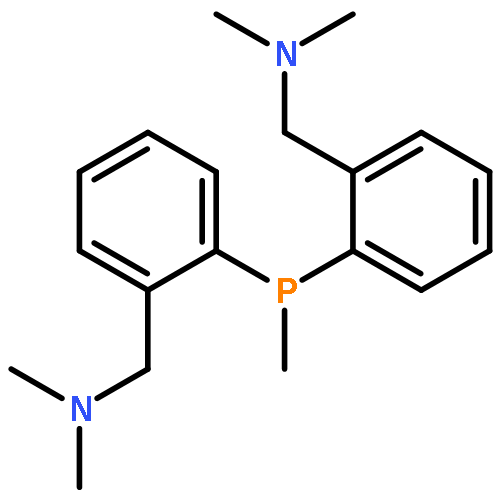
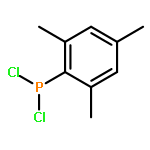
![PHOSPHONOUS DICHLORIDE, [2,4,6-TRIS(1-METHYLETHYL)PHENYL]-](http://img.cochemist.com/ccimg/53400/53356-73-3.png)
![PHOSPHONOUS DICHLORIDE, [2,4,6-TRIS(1-METHYLETHYL)PHENYL]-](http://img.cochemist.com/ccimg/53400/53356-73-3_b.png)
![PHOSPHINE SULFIDE, DIPROPYL[1-(TRIMETHYLSILYL)ETHENYL]-](http://img.cochemist.com/ccimg/686400/686353-35-5.png)
![PHOSPHINE SULFIDE, DIPROPYL[1-(TRIMETHYLSILYL)ETHENYL]-](http://img.cochemist.com/ccimg/686400/686353-35-5_b.png)
![PHOSPHINE, [BIS(TRIMETHYLSILYL)METHYL]BIS[2-(METHOXYMETHYL)PHENYL]-](http://img.cochemist.com/ccimg/855000/854916-54-4.png)
![PHOSPHINE, [BIS(TRIMETHYLSILYL)METHYL]BIS[2-(METHOXYMETHYL)PHENYL]-](http://img.cochemist.com/ccimg/855000/854916-54-4_b.png)
![SILANE, [(METHOXYDIMETHYLSILYL)METHYLENE]BIS[TRIMETHYL-](http://img.cochemist.com/ccimg/68300/68260-20-8.png)
![SILANE, [(METHOXYDIMETHYLSILYL)METHYLENE]BIS[TRIMETHYL-](http://img.cochemist.com/ccimg/68300/68260-20-8_b.png)
![SILANE, [(BROMODIMETHYLSILYL)METHYLENE]BIS[TRIMETHYL-](http://img.cochemist.com/ccimg/87000/86932-21-0.png)
![SILANE, [(BROMODIMETHYLSILYL)METHYLENE]BIS[TRIMETHYL-](http://img.cochemist.com/ccimg/87000/86932-21-0_b.png)
![Phosphine, [bis(trimethylsilyl)methyl]phenyl-](http://img.cochemist.com/ccimg/109200/109152-83-2.png)
![Phosphine, [bis(trimethylsilyl)methyl]phenyl-](http://img.cochemist.com/ccimg/109200/109152-83-2_b.png)
![PHOSPHONOUS DICHLORIDE, [BIS(TRIMETHYLSILYL)METHYL]-](http://img.cochemist.com/ccimg/76600/76505-20-9.png)
![PHOSPHONOUS DICHLORIDE, [BIS(TRIMETHYLSILYL)METHYL]-](http://img.cochemist.com/ccimg/76600/76505-20-9_b.png)
![PHOSPHINE, [1-[BIS(1-METHYLETHYL)PHOSPHINO]ETHENYL]DIPHENYL-](http://img.cochemist.com/ccimg/686400/686353-33-3.png)
![PHOSPHINE, [1-[BIS(1-METHYLETHYL)PHOSPHINO]ETHENYL]DIPHENYL-](http://img.cochemist.com/ccimg/686400/686353-33-3_b.png)
![PHOSPHINE, TRIS[BIS(TRIMETHYLSILYL)METHYL]-](http://img.cochemist.com/ccimg/478400/478364-98-6.png)
![PHOSPHINE, TRIS[BIS(TRIMETHYLSILYL)METHYL]-](http://img.cochemist.com/ccimg/478400/478364-98-6_b.png)
![Potassium, [(methoxydimethylsilyl)bis(trimethylsilyl)methyl]-](http://img.cochemist.com/ccimg/202200/202134-13-2.png)
![Potassium, [(methoxydimethylsilyl)bis(trimethylsilyl)methyl]-](http://img.cochemist.com/ccimg/202200/202134-13-2_b.png)


![Phosphine, [(diphenylphosphino)methyl]dimethyl-](http://img.cochemist.com/ccimg/62300/62263-64-3.png)
![Phosphine, [(diphenylphosphino)methyl]dimethyl-](http://img.cochemist.com/ccimg/62300/62263-64-3_b.png)
![Phosphine, bis[2,4,6-tris(1-methylethyl)phenyl]-](http://img.cochemist.com/ccimg/135700/135665-28-0.png)
![Phosphine, bis[2,4,6-tris(1-methylethyl)phenyl]-](http://img.cochemist.com/ccimg/135700/135665-28-0_b.png)

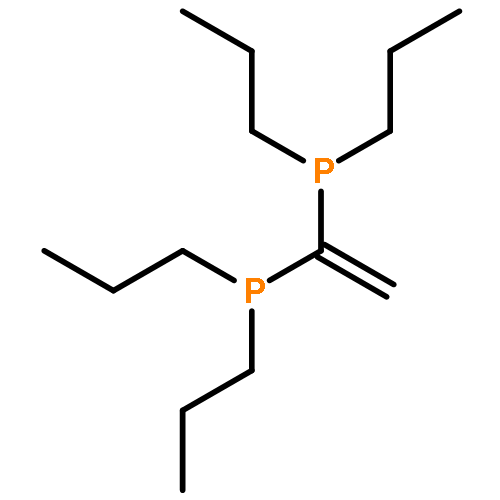
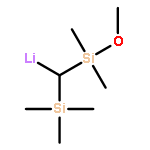
![Lithium, [2-(dimethylamino)phenyl]-](http://img.cochemist.com/ccimg/22700/22608-37-3.png)
![Lithium, [2-(dimethylamino)phenyl]-](http://img.cochemist.com/ccimg/22700/22608-37-3_b.png)


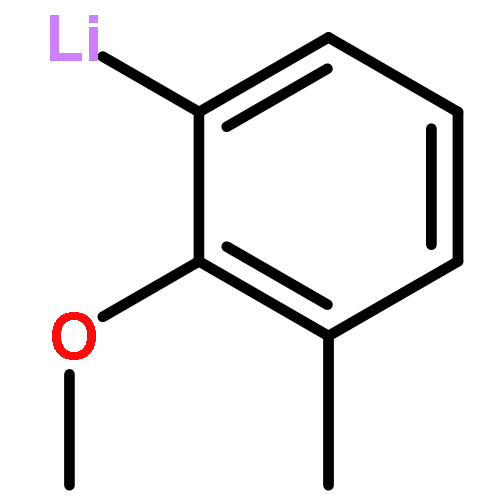
methyl-](http://img.cochemist.com/ccimg/679500/679406-54-3.png)
methyl-](http://img.cochemist.com/ccimg/679500/679406-54-3_b.png)
![Lanthanum, tris[bis(trimethylsilyl)methyl]-](http://img.cochemist.com/ccimg/121100/121086-85-9.png)
![Lanthanum, tris[bis(trimethylsilyl)methyl]-](http://img.cochemist.com/ccimg/121100/121086-85-9_b.png)


![Disiloxane, 1,3-bis[bis(trimethylsilyl)methyl]-1,1,3,3-tetramethyl-](http://img.cochemist.com/ccimg/110100/110086-49-2.png)
![Disiloxane, 1,3-bis[bis(trimethylsilyl)methyl]-1,1,3,3-tetramethyl-](http://img.cochemist.com/ccimg/110100/110086-49-2_b.png)


![Phosphine, [bis(trimethylsilyl)methyl][2-(methylthio)phenyl]-](http://img.cochemist.com/ccimg/496900/496868-40-7.png)
![Phosphine, [bis(trimethylsilyl)methyl][2-(methylthio)phenyl]-](http://img.cochemist.com/ccimg/496900/496868-40-7_b.png)
-,potassium salt](http://img.cochemist.com/ccimg/274700/274695-97-5.png)
-,potassium salt](http://img.cochemist.com/ccimg/274700/274695-97-5_b.png)
methyl-](http://img.cochemist.com/ccimg/274700/274695-93-1.png)
methyl-](http://img.cochemist.com/ccimg/274700/274695-93-1_b.png)
-, potassium salt](/data/chemimg/1172100/274695-90-8.png)
-, potassium salt](/data/chemimg/1172100/274695-90-8_b.png)




![Phosphine, [[bis(1-methylethyl)phosphino]methyl]diphenyl-](http://img.cochemist.com/ccimg/62300/62263-67-6.png)
![Phosphine, [[bis(1-methylethyl)phosphino]methyl]diphenyl-](http://img.cochemist.com/ccimg/62300/62263-67-6_b.png)
![Potassium, [(trimethylsilyl)methyl]-](http://img.cochemist.com/ccimg/53200/53127-82-5.png)
![Potassium, [(trimethylsilyl)methyl]-](http://img.cochemist.com/ccimg/53200/53127-82-5_b.png)