Co-reporter:Hagit Ben-Daat;Christopher L. Rock;Marco Flores;Thomas L. Groy;Amanda C. Bowman
Chemical Communications 2017 vol. 53(Issue 53) pp:7333-7336
Publication Date(Web):2017/06/29
DOI:10.1039/C7CC02281F
Addition of NaEt3BH to (Ph2PPrDI)CoCl2 affords the corresponding monohydride, (Ph2PPrDI)CoH. X-ray diffraction and DFT calculations indicate that this compound possesses a radical monoanion α-DI chelate and a Co(II) centre. Notably, (Ph2PPrDI)CoH catalyzes the hydroboration of alkynes and dihydroboration of nitriles under mild conditions.
Co-reporter:Chandrani Ghosh, Thomas L. Groy, Amanda C. Bowman and Ryan J. Trovitch
Chemical Communications 2016 vol. 52(Issue 24) pp:4553-4556
Publication Date(Web):26 Feb 2016
DOI:10.1039/C5CC09167E
Reduction of 6-coordinate (Ph2PPrDI)FeBr2 under N2 results in formation of the terminal dinitrogen complex, (Ph2PPrDI)FeN2. Heating this product to 75 °C allows for C–H and C–P activation of the chelate to generate the cisoid and transoid isomers of [(μ-PrPPh-κ5-P,N,N,Cγ,P-Ph2PPrDIPrPPh)Fe]2. Mechanistic possibilities for this transformation are discussed.
Co-reporter:Raja Pal, Brian R. Cherry, Marco Flores, Thomas L. Groy and Ryan J. Trovitch
Dalton Transactions 2016 vol. 45(Issue 24) pp:10024-10033
Publication Date(Web):13 Apr 2016
DOI:10.1039/C6DT00301J
Analysis of previously reported [(Ph2PPrPDI)MoI][I] by cyclic voltammetry revealed a reversible wave at −1.20 V vs. Fc+/0, corresponding to the Mo(II)/Mo(I) redox couple. Reduction of [(Ph2PPrPDI)MoI][I] using stoichiometric K/naphthalene resulted in ligand deprotonation rather than reduction to yield a Mo(II) monoiodide complex featuring a Mo–C bond to the α-position of one imine substituent, (κ6-P,N,N,N,C,P-Ph2PPrPDI)MoI. Successful isolation of the inner-sphere Mo(I) monoiodide complex, (Ph2PPrPDI)MoI, was achieved via reduction of [(Ph2PPrPDI)MoI][I] with equimolar Na/naphthalene. This complex was found to have a near octahedral coordination geometry by single crystal X-ray diffraction and electron paramagnetic resonance (EPR) spectroscopy revealed an unpaired Mo-based electron which is highly delocalized onto the PDI chelate core. Attempts to prepare a Mo(I) monohydride complex upon adding NaEt3BH to (Ph2PPrPDI)MoI resulted in disproportionation to yield an equimolar quantity of (κ6-P,N,N,N,C,P-Ph2PPrPDI)MoH and newly identified (Ph2PPrPDI)MoH2. Independent preparation of (Ph2PPrPDI)MoH2 was achieved by adding 2 equiv. NaEt3BH to [(Ph2PPrPDI)MoI][I] and a minimum hydride resonance T1 of 176 ms suggests that the Mo-bound H atoms are best described as classical hydrides. Interestingly, (Ph2PPrPDI)MoH2 can be converted to (κ6-P,N,N,N,C,P-Ph2PPrPDI)MoI upon iodomethane addition, while (Ph2PPrPDI)MoH2 is prepared from (κ6-P,N,N,N,C,P-Ph2PPrPDI)MoI in the presence of excess NaEt3BH. Similarly, (κ6-P,N,N,N,C,P-Ph2PPrPDI)MoI can be converted to (κ6-P,N,N,N,C,P-Ph2PPrPDI)MoH with 1 equiv. of NaEt3BH, while the opposite transformation occurs following iodomethane addition to (κ6-P,N,N,N,C,P-Ph2PPrPDI)MoH. Facile interconversion between [(Ph2PPrPDI)MoI][I], (κ6-P,N,N,N,C,P-Ph2PPrPDI)MoI, (κ6-P,N,N,N,C,P-Ph2PPrPDI)MoH, and (Ph2PPrPDI)MoH2 is expected to guide future reactivity studies on this unique set of compounds.
Co-reporter:Tufan K. Mukhopadhyay, Nicholas L. MacLean, Lu Gan, Daniel C. Ashley, Thomas L. Groy, Mu-Hyun Baik, Anne K. Jones, and Ryan J. Trovitch
Inorganic Chemistry 2015 Volume 54(Issue 9) pp:4475-4482
Publication Date(Web):April 22, 2015
DOI:10.1021/acs.inorgchem.5b00315
Heating a 1:1 mixture of (CO)5MnBr and the phosphine-substituted pyridine diimine ligand, Ph2PPrPDI, in THF at 65 °C for 24 h afforded the diamagnetic complex [(Ph2PPrPDI)Mn(CO)][Br] (1). Higher temperatures and longer reaction times resulted in bromide displacement of the remaining carbonyl ligand and the formation of paramagnetic (Ph2PPrPDI)MnBr (2). The molecular structure of 1 was determined by single crystal X-ray diffraction, and density functional theory (DFT) calculations indicate that this complex is best described as low-spin Mn(I) bound to a neutral Ph2PPrPDI chelating ligand. The redox properties of 1 and 2 were investigated by cyclic voltammetry (CV), and each complex was tested for electrocatalytic activity in the presence of both CO2 and Brønsted acids. Although electrocatalytic response was not observed when CO2, H2O, or MeOH was added to 1 individually, the addition of H2O or MeOH to CO2-saturated acetonitrile solutions of 1 afforded voltammetric responses featuring increased current density as a function of proton source concentration (icat/ip up to 2.4 for H2O or 4.2 for MeOH at scan rates of 0.1 V/s). Bulk electrolysis using 5 mM 1 and 1.05 M MeOH in acetonitrile at −2.2 V vs Fc+/0 over the course of 47 min gave H2 as the only detectable product with a Faradaic efficiency of 96.7%. Electrochemical experiments indicate that CO2 promotes 1-mediated H2 production by lowering apparent pH. While evaluating 2 for electrocatalytic activity, this complex was found to decompose rapidly in the presence of acid. Although modest H+ reduction activity was realized, the experiments described herein indicate that care must be taken when evaluating Mn complexes for electrocatalytic CO2 reduction.
Co-reporter:Raja Pal; Thomas L. Groy
Inorganic Chemistry 2015 Volume 54(Issue 15) pp:7506-7515
Publication Date(Web):July 23, 2015
DOI:10.1021/acs.inorgchem.5b01102
Using a multistep synthetic pathway, a bis(imino)pyridine (or pyridine diimine, PDI) molybdenum catalyst for the selective conversion of carbon dioxide into methanol has been developed. Starting from (Ph2PPrPDI)Mo(CO), I2 addition afforded [(Ph2PPrPDI)MoI(CO)][I], which features a seven-coordinate Mo(II) center. Heating this complex to 100 °C under vacuum resulted in CO loss and the formation of [(Ph2PPrPDI)MoI][I]. Reduction of [(Ph2PPrPDI)MoI][I] in the presence of excess K/Hg yielded (κ6-P,N,N,N,C,P-Ph2PPrPDI)MoH following methylene group C–H activation at the α-position of one PDI imine substituent. The addition of CO2 to (κ6-P,N,N,N,C,P-Ph2PPrPDI)MoH resulted in facile insertion to generate the respective η1-formate complex, (κ6-P,N,N,N,C,P-Ph2PPrPDI)Mo(OCOH). When low pressures of CO2 were added to solutions of (κ6-P,N,N,N,C,P-Ph2PPrPDI)MoH containing pinacolborane, the selective formation of H3COBPin and O(BPin)2 was observed along with precatalyst regeneration. When HBPin was limited, H2C(OBPin)2 was observed as an intermediate and (κ6-P,N,N,N,C,P-Ph2PPrPDI)Mo(OCOH) remained present throughout CO2 reduction. The hydroboration of CO2 to H3COBPin was optimized and 97% HBPin utilization by 0.1 mol % (κ6-P,N,N,N,C,P-Ph2PPrPDI)MoH was demonstrated over 8 h at 90 °C, resulting in a methoxide formation turnover frequency (TOF) of 40.4 h–1 (B–H utilization TOF = 121.2 h–1). Hydrolysis of the products and distillation at 65 °C allowed for MeOH isolation. The mechanism of (κ6-P,N,N,N,C,P-Ph2PPrPDI)MoH mediated CO2 hydroboration is presented in the context of these experimental observations. Notably, (κ6-P,N,N,N,C,P-Ph2PPrPDI)MoH is the first Mo hydroboration catalyst capable of converting CO2 to MeOH, and the importance of this study as it relates to previously described catalysts is discussed.
Co-reporter:Chandrani Ghosh; Tufan K. Mukhopadhyay; Marco Flores; Thomas L. Groy
Inorganic Chemistry 2015 Volume 54(Issue 21) pp:10398-10406
Publication Date(Web):October 19, 2015
DOI:10.1021/acs.inorgchem.5b01825
Heating (THF)2MnCl2 in the presence of the pyridine-substituted bis(imino)pyridine ligand, PyEtPDI, allowed preparation of the respective dihalide complex, (PyEtPDI)MnCl2. Reduction of this precursor using excess Na/Hg resulted in deprotonation of the chelate methyl groups to yield the bis(enamide)tris(pyridine)-supported product, (κ5-N,N,N,N,N-PyEtPDEA)Mn. This complex was characterized by single-crystal X-ray diffraction and found to possess an intermediate-spin (S = 3/2) Mn(II) center by the Evans method and electron paramagnetic resonance spectroscopy. Furthermore, (κ5-N,N,N,N,N-PyEtPDEA)Mn was determined to be an effective precatalyst for the hydrosilylation of aldehydes and ketones, exhibiting turnover frequencies of up to 2475 min–1 when employed under solvent-free conditions. This optimization allowed for isolation of the respective alcohols and, in two cases, the partially reacted silyl ethers, PhSiH(OR)2 [R = Cy and CH(Me)(nBu)]. The aldehyde hydrosilylation activity observed for (κ5-N,N,N,N,N-PyEtPDEA)Mn renders it one of the most efficient first-row transition metal catalysts for this transformation reported to date.
Co-reporter:Raja Pal, Thomas L. Groy, Amanda C. Bowman, and Ryan J. Trovitch
Inorganic Chemistry 2014 Volume 53(Issue 17) pp:9357-9365
Publication Date(Web):August 20, 2014
DOI:10.1021/ic501465v
Attempts to prepare low-valent molybdenum complexes that feature a pentadentate 2,6-bis(imino)pyridine (or pyridine diimine, PDI) chelate allowed for the isolation of two different products. Refluxing Mo(CO)6 with the pyridine-substituted PDI ligand, PyEtPDI, resulted in carbonyl ligand substitution and formation of the respective bis(ligand) compound (PyEtPDI)2Mo (1). This complex was investigated by single-crystal X-ray diffraction, and density functional theory calculations indicated that 1 possesses a Mo(0) center that back-bonds into the π*-orbitals of the unreduced PDI ligands. Heating an equimolar solution of Mo(CO)6 and the phosphine-substituted PDI ligand, Ph2PPrPDI, to 120 °C allowed for the preparation of (Ph2PPrPDI)Mo(CO) (2), which is supported by a κ5-N,N,N,P,P-Ph2PPrPDI chelate. Notably, 1 and 2 have been found to catalyze the hydrosilylation of benzaldehyde at 90 °C, and the optimization of 2-catalyzed aldehyde hydrosilylation at this temperature afforded turnover frequencies of up to 330 h–1. Considering additional experimental observations, the potential mechanism of 2-mediated carbonyl hydrosilylation is discussed.
Co-reporter:Tufan K. Mukhopadhyay, Marco Flores, Russell K. Feller, Brian L. Scott, R. Dean Taylor, Moshe Paz-Pasternak, Neil J. Henson, Francisca N. Rein, Nathan C. Smythe, Ryan J. Trovitch, and John C. Gordon
Organometallics 2014 Volume 33(Issue 24) pp:7101-7112
Publication Date(Web):December 1, 2014
DOI:10.1021/om500909h
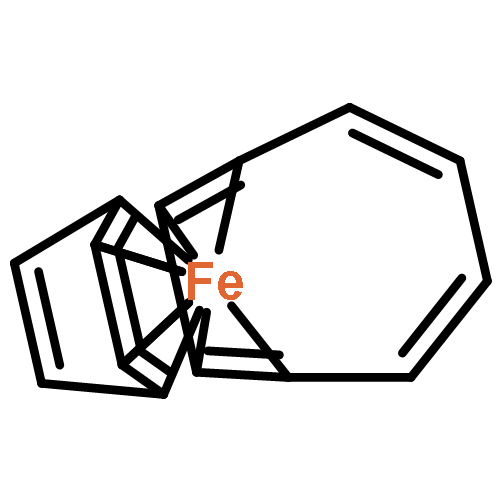
Formally zerovalent (κ3-phosphine)Fe(η4-COT) complexes supported by either Triphos (PhP(CH2CH2PPh2)2) or Triphos* (H3CC(CH2PPh2)3) have been prepared following chelate addition to (COT)2Fe (COT = 1,3,5,7-cyclooctatetraene) and by reduction of the respective dibromide complexes in the presence of excess COT. The solid-state structure of each complex was determined by single-crystal X-ray diffraction, and close inspection of the metrical parameters revealed significant COT ligand reduction, independent of the coordination geometry about iron. While the neutral and dianionic forms of the redox-active COT ligand have historically received a great deal of attention, a dearth of information regarding the often-evoked radical monoanion form of this ligand prompted the full electronic structure investigation of these complexes using a range of techniques. Comparison of the Mössbauer spectroscopic data collected for both (Triphos)Fe(η4-COT) complexes with data obtained for two appropriate reference compounds indicated that they possess a low-spin Fe(I) center that is antiferromagnetically coupled to a COT radical monoanion. Further evidence for this electronic structure determination by EPR spectroscopy and cyclic voltammetry is presented. A comparison of the solid-state metrical parameters determined in this study to those of related first-row transition-metal complexes has provided insight into the electronic structure analysis of related organometallic complexes.
Co-reporter:Tufan K. Mukhopadhyay ; Marco Flores ; Thomas L. Groy
Journal of the American Chemical Society 2013 Volume 136(Issue 3) pp:882-885
Publication Date(Web):December 24, 2013
DOI:10.1021/ja4116346
The reduction of (Ph2PPrPDI)MnCl2 allowed the preparation of the formally zerovalent complex, (Ph2PPrPDI)Mn, which features a pentadentate bis(imino)pyridine chelate. This complex is a highly active precatalyst for the hydrosilylation of ketones, exhibiting TOFs of up to 76,800 h–1 in the absence of solvent. Loadings as low as 0.01 mol % were employed, and (Ph2PPrPDI)Mn was found to mediate the atom-efficient utilization of Si–H bonds to form quaternary silane products. (Ph2PPrPDI)Mn was also shown to catalyze the dihydrosilylation of esters following cleavage of the substrate acyl C–O bond. Electronic structure investigation of (Ph2PPrPDI)Mn revealed that this complex possesses an unpaired electron on the metal center, rendering it likely that catalysis takes place following electron transfer to the incoming carbonyl substituent.
Co-reporter:Tyler M. Porter, Gabriel B. Hall, Thomas L. Groy and Ryan J. Trovitch
Dalton Transactions 2013 vol. 42(Issue 41) pp:14689-14692
Publication Date(Web):05 Sep 2013
DOI:10.1039/C3DT52419A
Although bis(α-diimine)Ni complexes were prepared when amine-substituted chelates were added to Ni(COD)2, the incorporation of strong-field phosphine donors allowed the isolation of (κ4-N,N,P,P-DI)Ni hydrosilylation catalysts. The crystallographic investigation of two different (κ4-N,N,P,P-DI)Ni compounds revealed that the geometry about nickel influences the observed degree of α-diimine reduction.
Co-reporter:Hagit Ben-Daat;Gabriel B. Hall;Thomas L. Groy
European Journal of Inorganic Chemistry 2013 Volume 2013( Issue 25) pp:4430-4442
Publication Date(Web):
DOI:10.1002/ejic.201300263
Abstract
The addition of aminoalkyl-substituted 2,6-bis(imino)pyridine (or pyridine diimine, PDI) ligands to [(COD)RhCl]2 (COD = 1,5-cyclooctadiene) resulted in the formation of rhodium monochloride complexes with the general formula (NPDI)RhCl (NPDI = iPr2NEtPDI or Me2NPrPDI). The investigation of (iPr2NEtPDI)RhCl and (Me2NPrPDI)RhCl by single-crystal X-ray diffraction verified the absence of amine arm coordination and a pseudo square-planar geometry about rhodium. Replacement of the chloride ligand with an outer-sphere anion was achieved by adding AgBF4 directly to (iPr2NEtPDI)RhCl to form [(iPr2NEtPDI)Rh][BF4]. Alternatively, this complex was prepared by chelate addition following the salt metathesis reaction between AgBF4 and [(COD)RhCl]2. Using the latter method, both [(NPDI)Rh][BF4] complexes were isolated and found to exhibit κ4-N,N,N,N-PDI coordination regardless of arm length or steric bulk. In contrast, the metallation of PPDI chelates featuring alkylphosphane imine substituents (PPDI = Ph2PEtPDI or Ph2PPrPDI) resulted in the formation of cationic complexes featuring κ5-N,N,N,P,P-PDI coordination in all instances, [(PPDI)Rh][X] (X = Cl, BF4). Adjusting the metallation stoichiometry allowed the preparation of [(Ph2PPrPDI)Rh][(COD)RhCl2], as validated by multinuclear NMR spectroscopy and single-crystal X-ray diffraction.
Co-reporter:Tyler M. Porter, Gabriel B. Hall, Thomas L. Groy and Ryan J. Trovitch
Dalton Transactions 2013 - vol. 42(Issue 41) pp:NaN14692-14692
Publication Date(Web):2013/09/05
DOI:10.1039/C3DT52419A
Although bis(α-diimine)Ni complexes were prepared when amine-substituted chelates were added to Ni(COD)2, the incorporation of strong-field phosphine donors allowed the isolation of (κ4-N,N,P,P-DI)Ni hydrosilylation catalysts. The crystallographic investigation of two different (κ4-N,N,P,P-DI)Ni compounds revealed that the geometry about nickel influences the observed degree of α-diimine reduction.
Co-reporter:Raja Pal, Brian R. Cherry, Marco Flores, Thomas L. Groy and Ryan J. Trovitch
Dalton Transactions 2016 - vol. 45(Issue 24) pp:NaN10033-10033
Publication Date(Web):2016/04/13
DOI:10.1039/C6DT00301J
Analysis of previously reported [(Ph2PPrPDI)MoI][I] by cyclic voltammetry revealed a reversible wave at −1.20 V vs. Fc+/0, corresponding to the Mo(II)/Mo(I) redox couple. Reduction of [(Ph2PPrPDI)MoI][I] using stoichiometric K/naphthalene resulted in ligand deprotonation rather than reduction to yield a Mo(II) monoiodide complex featuring a Mo–C bond to the α-position of one imine substituent, (κ6-P,N,N,N,C,P-Ph2PPrPDI)MoI. Successful isolation of the inner-sphere Mo(I) monoiodide complex, (Ph2PPrPDI)MoI, was achieved via reduction of [(Ph2PPrPDI)MoI][I] with equimolar Na/naphthalene. This complex was found to have a near octahedral coordination geometry by single crystal X-ray diffraction and electron paramagnetic resonance (EPR) spectroscopy revealed an unpaired Mo-based electron which is highly delocalized onto the PDI chelate core. Attempts to prepare a Mo(I) monohydride complex upon adding NaEt3BH to (Ph2PPrPDI)MoI resulted in disproportionation to yield an equimolar quantity of (κ6-P,N,N,N,C,P-Ph2PPrPDI)MoH and newly identified (Ph2PPrPDI)MoH2. Independent preparation of (Ph2PPrPDI)MoH2 was achieved by adding 2 equiv. NaEt3BH to [(Ph2PPrPDI)MoI][I] and a minimum hydride resonance T1 of 176 ms suggests that the Mo-bound H atoms are best described as classical hydrides. Interestingly, (Ph2PPrPDI)MoH2 can be converted to (κ6-P,N,N,N,C,P-Ph2PPrPDI)MoI upon iodomethane addition, while (Ph2PPrPDI)MoH2 is prepared from (κ6-P,N,N,N,C,P-Ph2PPrPDI)MoI in the presence of excess NaEt3BH. Similarly, (κ6-P,N,N,N,C,P-Ph2PPrPDI)MoI can be converted to (κ6-P,N,N,N,C,P-Ph2PPrPDI)MoH with 1 equiv. of NaEt3BH, while the opposite transformation occurs following iodomethane addition to (κ6-P,N,N,N,C,P-Ph2PPrPDI)MoH. Facile interconversion between [(Ph2PPrPDI)MoI][I], (κ6-P,N,N,N,C,P-Ph2PPrPDI)MoI, (κ6-P,N,N,N,C,P-Ph2PPrPDI)MoH, and (Ph2PPrPDI)MoH2 is expected to guide future reactivity studies on this unique set of compounds.
Co-reporter:Chandrani Ghosh, Thomas L. Groy, Amanda C. Bowman and Ryan J. Trovitch
Chemical Communications 2016 - vol. 52(Issue 24) pp:NaN4556-4556
Publication Date(Web):2016/02/26
DOI:10.1039/C5CC09167E
Reduction of 6-coordinate (Ph2PPrDI)FeBr2 under N2 results in formation of the terminal dinitrogen complex, (Ph2PPrDI)FeN2. Heating this product to 75 °C allows for C–H and C–P activation of the chelate to generate the cisoid and transoid isomers of [(μ-PrPPh-κ5-P,N,N,Cγ,P-Ph2PPrDIPrPPh)Fe]2. Mechanistic possibilities for this transformation are discussed.
Co-reporter:Hagit Ben-Daat, Christopher L. Rock, Marco Flores, Thomas L. Groy, Amanda C. Bowman and Ryan J. Trovitch
Chemical Communications 2017 - vol. 53(Issue 53) pp:NaN7336-7336
Publication Date(Web):2017/05/05
DOI:10.1039/C7CC02281F
Addition of NaEt3BH to (Ph2PPrDI)CoCl2 affords the corresponding monohydride, (Ph2PPrDI)CoH. X-ray diffraction and DFT calculations indicate that this compound possesses a radical monoanion α-DI chelate and a Co(II) centre. Notably, (Ph2PPrDI)CoH catalyzes the hydroboration of alkynes and dihydroboration of nitriles under mild conditions.
.jpg)

















![Silane, phenyl[(1E)-2-phenylethenyl]-](http://img.cochemist.com/ccimg/168900/168842-96-4.png)
![Silane, phenyl[(1E)-2-phenylethenyl]-](http://img.cochemist.com/ccimg/168900/168842-96-4_b.png)

