Co-reporter:Min Huang
Science China Life Sciences 2017 Volume 60( Issue 1) pp:94-97
Publication Date(Web):2017 January
DOI:10.1007/s11427-016-0300-y
Co-reporter:Yang Liu, Xia Peng, Xiaocong Guan, Dong Lu, Yong Xi, Shiyu Jin, Hui Chen, Limin Zeng, Jing Ai, Meiyu Geng, Youhong Hu
European Journal of Medicinal Chemistry 2017 Volume 126(Volume 126) pp:
Publication Date(Web):27 January 2017
DOI:10.1016/j.ejmech.2016.10.003
•Novel Ponatinib analogues as selective FGFRs inhibitors were synthesized.•The SAR of these novel Ponatinib analogues were investigated.•The target selectivity for FGFR1 over KDR was improved by structural optimization.•Lead compound with both high potent enzymatic and cellular activity was achieved.•The evaluation of the lead compound 4 in vivo was reported.FGF receptors (FGFRs) are tyrosine kinases that are overexpressed in diverse tumors by genetic alterations such as gene amplifications, somatic mutations and translocations. Owing to this characteristic, FGFRs are attractive targets for cancer treatment. It has been demonstrated that most multi-targeted, ATP competitive tyrosine kinase inhibitors are active against FGFRs as well as other kinases. The design of new and more selective inhibitors of FGFRs, which might be reduced off-target and side effects, is a difficult yet significant challenge. The results of the current investigation, show that novel Ponatinib analogues are highly active as FGFR inhibitors and that they possess reduced kinase insert domain receptor (KDR) activities. Observations made in a structure and activity relationship (SAR) investigation led to the development of a promising, orally available lead compound 4, which displays a 50–100 fold in vitro selectivity for inhibition of FGFR1-3 over KDR. In addition, biological evaluation of compound 4 showed that it displays significant antitumor activities in FGFR1-amplificated H1581 and FGFR2-amplificated SNU-16 xenograft models.A series of novel Ponatinib analogues were synthesized. Observations made in a structure and activity relationship (SAR) investigation led to compound 4 as a predominant FGFR inhibitor.Download high-res image (199KB)Download full-size image
Co-reporter:Zuoquan Xie;Hong Liu
Science China Life Sciences 2017 Volume 60( Issue 6) pp:585-600
Publication Date(Web):27 May 2017
DOI:10.1007/s11427-017-9046-6
Sphingosine-1-phosphate (S1P) is a potent pleotropic bioactive lipid mediator involved in immune cell trafficking, cell survival, cell proliferation, cell migration, angiogenesis and many other cellular processes. S1P either activates S1P receptors (S1PR1-5) through “inside-out signaling” or acts directly on intracellular targets to regulate various cellular processes. In the past two decades, much progress has been made in exploring S1P signaling and its pathogenic roles in diseases as well as in developing modulators of S1P signaling, including S1P agonists, S1P antagonists and sphingosine kinase (SphK) inhibitors. Ceramide and S1P have been defined as reciprocal regulators of cell fate, and S1P signaling has been shown to be crucial for the pathogenesis of various diseases, including autoimmune diseases, inflammation and cancer; therefore, targeting S1P signaling may curtail the process of pathogenesis and serve as a potential therapeutic target for the treatment of these diseases. In this review, we describe recent advances in our understanding of S1P signaling in cancer development (particularly in inflammation- associated cancer) as well as in innate and adaptive immunity, and we also discuss modulators of S1P signaling in cancer treatment.
Co-reporter:Danni Sun, Hongchun Liu, Xiaoyang Dai, Xingling Zheng, Juan Yan, Rongrui Wei, Xuhong Fu, Min Huang, Aijun Shen, Xun Huang, Jian Ding, Meiyu Geng
Cancer Letters 2017 Volume 406(Volume 406) pp:
Publication Date(Web):10 October 2017
DOI:10.1016/j.canlet.2017.06.029
•Aspirin inhibits mTORC1 signaling in an AMPK-independent manner.•Aspirin disrupts the mTOR-raptor interaction.•mTORC1 inhibition contributed to the synergistic effect of aspirin combining with sorafenib.Aspirin is associated with a reduced risk of cancer and delayed progression of malignant disease. Adenosine 5‘-monophosphate (AMP)-activated protein kinase (AMPK)-mTOR signaling is believed to partially contribute to these anticancer effects, although the mechanism is unclear. In this study, we revealed the mechanism underlying the effects of aspirin on AMPK-mTOR signaling, and described a mechanism-based rationale for the use of aspirin in cancer therapy. We found that aspirin inhibited mTORC1 signaling through AMPK-dependent and -independent manners. Aspirin inhibited the AMPK-TSC pathway, thus resulting in the suppression of mTORC1 activity. In parallel, it directly disrupted the mTOR-raptor interaction. Additionally, the combination of aspirin and sorafenib showed synergetic effects via inhibiting mTORC1 signaling and the PI3K/AKT, MAPK/ERK pathways. Aspirin and sorafenib showed synergetic anticancer efficacy in the SMMC-7721 model. Our study provides mechanistic insights and a mechanism-based rationale for the roles of aspirin in cancer treatment.
Co-reporter:Meining Wang; Aijun Shen; Chi Zhang; Zilan Song; Jing Ai; Hongchun Liu; Liping Sun; Jian Ding; Meiyu Geng;Ao Zhang
Journal of Medicinal Chemistry 2016 Volume 59(Issue 12) pp:5563-5586
Publication Date(Web):February 4, 2016
DOI:10.1021/acs.jmedchem.5b01106
Heat shock protein 90 (Hsp90) is a ubiquitous chaperone of all of the oncogenic tyrosine kinases. Many Hsp90 inhibitors, alone or in combination, have shown significant antitumor efficacy against the kinase-positive naïve and mutant models. However, clinical trials of these inhibitors are unsuccessful due to insufficient clinical benefits and nonoptimal safety profiles. Recently, much progress has been reported on the Hsp90–cochaperone–client complex, which will undoubtedly assist in the understanding of the interactions between Hsp90 and its clients. Meanwhile, Hsp90 inhibitors have shown promise against patients’ resistance caused by early generation tyrosine kinase inhibitors (TKIs), and at least 13 Hsp90 inhibitors are being reevaluated in the clinic. In this regard, the objectives of the current perspective are to summarize the structure and function of the Hsp90–cochaperone–client complex, to analyze the structural and functional insights into the Hsp90–client interactions to address several existing unresolved problems with Hsp90 inhibitors, and to highlight the preclinical and clinical studies of Hsp90 inhibitors as an effective treatment against resistance to tyrosine kinase inhibitors.
Co-reporter:Wei Yan; Xinyi Wang; Yang Dai; Bin Zhao; Xinying Yang; Jun Fan; Yinglei Gao; Fanwang Meng; Yuming Wang; Cheng Luo; Jing Ai; Meiyu Geng;Wenhu Duan
Journal of Medicinal Chemistry 2016 Volume 59(Issue 14) pp:6690-6708
Publication Date(Web):June 27, 2016
DOI:10.1021/acs.jmedchem.6b00056
Fibroblast growth factor receptor (FGFR) represents an attractive oncology target for cancer therapy in view of its critical role in promoting cancer formation and progression, as well as causing resistance to approved therapies. In this article, we describe the identification of the potent pan-FGFR inhibitor (R)-21c (FGFR1–4 IC50 values of 0.9, 2.0, 2.0, and 6.1 nM, respectively). Compound (R)-21c exhibited excellent in vitro inhibitory activity against a panel of FGFR-amplified cell lines. Western blot analysis demonstrated that (R)-21c suppressed FGF/FGFR and downstream signaling pathways at nanomolar concentrations. Moreover, (R)-21c provided nearly complete inhibition of tumor growth (96.9% TGI) in NCI-H1581 (FGFR1-amplified) xenograft mice model at the dose of 10 mg/kg/qd via oral administration.
Co-reporter:Yang Liu, Shiyu Jin, Xia Peng, Dong Lu, Limin Zeng, Yiming Sun, Jing Ai, Meiyu Geng, Youhong Hu
European Journal of Medicinal Chemistry 2016 Volume 108() pp:322-333
Publication Date(Web):27 January 2016
DOI:10.1016/j.ejmech.2015.11.042
•Pyridazinone derivatives as novel c-Met inhibitors were designed and synthesized via a cyclization strategy.•Lead compound 19 with both potent enzyme and cellular activity was achieved.•Docking study of lead 19 was reported.•In vitro DMPK profiles and in vivo antitumor activity of lead 19 were evaluated.Over activation of c-Met tyrosine kinase is known to promote tumorigenesis and metastasis, as well as to cause therapeutic resistance. Herein we describe the design, synthesis and biological activities of novel, ATP-competitive, c-Met tyrosine kinase inhibitors that are members of the 6-aryl-2-(3-(heteroarylamino)benzyl)pyridazinone family. A structure-activity relationship (SAR) study of these substances led to identification of pyridazinone 19 as a highly selective and potent c-Met tyrosine inhibitor, which displays favorable pharmacokinetic properties in mice and significant antitumor activity against a c-Met driven EBC-1 tumor xenograft.Pyridazinone derivatives as novel c-Met inhibitors were designed and synthesized via a cyclization strategy. 19 was found with favorable pharmacokinetic properties and significant antitumor activity.
Co-reporter:Zhengsheng Zhan, Xia Peng, Qiufeng Liu, Fang Chen, Yinchun Ji, Shanyan Yao, Yong Xi, Yipeng Lin, Tiantian Chen, Yechun Xu, Jing Ai, Meiyu Geng, Wenhu Duan
European Journal of Medicinal Chemistry 2016 Volume 116() pp:239-251
Publication Date(Web):30 June 2016
DOI:10.1016/j.ejmech.2016.03.076
•A series of CH2-/CF2-linked triazolotriazines as c-Met inhibitors were reported.•The compounds were assayed c-Met activities in both enzymatic and cellular level.•Based on good antitumor potency and PK profiles, 23 was selected as a drug candidate.c-Met/HGF overexpression has been detected in many human malignancies including tumors which are resistant to anticancer therapy. Disrupting the aberrant c-Met/HGF axis has enjoyed significant progress in both preclinical and clinical antitumor campaign. To eliminate the OCH2-related metabolic deficiency of our previously reported triazolotriazine 2, we synthesized a series of CH2-/CF2-linked triazolotriazines and assessed their c-Met activities, leading to the highly potent compound 23 with IC50 values of 0.24 nM of enzymatic activity in c-Met and 0.85 nM of cellular activity in EBC-1 cancer cell line, as well as with complete tumor regression in EBC-1 xenograft mice model at dose of 25 mg/kg via oral administration. Based on its potent anti-proliferative activities and favorable pharmacokinetic properties, 23 has been selected as a drug candidate for preclinical investigation.
Co-reporter:Liandi Chi; Delin Wu; Zhuo Li; Minmin Zhang; Hongchun Liu; Caifen Wang; Shuangying Gui; Meiyu Geng; Haiyan Li;Jiwen Zhang
Molecular Pharmaceutics 2016 Volume 13(Issue 1) pp:113-124
Publication Date(Web):November 16, 2015
DOI:10.1021/acs.molpharmaceut.5b00566
In answering to the challenge of enzymatic unstability of Biopharmaceutics Classification System (BCS) class II drugs, an effective remote loading strategy was developed to successfully incorporate the drug–cyclodextrin (CD) complex into niosomes to modify the release and stability of a drug candidate, pseudolaric acid B (PAB). Judged by binding constants, and combined solubilization effects of pH and CD complexation on PAB at different pH, the complex internalization driven by a transmembrane pH gradient (from 2.0 to 7.4) and the dynamic shifting of PAB–CD complexation equilibrium at this gradient were introduced. The transfer of PAB–CD complex into the internal aqueous phase of niosomes at 60 °C was primarily verified by synchrotron radiation Fourier transform infrared spectroscopy. The remote loading samples behaved as retarded release at pH 5.8, 6.8, and 7.4, for which the stability of PAB in rat plasma was significantly enhanced (about 8.1-fold), in comparison with niosomes prepared by the passive and lipid bilayer loading of PAB. The drug–carrier interaction based release modeling was further fitted, and the convection rate constant (ks) and free energy difference between free and bound states (ΔG) indicated the strongest PAB–carrier interactions in remote loading niosomes. The remote loading strategy also reduced the CD–cholesterol interaction and provided better physical stability of the system. In conclusion, the remote loading of drug–CD complex into niosomes provides advantages to modify the release and enhance the stability of unstable BCS class II drug.
Co-reporter:Zilan Song, Zongjun Xia, Yinchun Ji, Li Xing, Yinglei Gao, Jing Ai, Meiyu Geng, Ao Zhang
European Journal of Medicinal Chemistry 2016 Volume 118() pp:244-249
Publication Date(Web):8 August 2016
DOI:10.1016/j.ejmech.2016.04.046
•Our early SAR has identified 6 as a high potency orally bioavailable ALK inhibitor.•Further lead profiling disclosed that 6 is also a highly potent RET inhibitor.•6 is active against both ALK resistant and hot spot-activating mutants.•Tumor stasis and partial tumor regression were achieved with 6 in xenograft models.•6 is now being profiled further in our preclinical settings as a new ALK inhibitor.Our early structure–activity relationship study has identified benzo[b]carbazolone 6 as a high potency orally bioavailable ALK inhibitor. Further lead profiling disclosed that 6 is active against both ALK resistant and hot spot-activating mutants, and is also highly potent against RET kinase. Tumor stasis and partial tumor regression were achieved with 6 in both NIH/3T3-EML4-ALK and NIH/3T3-EML4-ALK L1196M xenograft models. Based on the optimal in vitro and in vivo antitumor efficacy, compound 6 is now being profiled further in our preclinical settings as a new orally available ALK/RET dual inhibitor.
Co-reporter:Dong Lu, Aijun Shen, Yang Liu, Xia Peng, Weiqiang Xing, Jing Ai, Meiyu Geng, Youhong Hu
European Journal of Medicinal Chemistry 2016 Volume 115() pp:191-200
Publication Date(Web):10 June 2016
DOI:10.1016/j.ejmech.2016.03.027
•Docking studies with 1 and 7a with c-Met protein were carried out.•A molecular hybridization strategy was employed to design c-Met inhibitors.•Novel benzo[d]oxazol-2(3H)-one-quinolone derivatives were synthesized.•13 displayed excellent inhibition against c-Met kinase and the EBC-1 cell line.Analysis of the results of studies of docking 1 and 7a with c-Met kinase led to the identification of benzo[d]oxazol-2(3H)-one-quinolone derivatives as potential inhibitors of this enzyme. A molecular hybrid strategy, using a 4-ethoxy-7-substituted-quinoline core and a benzo[d]oxazol-2(3H)-one scaffold, was employed to design members of this family for study as inhibitors of the kinase and proliferation of EBC-1 cells. Most of the substances were found to display good to excellent c-Met kinase inhibitory activities. The results of a structure–activity relationship (SAR) study led to the discovery of benzo[d]oxazol-2(3H)-one-quinolone 13, which has IC50 values of 1 nM against c-Met kinase and 5 nM against proliferation of the EBC-1 cell line.Members of a series of benzo[d]oxazol-2(3H)-one-quinolones were designed to serve as c-Met kinase inhibitors. The results of a SAR studies identified 13 as a highly potent inhibitor of this kinase.
Co-reporter:Bin Zhao, Yixuan Li, Pan Xu, Yang Dai, Cheng Luo, Yiming Sun, Jing Ai, Meiyu Geng, and Wenhu Duan
ACS Medicinal Chemistry Letters 2016 Volume 7(Issue 6) pp:629
Publication Date(Web):April 20, 2016
DOI:10.1021/acsmedchemlett.6b00066
Fibroblast growth factor receptors (FGFRs) are important targets for cancer therapy. Herein, we describe the design, synthesis, and biological evaluation of a novel series of 1H-pyrazolo[3,4-b]pyridine derivatives as potent and selective FGFR kinase inhibitors. On the basis of its excellent in vitro potency and favorable pharmacokinetic properties, compound 7n was selected for in vivo evaluation and showed significant antitumor activity in a FGFR1-driven H1581 xenograft model. These results indicated that 7n would be a promising candidate for further drug development.Keywords: Cancer; FGFR; inhibitor; pyrazolo[3,4-b]pyridine
Co-reporter:Mei Han, Chengyan Wang, Yinchun Ji, Zilan Song, Li Xing, Yi Su, Xisheng Wang, Ao Zhang, Jing Ai, Meiyu Geng
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 22) pp:5399-5402
Publication Date(Web):15 November 2016
DOI:10.1016/j.bmcl.2016.10.039
A metabolism-based fine-tuning structure-optimization was conducted to address the oxidative metabolism and hERG blockade of our early ALK inhibitor. Compound 8 was identified showing high potency against both ALK wild type and gatekeeper mutant. In addition to the optimal PK properties and significant cell antiproliferative effects, 8 showed complete tumor growth inhibition at doses of 50 or 10 mg/kg once daily in the Karpas299 xenograft model. All these results encouraged the further development of 8 as a potent and orally bioavailable ALK inhibitor.
Co-reporter:Hong Cui, Xia Peng, Jian Liu, Chunhua Ma, Yinchun Ji, Wei Zhang, Meiyu Geng, Yingxia Li
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 18) pp:4483-4486
Publication Date(Web):15 September 2016
DOI:10.1016/j.bmcl.2016.07.077
A series of 2-oxo-1,2-dihydroquinoline-containing c-Met inhibitors were designed, synthesized and evaluated for their in vitro activities targeting c-Met. Most compounds showed high potency against c-Met with IC50 values in the single-digit nM range. Among these compounds, two target compounds, namely 1h and 1n, stood out as the most potent c-Met inhibitors with IC50s of 0.6 and 0.7 nM, respectively. And 1a exhibited higher potency than BMS-777607 did with respect to the inhibition of cell proliferation. The introduction of electron-donating substituent was favorable for the activities of the compounds to some extent. Furthermore, molecular docking studies also gave encouraging results that supported this work.
Co-reporter:Jun Fan, Yang Dai, Jingwei Shao, Xia Peng, Chen Wang, Sufen Cao, Bin Zhao, Jing Ai, Meiyu Geng, Wenhu Duan
Bioorganic & Medicinal Chemistry Letters 2016 26(11) pp: 2594-2599
Publication Date(Web):1 June 2016
DOI:10.1016/j.bmcl.2016.04.028
Fibroblast growth factor receptors (FGFRs) are important oncology targets due to the dysregulation of this signaling pathway in a wide variety of human cancers. We identified a series of pyrazolylaminoquinazoline derivatives as potent FGFR inhibitors with low nanomolar potency. The representative compound 29 strongly inhibited FGFR1–3 kinase activity and suppressed FGFR signaling transduction in FGFR-addicted cancer cells; FGFRs-driven cell proliferation was also strongly inhibited regardless of mechanistic complexity implicated in FGFR activation, which further confirmed that 29 was a potent pan-FGFR inhibitor. The flexibility of our structure offered the potential to preserve good affinity for mutant FGFR, which is important for developing TKIs with long-term efficacy.
Co-reporter:Yuchi Ma; Guangqiang Sun; Danqi Chen; Xia Peng; Yue-Lei Chen; Yi Su; Yinchun Ji; Jin Liang; Xin Wang; Lin Chen; Jian Ding; Bing Xiong; Jing Ai; Meiyu Geng;Jingkang Shen
Journal of Medicinal Chemistry 2015 Volume 58(Issue 5) pp:2513-2529
Publication Date(Web):February 10, 2015
DOI:10.1021/jm502018y
c-Met has emerged as an attractive target for targeted cancer therapy because of its abnormal activation in many cancer cells. To identify high potent and selective c-Met inhibitors, we started with profiling the potency and in vitro metabolic stability of a reported hit 7. By rational design, a novel sulfonylpyrazolo[4,3-b]pyridine 9 with improved DMPK properties was discovered. Further elaboration of π–π stacking interactions and solvent accessible polar moieties led to a series of highly potent and selective type I c-Met inhibitors. On the basis of in vitro and in vivo pharmacological and pharmacokinetics studies, compound 46 was selected as a preclinical candidate for further anticancer drug development.
Co-reporter:Zilan Song; Yanhong Yang; Zhiqing Liu; Xia Peng; Junfeng Guo; Xinying Yang; Kui Wu; Jing Ai; Jian Ding; Meiyu Geng;Ao Zhang
Journal of Medicinal Chemistry 2015 Volume 58(Issue 1) pp:197-211
Publication Date(Web):April 30, 2014
DOI:10.1021/jm5005144
We have developed a series of new 2,4-diarylaminopyrimidine analogues (DAAPalogues) bearing a flexible amino acid side chain, different from the majority of the literature reported ALK inhibitors that often possess a structurally constrained arylpiperazine fragment or its equivalents in the solvent-interaction region. Extensive structural elaboration led to compound 15 possessing IC50 values of 2.7 and 15.3 nM, respectively, in the ALK wild-type and gate-keeper mutant L1196M enzymatic assays. This compound not only showed high proliferative inhibition against ALK-addicted cells across different oncogenic forms but also effectively suppressed several ALK secondary mutant cells, including the gate-keeper L1196M and F1174L. Significant antitumor efficacy was achieved in the ALK-driven SUP-M2 xenograft model.
Co-reporter:Xiaolong Jiang, Ji Zhou, Jing Ai, Zilan Song, Xia Peng, Li Xing, Yong Xi, Junfeng Guo, Qizheng Yao, Jian Ding, Meiyu Geng, Ao Zhang
European Journal of Medicinal Chemistry 2015 Volume 105() pp:39-56
Publication Date(Web):13 November 2015
DOI:10.1016/j.ejmech.2015.10.005
•New benzo[b]carbazolone ALK inhibitors were designed, synthesized and evaluated.•New compounds possess more rotatable bonds and higher molecular flexibility.•Extensive structure—activity, anti-resistance, and pharmacokinetics study was conducted.•15b showed favorable PK profile and better efficacy in vivo than Crizotinib.•15b was identified as a potent second-generation benzo[b]carbazolone-bearing ALK inhibitor.Four series of tetracyclic benzo[b]carbazolone compounds possessing more rotatable bonds and higher molecular flexibility were designed by either inserting a linker within the C8-side chain or by opening the middle ketone ring on the basis of compound 5 (Alectinib, CH5424802). Compound 15b was identified showing nearly identical high potency against both wild-type and the gatekeeper mutant ALK kinase (3.4 vs 3.9 nM). This compound has favorable PK profile with an oral bioavailability of 67.1% in rats. Moreover, compound 15b showed significant growth inhibition against ALK driven cancer cells and KARPAS-299 xenograft model.
Co-reporter:Chunpu Li, Jing Ai, Dengyou Zhang, Xia Peng, Xi Chen, Zhiwei Gao, Yi Su, Wei Zhu, Yinchun Ji, Xiaoyan Chen, Meiyu Geng, and Hong Liu
ACS Medicinal Chemistry Letters 2015 Volume 6(Issue 5) pp:507
Publication Date(Web):March 2, 2015
DOI:10.1021/ml5004876
A series of imidazo[1,2-a]pyridine derivatives against c-Met was designed by means of bioisosteric replacement. In this study, a selective, potent c-Met inhibitor, 22e was identified, with IC50 values of 3.9 nM against c-Met kinase and 45.0 nM against c-Met-addicted EBC-1 cell proliferation, respectively. Compound 22e inhibited c-Met phosphorylation and downstream signaling across different oncogenic forms in c-Met overactivated cancer cells and model cells. Compound 22e significantly inhibited tumor growth (TGI = 75%) with good oral bioavailability (F = 29%) and no significant hERG inhibition. On the basis of systematic metabolic study, the pathway of all possible metabolites of 22e in liver microsomes of different species has been proposed, and a major NADPH-dependent metabolite 33 was generated by liver microsomes. To block the metabolic site, 42 was designed and synthesized for further evaluation. Taken together, the imidazo[1,2-a]pyridine scaffold showed promising pharmacological inhibition of c-Met and warrants further investigation.Keywords: c-Met inhibitor; imidazo[1,2-a]pyridine; metabolic stability; Receptor tyrosine kinase
Co-reporter:Xiao-De An, Hongyan Liu, Zhong-Liang Xu, Yi Jin, Xia Peng, Ying-Ming Yao, Meiyu Geng, Ya-Qiu Long
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 3) pp:708-716
Publication Date(Web):1 February 2015
DOI:10.1016/j.bmcl.2014.11.070
Starting from our previously identified novel c-Met kinase inhibitors bearing 1H-imidazo[4,5-h][1,6]naphthyridin-2(3H)-one scaffold, a global structural exploration was conducted to furnish an optimal binding motif for further development, directed by the enzyme inhibitory mechanism. First round SAR study picked two imidazonaphthyridinone frameworks with 1,8- and 3,5-disubstitution pattern as class I and class II c-Met kinase inhibitors, respectively. Further structural optimization on type II inhibitors by truncation of the imidazonaphthyridinone core and incorporation of an N-phenyl cyclopropane-1,1-dicarboxamide pharmacophore led to the discovery of novel imidazopyridine-based c-Met kinase inhibitors, displaying nanomolar enzyme inhibitory activity and improved Met kinase selectivity. More significantly, the new chemotype c-Met kinase inhibitors effectively inhibited Met phosphorylation and its downstream signaling as well as the proliferation of Met-dependent EBC-1 human lung cancer cells at submicromolar concentrations.A new class of imidazopyridine-based c-Met kinase inhibitors exhibited nanomolar inhibition against the enzyme and the growth of Met-driven EBC-1 human lung cancer cells.
Co-reporter:Weiqiang Xing, Jing Ai, Shiyu Jin, Zhangxing Shi, Xia Peng, Lang Wang, Yinchun Ji, Dong Lu, Yang Liu, Meiyu Geng, Youhong Hu
European Journal of Medicinal Chemistry 2015 95() pp: 302-312
Publication Date(Web):
DOI:10.1016/j.ejmech.2015.03.041
Co-reporter:Tao Meng ; Dadong Zhang ; Zuoquan Xie ; Ting Yu ; Shuchao Wu ; Lorenza Wyder ; Urs Regenass ; Kurt Hilpert ; Min Huang ; Meiyu Geng ;Jingkang Shen
Journal of Medicinal Chemistry 2014 Volume 57(Issue 23) pp:9832-9843
Publication Date(Web):November 10, 2014
DOI:10.1021/jm5010144
Upregulation of pyruvate dehydrogenase kinase (PDHK) has been observed in a variety of cancers. Inhibition of PDHK offers an attractive opportunity for the development of novel cancer therapies. To obtain novel PDHK inhibitors, we took advantage of the homology of the ATP-binding pocket between Heat Shock Protein 90 (HSP90) and PDHK, and utilized 4,5-diarylisoxazole based HSP90 inhibitor for structural design. Our efforts led to the identification of 5k that inhibited PDHK1 with an IC50 value of 17 nM, which, however, showed marginal cellular activity. Further structural optimization resulted in compound 11a with improved cellular activity which could effectively modulate the metabolic profile of cancer cells and lead to the inhibition of cancer cell proliferation, evidenced by the increased oxidative phosphorylation and decreased glycolysis and associated oxidative stress. Our results suggested 11a as an excellent lead compound and a favorable biological tool to further evaluate the therapeutic potential of PDHK and HSP90 dual inhibitors in the treatment of cancer.
Co-reporter:Zhiqing Liu, Xihua Yue, Zilan Song, Xia Peng, Junfeng Guo, Yinchun Ji, Zhen Cheng, Jian Ding, Jing Ai, Meiyu Geng, Ao Zhang
European Journal of Medicinal Chemistry 2014 Volume 86() pp:438-448
Publication Date(Web):30 October 2014
DOI:10.1016/j.ejmech.2014.09.003
•ALK has been recognized as an emergent therapeutic target for cancer treatment.•New 2,4-diarylaminopyrimidines bearing an 2-aminothiazole motif was developed.•Compound 5i showed highest potency of 12.4 nM against ALK.•Compound 5i also showed 24.1 nM against ALK gatekeeper mutation L1196M.•5i dose-dependently inhibited phosphorylation of ALK and its down-stream pathways.A series of new 2,4-diarylaminopyrimidine analogues (DAAPalogues) was developed by incorporation of a substituted 2-aminothiazole component as the C-2 substituent of the center pyrimidine core. Compound 5i showed highest potency of 12.4 nM against ALK and 24.1 nM against ALK gatekeeper mutation L1196M. Although only having moderate cellular potency in the SUP-M2 cells harboring NPM-ALK, compound 5i showed good kinase selectivity and dose-dependently inhibited phosphorylation of ALK and its down-stream signaling pathways.
Co-reporter:Danqi Chen, Aijun Shen, Jian Li, Feng Shi, Wuyan Chen, Jing Ren, Hongchun Liu, Yechun Xu, Xin Wang, Xinying Yang, Yiming Sun, Min Yang, Jianhua He, Yueqin Wang, Liping Zhang, Min Huang, Meiyu Geng, Bing Xiong, Jingkang Shen
European Journal of Medicinal Chemistry 2014 Volume 87() pp:765-781
Publication Date(Web):24 November 2014
DOI:10.1016/j.ejmech.2014.09.065
•A new scaffold of HSP90 inhibitors was developed with aids of fragment screening.•Comprehensive assessment was taken.•Compound 108 exhibited excellent in vivo activities and pharmacokinetic properties.HSP90 is ubiquitously overexpressed in a broad spectrum of human cancers and has been recognized as an attractive target for cancer treatment. Here, we described the fragment screening, synthesis and structure-activity relationship studies of small molecule inhibitors with 4,5-diarylisoxazole scaffold targeting HSP90. Among them, the compound N-(3-(2,4-dihydroxy-5-isopropylphenyl)-4-(4-((4-morpholinopiperidin-1-yl)methyl)phenyl)isoxazol-5-yl)cyclopropanecarboxamide (108) showed high affinity for binding to HSP90 (FP binding assay, IC50 = 0.030 μM) and inhibited the proliferation of various human cancer cell lines with averaging GI50 about 88 nM. Compound 108 exhibited its functional inhibition of HSP90 by depleting key signaling pathways and concomitantly elevating of HSP70 and HSP27 in U-87MG cells. Further in vivo studies showed that compound 108 strongly suppressed the tumor growth of human glioblastoma xenograft model U-87MG with T/C = 18.35% at 50 mg/kg q3w/2.5w. Moreover, compound 108 also exhibited good pharmacokinetic properties. Together, our study implicates that compound 108 is a promising candidate of HSP90 inhibitor and is currently advanced to preclinical study.By fragment screening and x-ray crystallography techniques a scaffold of 4,5-diarylisoxazole was selected and developed to a novel series of HSP90 inhibitors with excellent in vivo activities.
Co-reporter:Jimin Xu, Jing Ai, Sheng Liu, Xia Peng, Linqian Yu, Meiyu Geng and Fajun Nan
Organic & Biomolecular Chemistry 2014 vol. 12(Issue 22) pp:3721-3734
Publication Date(Web):03 Apr 2014
DOI:10.1039/C4OB00364K
A library of biscoumarin-based c-Met inhibitors was synthesized, based on optimization of 3,3′-biscoumarin hit 3, which was identified as a non-ATP competitive inhibitor of c-Met from a diverse library of coumarin derivatives. Among these compounds, 38 and 40 not only showed potent enzyme activities with IC50 values of 107 nM and 30 nM, respectively, but also inhibited c-Met phosphorylation in BaF3/TPR-Met and EBC-1 cells.
Co-reporter:Zhiqing Liu, Jing Ai, Xia Peng, Zilan Song, Kui Wu, Jing Zhang, Qizheng Yao, Yi Chen, Yinchun Ji, Yanhong Yang, Meiyu Geng, and Ao Zhang
ACS Medicinal Chemistry Letters 2014 Volume 5(Issue 4) pp:304-308
Publication Date(Web):February 8, 2014
DOI:10.1021/ml400373j
By repurposing a typical dopamine D1/D5 receptor agonist motif, C1-substituted-N3-benzazepine or benzazecine, into the classical RTK inhibitor 2,4-diaminopyrimidine skeleton, a series of new 2,4-diarylaminopyrimidine analogues (DAAPalogues) were developed. Compounds 7 and 8a were identified possessing high potency against both c-Met and ALK kinases. Compound 8a displayed appreciable antitumor efficacy at the dose of 1 mg/kg in the ALK-driven BF3/EML4-ALK xenograft mice model.Keywords: 2,4-diarylaminopyrimidine analogues; c-Met/ALK dual inhibitor; C1-Substituted-N3-benzazepine; structure repurposing;
Co-reporter:Zhengsheng Zhan, Jing Ai, Qiufeng Liu, Yinchun Ji, Tiantian Chen, Yechun Xu, Meiyu Geng, and Wenhu Duan
ACS Medicinal Chemistry Letters 2014 Volume 5(Issue 6) pp:673
Publication Date(Web):March 26, 2014
DOI:10.1021/ml500066m
Both c-Met and VEGFR-2 are important targets for cancer therapies. Here we report a series of potent dual c-Met and VEGFR-2 inhibitors bearing an anilinopyrimidine scaffold. Two novel synthetic protocols were employed for rapid analoguing of the designed molecules for structure–activity relationship (SAR) exploration. Some analogues displayed nanomolar potency against c-Met and VEGFR-2 at enzymatic level. Privileged compounds 3a, 3b, 3g, 3h, and 18a exhibited potent antiproliferative effect against c-Met addictive cell lines with IC50 values ranged from 0.33 to 1.7 μM. In addition, a cocrystal structure of c-Met in complex with 3h has been determined, which reveals the binding mode of c-Met to its inhibitor and helps to interpret the SAR of the analogues.Keywords: Anilinopyrimidine; c-Met; dual inhibitor; SAR; VEGFR-2
Co-reporter:Jing Ren, Jian Li, Yueqin Wang, Wuyan Chen, Aijun Shen, Hongchun Liu, Danqi Chen, Danyan Cao, Yanlian Li, Naixia Zhang, Yechun Xu, Meiyu Geng, Jianhua He, Bing Xiong, Jingkang Shen
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 11) pp:2525-2529
Publication Date(Web):1 June 2014
DOI:10.1016/j.bmcl.2014.03.100
Heat shock protein 90 (HSP90) is a molecular chaperone to fold and maintain the proper conformation of many signaling proteins, especially some oncogenic proteins and mutated unstable proteins. Inhibition of HSP90 was recognized as an effective approach to simultaneously suppress several aberrant signaling pathways, and therefore it was considered as a novel target for cancer therapy. Here, by integrating several techniques including the fragment-based drug discovery method, fragment merging, computer aided inhibitor optimization, and structure-based drug design, we were able to identify a series of HSP90 inhibitors. Among them, inhibitors 13, 32, 36 and 40 can inhibit HSP90 with IC50 about 20–40 nM, which is at least 200-fold more potent than initial fragments in the protein binding assay. These new HSP90 inhibitors not only explore interactions with an under-studied subpocket, also offer new chemotypes for the development of novel HSP90 inhibitors as anticancer drugs.
Co-reporter:Danqi Chen, Aijun Shen, Guanghua Fang, Hongchun Liu, Minmin Zhang, Shuai Tang, Bing Xiong, Lanping Ma, Meiyu Geng, Jingkang Shen
Acta Pharmaceutica Sinica B (January 2016) Volume 6(Issue 1) pp:93-99
Publication Date(Web):January 2016
DOI:10.1016/j.apsb.2015.11.002
Co-reporter:Min Huang, Aijun Shen, Jian Ding, Meiyu Geng
Trends in Pharmacological Sciences (January 2014) Volume 35(Issue 1) pp:41-50
Publication Date(Web):1 January 2014
DOI:10.1016/j.tips.2013.11.004
•Tumor heterogeneity is the key factor underlying limited response rate to targeted therapy.•Personalized medicines depend on biomarkers for selecting patients and directing therapy.•Co-development of predictive and response biomarkers is required for drug development.•PDX model-based trials mimicking human patients may improve biomarker discovery.The tremendous advances achieved in the understanding of cancer biology have delivered unprecedented progress in molecularly targeted cancer therapy in the past decade. The fast growing category of targeted anticancer agents available for clinical use is accompanied by a conceptual revolution in anticancer drug development. Nevertheless, molecularly targeted cancer therapy remains challenged by a high failure rate and an extremely small proportion of patients that can benefit. It is pivotal to take lessons from the past and seek new solutions. This review discusses conceptual progress and remaining challenges in molecularly targeted cancer therapy, and proposes feasible alternatives to increase chances of clinical success in the future.
Co-reporter:Mei Han, Shan Li, Jing Ai, Rong Sheng, Yongzhou Hu, Youhong Hu, Meiyu Geng
Bioorganic & Medicinal Chemistry Letters (1 December 2016) Volume 26(Issue 23) pp:
Publication Date(Web):1 December 2016
DOI:10.1016/j.bmcl.2016.10.061
A series of novel 4-chloro-benzamides derivatives containing substituted five-membered heteroaryl ring were designed, synthesized and evaluated as RET kinase inhibitors for cancer therapy. Most of compounds exhibited moderate to high potency in ELISA-based kinase assay. In particular, compound I-8 containing 1,2,4-oxadiazole strongly inhibited RET kinase activity both in molecular and cellular level. In turn, I-8 inhibited cell proliferation driven by RET wildtype and gatekeeper mutation. The results implied that 4-chloro-3-(5-(pyridin-3-yl)-1,2,4-oxadiazole-3-yl)benzamides are promising lead compounds as novel RET kinase inhibitor for further investigation.
Co-reporter:Mei Han, Shan Li, Jing Ai, Rong Sheng, Yongzhou Hu, Youhong Hu, Meiyu Geng
Bioorganic & Medicinal Chemistry Letters (1 December 2016) Volume 26(Issue 23) pp:5679-5684
Publication Date(Web):1 December 2016
DOI:10.1016/j.bmcl.2016.10.061
A series of novel 4-chloro-benzamides derivatives containing substituted five-membered heteroaryl ring were designed, synthesized and evaluated as RET kinase inhibitors for cancer therapy. Most of compounds exhibited moderate to high potency in ELISA-based kinase assay. In particular, compound I-8 containing 1,2,4-oxadiazole strongly inhibited RET kinase activity both in molecular and cellular level. In turn, I-8 inhibited cell proliferation driven by RET wildtype and gatekeeper mutation. The results implied that 4-chloro-3-(5-(pyridin-3-yl)-1,2,4-oxadiazole-3-yl)benzamides are promising lead compounds as novel RET kinase inhibitor for further investigation.
Co-reporter:Jimin Xu, Jing Ai, Sheng Liu, Xia Peng, Linqian Yu, Meiyu Geng and Fajun Nan
Organic & Biomolecular Chemistry 2014 - vol. 12(Issue 22) pp:NaN3734-3734
Publication Date(Web):2014/04/03
DOI:10.1039/C4OB00364K
A library of biscoumarin-based c-Met inhibitors was synthesized, based on optimization of 3,3′-biscoumarin hit 3, which was identified as a non-ATP competitive inhibitor of c-Met from a diverse library of coumarin derivatives. Among these compounds, 38 and 40 not only showed potent enzyme activities with IC50 values of 107 nM and 30 nM, respectively, but also inhibited c-Met phosphorylation in BaF3/TPR-Met and EBC-1 cells.

![(2S,5R,6R)-3,3-DIMETHYL-7-OXO-6-[[(2R)-2-PHENYL-2-[[2-[4-(1,4,5,6-TETRAHYDROPYRIMIDIN-2-YL)PHENYL]ACETYL]AMINO]ACETYL]AMINO]-4-THIA-1-AZABICYCLO[3.2.0]HEPTANE-2-CARBOXYLIC ACID](/data/chemimg/415200/1265848-98-3.png)
![(2S,5R,6R)-3,3-DIMETHYL-7-OXO-6-[[(2R)-2-PHENYL-2-[[2-[4-(1,4,5,6-TETRAHYDROPYRIMIDIN-2-YL)PHENYL]ACETYL]AMINO]ACETYL]AMINO]-4-THIA-1-AZABICYCLO[3.2.0]HEPTANE-2-CARBOXYLIC ACID](/data/chemimg/415200/1265848-98-3_b.png)
![9-ethyl-8-iodo-6,6-dimethyl-11-oxo-6,11-dihydro-5h-benzo[b]carbaz Ole-3-carbonitrile](http://img.cochemist.com/ccimg/1256600/1256584-80-1.png)
![9-ethyl-8-iodo-6,6-dimethyl-11-oxo-6,11-dihydro-5h-benzo[b]carbaz Ole-3-carbonitrile](http://img.cochemist.com/ccimg/1256600/1256584-80-1_b.png)






![(2S)-2-[(4-METHOXYPHENYL)SULFANYLCARBONYLAMINO]PENTANEDIOIC ACID](http://img.cochemist.com/ccimg/1256600/1256584-75-4.png)
![(2S)-2-[(4-METHOXYPHENYL)SULFANYLCARBONYLAMINO]PENTANEDIOIC ACID](http://img.cochemist.com/ccimg/1256600/1256584-75-4_b.png)






![Benzamide, 2-fluoro-N-methyl-4-[7-(6-quinolinylmethyl)imidazo[1,2-b][1,2,4]triazin-2-yl]-](/data/chemimg/315100/1029712-80-8.png)
![Benzamide, 2-fluoro-N-methyl-4-[7-(6-quinolinylmethyl)imidazo[1,2-b][1,2,4]triazin-2-yl]-](/data/chemimg/315100/1029712-80-8_b.png)




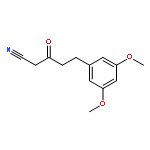
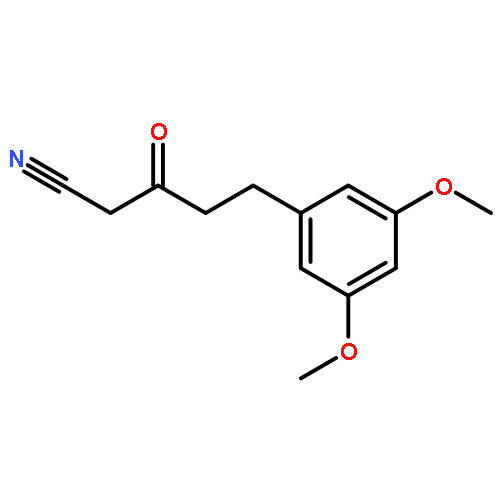
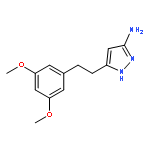
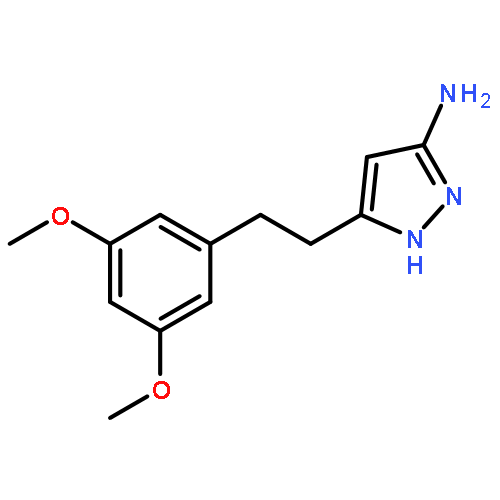
![Thieno[3,2-d]pyrimidine, 2-(1H-indazol-4-yl)-6-[[4-(methylsulfonyl)-1-piperazinyl]methyl]-4-(4-morpholinyl)-](http://img.cochemist.com/ccimg/957100/957054-30-7.png)
![Thieno[3,2-d]pyrimidine, 2-(1H-indazol-4-yl)-6-[[4-(methylsulfonyl)-1-piperazinyl]methyl]-4-(4-morpholinyl)-](http://img.cochemist.com/ccimg/957100/957054-30-7_b.png)
![6-Methoxyimidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/955400/955376-51-9.png)
![6-Methoxyimidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/955400/955376-51-9_b.png)



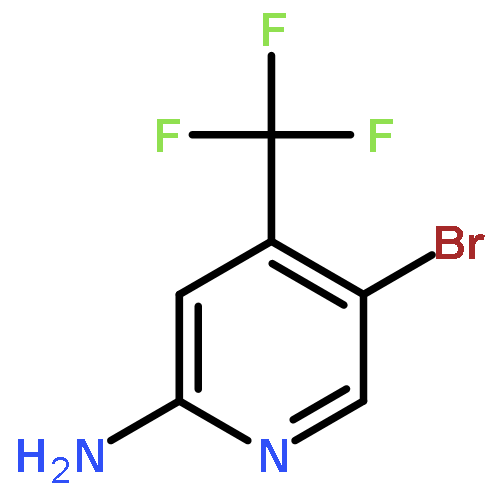
![3-iodo-4-methyl-n-{4-[(4-methyl-1-piperazinyl)methyl]-3-(trifluor Omethyl)phenyl}benzamide](http://img.cochemist.com/ccimg/943400/943320-50-1.png)
![3-iodo-4-methyl-n-{4-[(4-methyl-1-piperazinyl)methyl]-3-(trifluor Omethyl)phenyl}benzamide](http://img.cochemist.com/ccimg/943400/943320-50-1_b.png)
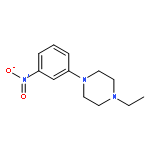
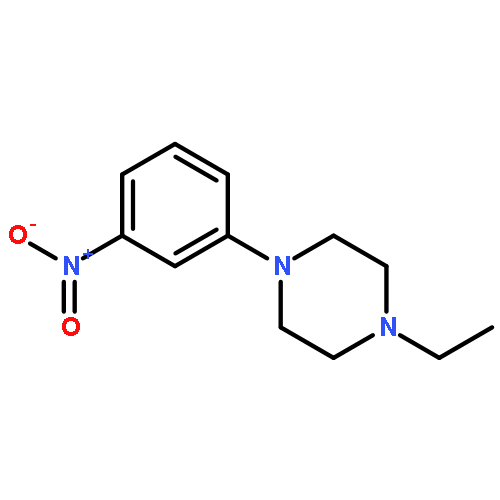
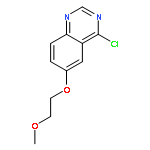
![Morpholine, 4-[6-[2-(trimethylsilyl)ethynyl]-3-pyridinyl]-](http://img.cochemist.com/ccimg/926100/926009-41-8.png)
![Morpholine, 4-[6-[2-(trimethylsilyl)ethynyl]-3-pyridinyl]-](http://img.cochemist.com/ccimg/926100/926009-41-8_b.png)




![9-[[[(2R)-1,4-dioxan-2-yl]methyl-methylsulfamoyl]amino]-2-(1-methylpyrazol-4-yl)-11-oxobenzo[1,2]cyclohepta[2,4-b]pyridine](http://img.cochemist.com/ccimg/917900/917879-39-1.png)
![9-[[[(2R)-1,4-dioxan-2-yl]methyl-methylsulfamoyl]amino]-2-(1-methylpyrazol-4-yl)-11-oxobenzo[1,2]cyclohepta[2,4-b]pyridine](http://img.cochemist.com/ccimg/917900/917879-39-1_b.png)
![2-Methyl-2-(4-(3-methyl-2-oxo-8-(quinolin-3-yl)-2,3-dihydro-1H-imidazo[4,5-c]quinolin-1-yl)phenyl)propanenitrile](http://img.cochemist.com/ccimg/915100/915019-65-7.png)
![2-Methyl-2-(4-(3-methyl-2-oxo-8-(quinolin-3-yl)-2,3-dihydro-1H-imidazo[4,5-c]quinolin-1-yl)phenyl)propanenitrile](http://img.cochemist.com/ccimg/915100/915019-65-7_b.png)
![(3R,4R)-3-(5,6-Dihydro-4H-pyrrolo[3,2,1-ij]quinolin-1-yl)-4-(1H-indol-3-yl)pyrrolidine-2,5-dione](http://img.cochemist.com/ccimg/905900/905854-02-6.png)
![(3R,4R)-3-(5,6-Dihydro-4H-pyrrolo[3,2,1-ij]quinolin-1-yl)-4-(1H-indol-3-yl)pyrrolidine-2,5-dione](http://img.cochemist.com/ccimg/905900/905854-02-6_b.png)
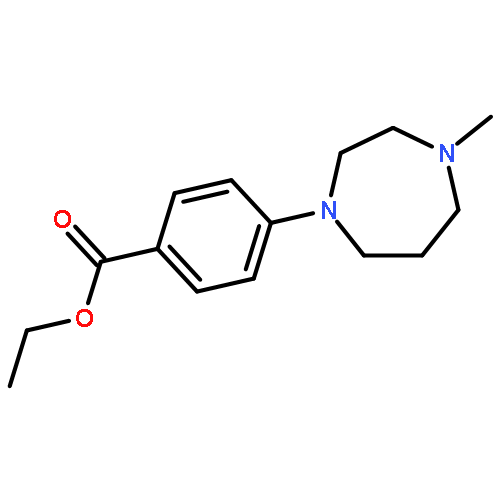
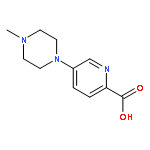
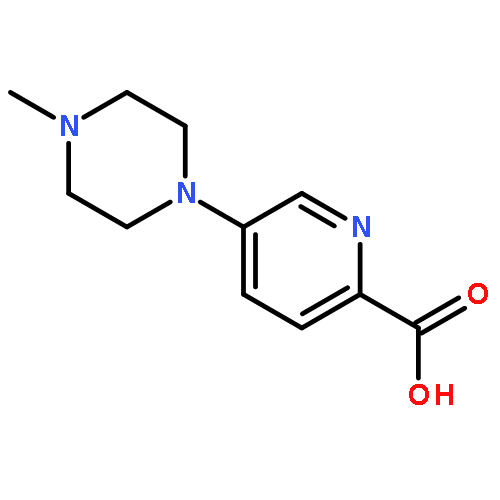

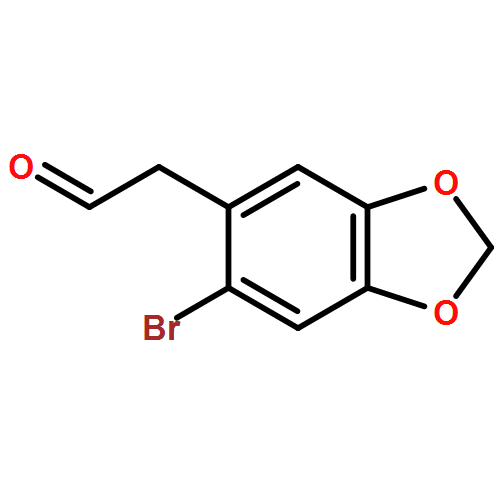
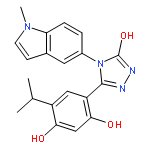
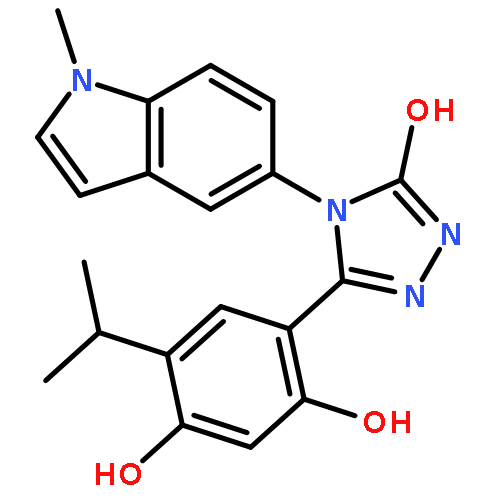
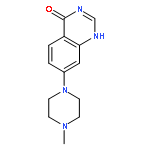
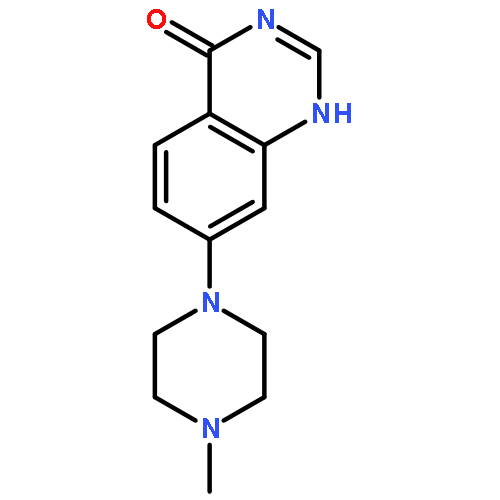
![Benzenamine, 4-[(2-chloro-4-pyridinyl)oxy]-3-fluoro-](http://img.cochemist.com/ccimg/868800/868733-61-3.png)
![Benzenamine, 4-[(2-chloro-4-pyridinyl)oxy]-3-fluoro-](http://img.cochemist.com/ccimg/868800/868733-61-3_b.png)

![Pyridine, 3-methoxy-5-[(trimethylsilyl)ethynyl]-](http://img.cochemist.com/ccimg/823200/823199-09-3.png)
![Pyridine, 3-methoxy-5-[(trimethylsilyl)ethynyl]-](http://img.cochemist.com/ccimg/823200/823199-09-3_b.png)




![(1S,11S,13S,14R,15R)-14,15-DIMETHOXY-20-METHYL-5,7,21-TRIOXA-20-AZAHEXACYCLO[11.4.3.1<SUP>11,14</SUP>.0<SUP>1,13</SUP>.0<SUP>2,10</SUP>.0<SUP>4,8</SUP>]HENICOSA-2(10),3,8-TRIEN-16-ONE](http://img.cochemist.com/ccimg/747500/747414-16-0.png)
![(1S,11S,13S,14R,15R)-14,15-DIMETHOXY-20-METHYL-5,7,21-TRIOXA-20-AZAHEXACYCLO[11.4.3.1<SUP>11,14</SUP>.0<SUP>1,13</SUP>.0<SUP>2,10</SUP>.0<SUP>4,8</SUP>]HENICOSA-2(10),3,8-TRIEN-16-ONE](http://img.cochemist.com/ccimg/747500/747414-16-0_b.png)


![1,3,2-Dioxaborolane, 4,4,5,5-tetramethyl-2-[4-(1-methylethoxy)phenyl]-](http://img.cochemist.com/ccimg/502700/502649-34-5.png)
![1,3,2-Dioxaborolane, 4,4,5,5-tetramethyl-2-[4-(1-methylethoxy)phenyl]-](http://img.cochemist.com/ccimg/502700/502649-34-5_b.png)
![Imidazo[1,2-a]pyridine-3-sulfonyl chloride](http://img.cochemist.com/ccimg/499800/499770-78-4.png)
![Imidazo[1,2-a]pyridine-3-sulfonyl chloride](http://img.cochemist.com/ccimg/499800/499770-78-4_b.png)





![3-(1H-Indol-3-yl)-4-[2-(4-methylpiperazin-1-yl)quinazolin-4-yl]pyrrole-2,5-dione](http://img.cochemist.com/ccimg/425700/425637-18-9.png)
![3-(1H-Indol-3-yl)-4-[2-(4-methylpiperazin-1-yl)quinazolin-4-yl]pyrrole-2,5-dione](http://img.cochemist.com/ccimg/425700/425637-18-9_b.png)
















![Benzenamine, 3-[(methylsulfonyl)methyl]-](http://img.cochemist.com/ccimg/262000/261925-02-4.png)
![Benzenamine, 3-[(methylsulfonyl)methyl]-](http://img.cochemist.com/ccimg/262000/261925-02-4_b.png)
![2-Oxa-6-azaspiro[3.4]octane](http://img.cochemist.com/ccimg/220300/220290-68-6.png)
![2-Oxa-6-azaspiro[3.4]octane](http://img.cochemist.com/ccimg/220300/220290-68-6_b.png)
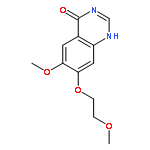
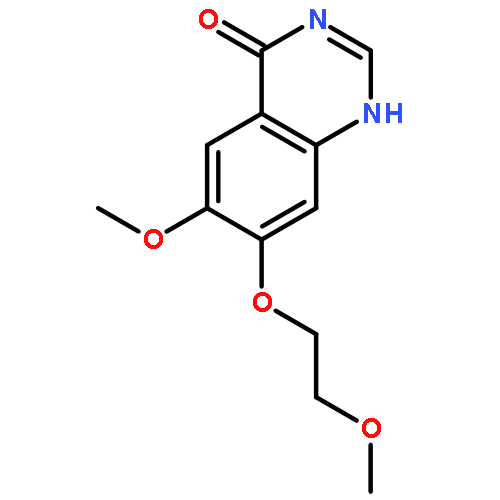








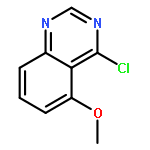
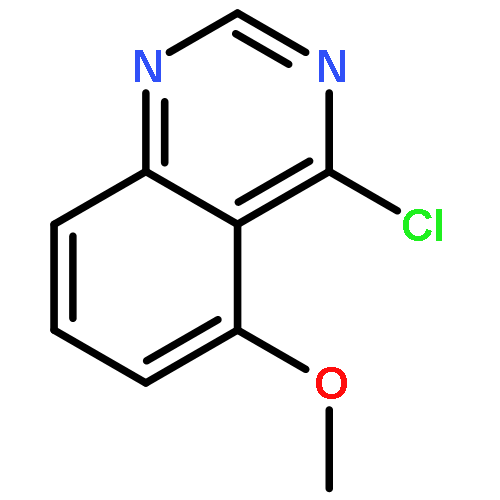


![(3aR,6aS)-2-Methyloctahydropyrrolo[3,4-c]pyrrole](http://img.cochemist.com/ccimg/172800/172739-03-6.png)
![(3aR,6aS)-2-Methyloctahydropyrrolo[3,4-c]pyrrole](http://img.cochemist.com/ccimg/172800/172739-03-6_b.png)










![4-(4-Fluorophenyl)-2-[4-(methylsulfinyl)phenyl]-5-(4-pyridyl)-1H-imidazole](http://img.cochemist.com/ccimg/152200/152121-47-6.png)
![4-(4-Fluorophenyl)-2-[4-(methylsulfinyl)phenyl]-5-(4-pyridyl)-1H-imidazole](http://img.cochemist.com/ccimg/152200/152121-47-6_b.png)








![CYCLOPROPANECARBOXYLIC ACID, 1-[[(3-CHLOROPHENYL)AMINO]CARBONYL]-](http://img.cochemist.com/ccimg/146700/146653-80-7.png)
![CYCLOPROPANECARBOXYLIC ACID, 1-[[(3-CHLOROPHENYL)AMINO]CARBONYL]-](http://img.cochemist.com/ccimg/146700/146653-80-7_b.png)


![Cyclopropanecarboxylic acid, 1-[(phenylamino)carbonyl]-](http://img.cochemist.com/ccimg/145600/145591-80-6.png)
![Cyclopropanecarboxylic acid, 1-[(phenylamino)carbonyl]-](http://img.cochemist.com/ccimg/145600/145591-80-6_b.png)










![2,7-diazaspiro[3.5]nonane](http://img.cochemist.com/ccimg/136100/136098-14-1.png)
![2,7-diazaspiro[3.5]nonane](http://img.cochemist.com/ccimg/136100/136098-14-1_b.png)


![5-Boc-hexahydropyrrolo[3,4-b]pyrrole](http://img.cochemist.com/ccimg/132500/132414-81-4.png)
![5-Boc-hexahydropyrrolo[3,4-b]pyrrole](http://img.cochemist.com/ccimg/132500/132414-81-4_b.png)




![Cyclopropanecarboxylic acid, 1-[[[4-(trifluoromethyl)phenyl]amino]carbonyl]-](http://img.cochemist.com/ccimg/113200/113136-89-3.png)
![Cyclopropanecarboxylic acid, 1-[[[4-(trifluoromethyl)phenyl]amino]carbonyl]-](http://img.cochemist.com/ccimg/113200/113136-89-3_b.png)



![4,5,6,7-Tetrahydrothiazolo[5,4-c]pyridin-2-amine](http://img.cochemist.com/ccimg/97900/97817-23-7.png)
![4,5,6,7-Tetrahydrothiazolo[5,4-c]pyridin-2-amine](http://img.cochemist.com/ccimg/97900/97817-23-7_b.png)




![2-Methyl-octahydro-pyrrolo[3,4-c]pyrrole](http://img.cochemist.com/ccimg/86800/86732-28-7.png)
![2-Methyl-octahydro-pyrrolo[3,4-c]pyrrole](http://img.cochemist.com/ccimg/86800/86732-28-7_b.png)


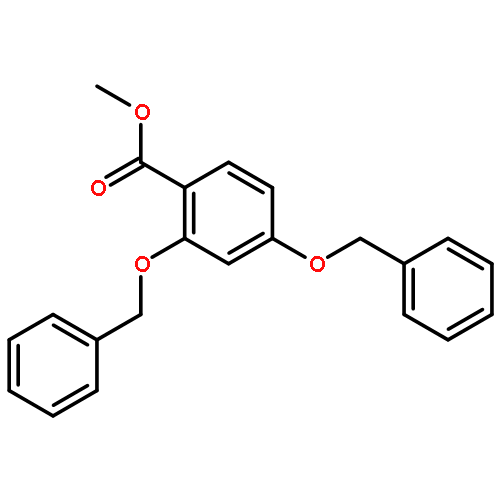
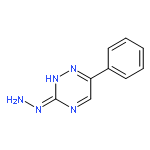
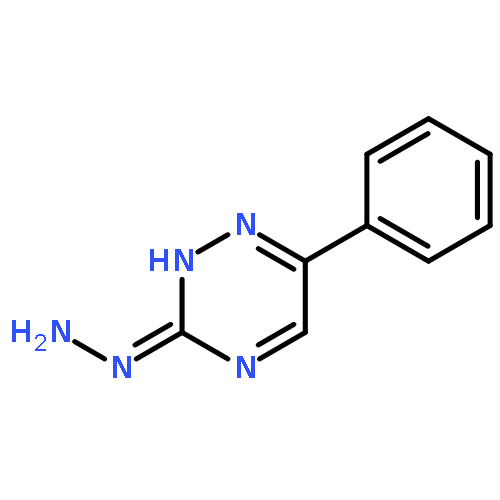
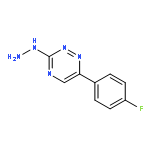


![Ethanone, 1-[2-hydroxy-4,6-bis(methoxymethoxy)phenyl]-](http://img.cochemist.com/ccimg/65500/65490-09-7.png)
![Ethanone, 1-[2-hydroxy-4,6-bis(methoxymethoxy)phenyl]-](http://img.cochemist.com/ccimg/65500/65490-09-7_b.png)







![Ethanone, 1-[2,3,4-tris(phenylmethoxy)phenyl]-](http://img.cochemist.com/ccimg/50500/50439-63-9.png)
![Ethanone, 1-[2,3,4-tris(phenylmethoxy)phenyl]-](http://img.cochemist.com/ccimg/50500/50439-63-9_b.png)


![Benzene, 1-[(methylsulfonyl)methyl]-2-nitro-](http://img.cochemist.com/ccimg/25200/25195-60-2.png)
![Benzene, 1-[(methylsulfonyl)methyl]-2-nitro-](http://img.cochemist.com/ccimg/25200/25195-60-2_b.png)


![{4-[(Methylsulfonyl)methyl]phenyl}amine hydrochloride](http://img.cochemist.com/ccimg/24200/24176-70-3.png)
![{4-[(Methylsulfonyl)methyl]phenyl}amine hydrochloride](http://img.cochemist.com/ccimg/24200/24176-70-3_b.png)




![5-Methyl-4,5,6,7-tetrahydrothiazolo[5,4-c]pyridin-2-amine](http://img.cochemist.com/ccimg/17900/17899-48-8.png)
![5-Methyl-4,5,6,7-tetrahydrothiazolo[5,4-c]pyridin-2-amine](http://img.cochemist.com/ccimg/17900/17899-48-8_b.png)









![2-Chloro-1H-benzo[d]imidazole](http://img.cochemist.com/ccimg/4900/4857-06-1.png)
![2-Chloro-1H-benzo[d]imidazole](http://img.cochemist.com/ccimg/4900/4857-06-1_b.png)




![6-Fluorobenzo[d]oxazol-2(3H)-one](http://img.cochemist.com/ccimg/3000/2923-94-6.png)
![6-Fluorobenzo[d]oxazol-2(3H)-one](http://img.cochemist.com/ccimg/3000/2923-94-6_b.png)
![1-[2-HYDROXY-3,4-BIS(PHENYLMETHOXY)PHENYL]ETHANONE](http://img.cochemist.com/ccimg/2700/2652-27-9.png)
![1-[2-HYDROXY-3,4-BIS(PHENYLMETHOXY)PHENYL]ETHANONE](http://img.cochemist.com/ccimg/2700/2652-27-9_b.png)




![8-Oxa-3-azabicyclo[3.2.1]octane](http://img.cochemist.com/ccimg/300/280-13-7.png)
![8-Oxa-3-azabicyclo[3.2.1]octane](http://img.cochemist.com/ccimg/300/280-13-7_b.png)









![5-Chloro-N2-[2-methoxy-4-[4-(4-methyl-1-piperazinyl)-1-piperidinyl]phenyl]-N4-[2-[(1-methylethyl)sulfonyl]phenyl]-2,4-pyrimidinediamine](http://img.cochemist.com/ccimg/761500/761439-42-3.png)
![5-Chloro-N2-[2-methoxy-4-[4-(4-methyl-1-piperazinyl)-1-piperidinyl]phenyl]-N4-[2-[(1-methylethyl)sulfonyl]phenyl]-2,4-pyrimidinediamine](http://img.cochemist.com/ccimg/761500/761439-42-3_b.png)























