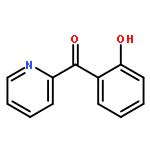
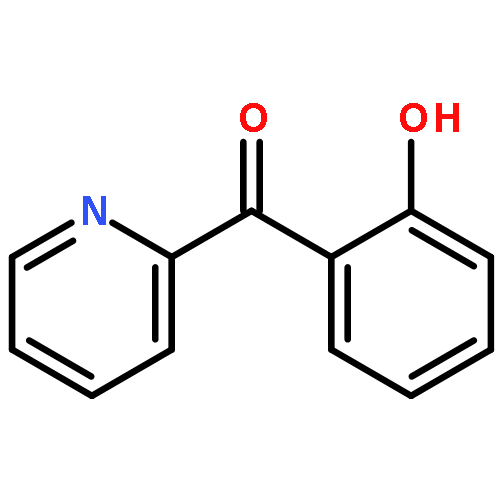
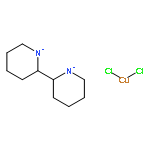
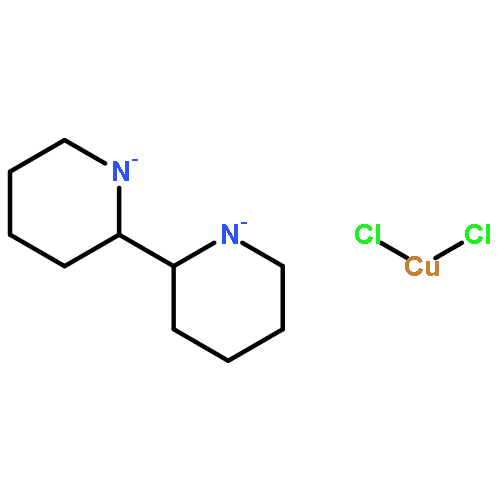
Co-reporter:Elikplim Abada, Peter Y. Zavalij, and Andrei N. Vedernikov
Journal of the American Chemical Society 2017 Volume 139(Issue 2) pp:643-646
Publication Date(Web):January 3, 2017
DOI:10.1021/jacs.6b12648
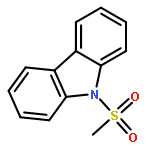
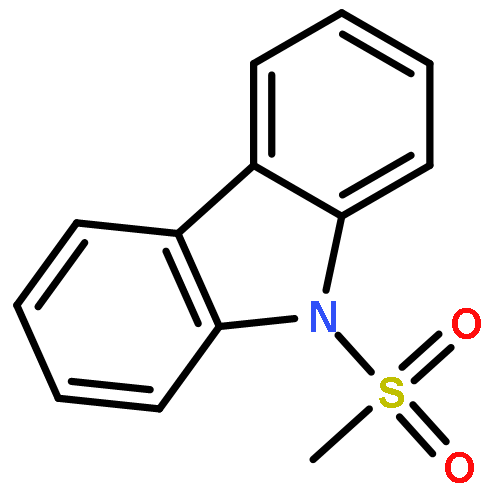
Di-2-pyridyl ketone (dpk)-supported amidoarylpallada(II)cycles derived from various 2-(N-R-amino)biphenyls (R = H, Me, CF3CO, MeSO2, CF3SO2) react with hydrogen peroxide in MeOH, THF, MeCN or AcOH to form the corresponding C–N coupled products, N-R-substituted carbazoles, in 82–98% yield. For R = MeSO2 and CF3SO2, the corresponding reaction intermediates, amidoaryl Pd(IV) complexes were isolated and characterized by single crystal X-ray diffraction and/or NMR spectroscopy. For the first time, the C(sp2)–N reductive elimination from isolated amidoaryl Pd(IV) complexes has been studied in detail.
Co-reporter:David Watts, Daoyong Wang, Mackenzie Adelberg, Peter Y. Zavalij, and Andrei N. Vedernikov
Organometallics 2017 Volume 36(Issue 1) pp:207-219
Publication Date(Web):September 21, 2016
DOI:10.1021/acs.organomet.6b00613
A novel sulfonated CNN pincer ligand has been designed to support CH and O2 activation at a Pt(II) center. The derived cycloplatinated aqua complex 7 was found to be one of the most active reported homogeneous Pt catalysts for H/D exchange between studied arenes (benzene, benzene-d6, toluene-d8, p-xylene, and mesitylene) and 2,2,2-trifluoroethanol (TFE) or 2,2,2-trifluoroethanol-d; the TON for C6D6 as a substrate is >250 after 48 h at 80 °C. The reaction is very selective; no benzylic CH bond activation was observed. The per-CH-bond reactivity diminishes in the series benzene (19) > toluene (p-CH:m-CH:o-CH = 1:0.9:0.2) > xylene (2.9) > mesitylene (1.1). The complex 7 reacts slowly in TFE solutions under ambient light but not in the dark with O2 to selectively produce a Pt(IV) trifluoroethoxo derivative. The H/D exchange reaction kinetics and results of the DFT study suggest that complex 7, and not its TFE derivatives, is the major species responsible for the arene CH bond activation. The reaction deuterium kinetic isotope effect, kH/kD = 1.7, the reaction selectivity, and reaction kinetics modeling suggest that the CH bond cleavage step is rate-determining.
Co-reporter:Anna V. Sberegaeva; Peter Y. Zavalij
Journal of the American Chemical Society 2016 Volume 138(Issue 4) pp:1446-1455
Publication Date(Web):January 14, 2016
DOI:10.1021/jacs.5b12832
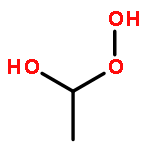
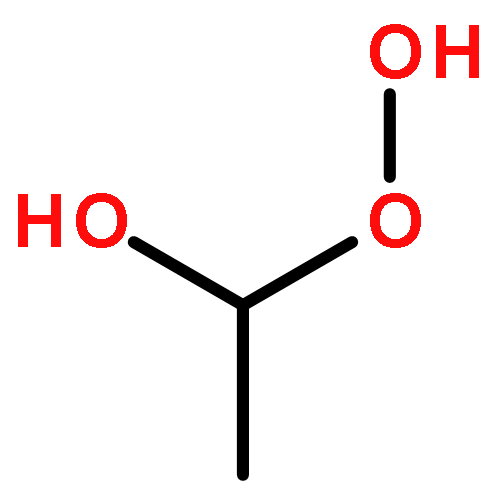
Photochemical aerobic oxidation of n-Pr4N[(dpms)PdIIMe(OH)] (5) and (dpms)PdIIMe(OH2) (8) (dpms = di(2-pyridyl)methanesulfonate) in water in the pH range of 6–14 at 21 °C was studied and found to produce, in combined high yield, a mixture of MeOH, C2H6, and MeOOH along with water-soluble n-Pr4N[(dpms)PdII(OH)2] (9). By changing the reaction pH and concentration of the substrate, the oxidation reaction can be directed toward selective production of ethane (up to 94% selectivity) or methanol (up to 54% selective); the yield of MeOOH can be varied in the range of 0–40%. The source of ethane was found to be an unstable dimethyl PdIV complex (dpms)PdIVMe2(OH) (7), which could be generated from 5 and MeI. For shedding light on the role of MeOOH in the aerobic reaction, oxidation of 5 and 8 with a range of hydroperoxo compounds, including MeOOH, t-BuOOH, and H2O2, was carried out. The proposed mechanism of aerobic oxidation of 5 or 8 involves predominant direct reaction of excited methylpalladium(II) species with O2 to produce a highly electrophilic monomethyl PdIV transient that is involved in subsequent transfer of its methyl group to 5 or 8, H2O, and other nucleophilic components of the reaction mixture.
Co-reporter:Dominik Munz, Daoyong Wang, Megan M. Moyer, Michael S. Webster-Gardiner, Pranaw Kunal, David Watts, Brian G. Trewyn, Andrei N. Vedernikov, and T. Brent Gunnoe
ACS Catalysis 2016 Volume 6(Issue 7) pp:4584
Publication Date(Web):June 15, 2016
DOI:10.1021/acscatal.6b01532
We report platinum catalysts for the efficient aerobic oxidation of olefins to form epoxides and/or derived glycol monoethers. The catalysts—diaqua and dichloro PtII complexes supported by the ligand di(2-pyridine)methanesulfonate (dpms)—are most active when they are covalently tethered to mesoporous silica nanoparticles (MSNs). Supporting the molecular Pt complexes on the MSNs prevents bimolecular catalyst deactivation. Using this strategy, >40 000 turnovers are achieved for the aerobic oxidation of norbornene in 2,2,2-trifluoroethanol. The position of the tether and the nature of other ligands in the metal coordination sphere (aqua, hydroxo, or chloro) are shown to affect the catalyst activity. The new MSN-supported Pt materials were characterized by nuclear magnetic resonance (NMR) spectroscopy, nitrogen physisorption, powder X-ray diffraction (PXRD), scanning electron microscopy (SEM), transmission electron microscopy (TEM), and thermogravimetric analysis (TGA).Keywords: epoxidation; immobilized catalysts; olefins; oxygen; platinum
Co-reporter:Shrinwantu Pal, Peter Y. Zavalij, and Andrei N. Vedernikov
Organometallics 2015 Volume 34(Issue 20) pp:5183-5190
Publication Date(Web):October 9, 2015
DOI:10.1021/acs.organomet.5b00737
A new anionic methoxy methylbis(2-pyridyl)borate ligand enables facile oxidation of the derived dimethylplatinum(II) complex, 10, with O2 in methanol to produce a mixture of two platinum(IV) complexes in combined quantitative yield, one supported by a modified ligand, dimethoxybis(2-pyridyl)borate, and the second supported by the original methoxy methylbis(2-pyridyl)borate. The former product results from the aerobic PtII-to-PtIV oxidation and B-to-PtIV methyl migration with subsequent bridging of the boron and PtIV centers by a methoxy group derived from the solvent. A similar reactivity was also demonstrated for 11, the methyl methoxo platinum(II) analogue of 10. Factors responsible for the product distribution are discussed.
Co-reporter:Anna V. Sberegaeva ; Wei-Guang Liu ; Robert J. Nielsen ; William A. Goddard ; III
Journal of the American Chemical Society 2014 Volume 136(Issue 12) pp:4761-4768
Publication Date(Web):March 5, 2014
DOI:10.1021/ja501213w
The mechanism of oxidation by O2 of (dpms)PtIIMe(OH2) (1) and (dpms)PtIIMe(OH)− (2) [dpms = di(2-pyridyl)methanesulfonate] in water in the pH range of 4–14 at 21 °C was explored using kinetic and isotopic labeling experiments. At pH ≤ 8, the reaction leads to a C1-symmetric monomethyl PtIV complex (dpms)PtIVMe(OH)2 (5) with high selectivity ≥97%; the reaction rate is first-order in [PtIIMe] and fastest at pH 8.0. This behavior was accounted for by assuming that (i) the O2 activation at the PtII center to form a PtIV hydroperoxo species 4 is the reaction rate-limiting step and (ii) the anionic complex 2 is more reactive toward O2 than neutral complex 1 (pKa = 8.15 ± 0.02). At pH ≥ 10, the oxidation is inhibited by OH– ions; the reaction order in [PtIIMe] changes to 2, consistent with a change of the rate-limiting step, which now involves oxidation of complex 2 by PtIV hydroperoxide 4. At pH ≥ 12, formation of a C1-symmetric dimethyl complex 6, (dpms)PtIVMe2(OH), along with [(dpms)PtII(OH)2]− (7) becomes the dominant reaction pathway (50–70% selectivity). This change in the product distribution is explained by the formation of a Cs-symmetric intermediate (dpms)PtIVMe(OH)2 (8), a good methylating agent. The secondary deuterium kinetic isotope effect in the reaction leading to complex 6 is negligible; kH/kD = 0.98 ± 0.02. This observation and experiments with a radical scavenger TEMPO do not support a homolytic mechanism. A SN2 mechanism was proposed for the formation of complex 6 that involves complex 2 as a nucleophile and intermediate 8 as an electrophile.
Co-reporter:S. Pal, P. Y. Zavalij and A. N. Vedernikov
Chemical Communications 2014 vol. 50(Issue 40) pp:5376-5378
Publication Date(Web):11 Nov 2013
DOI:10.1039/C3CC47445C
Dimethyl- and diphenylplatinum(II) fragments PtIIR2 (R = Me, Ph) enable facile and efficient oxidative C(sp3)–H bond cleavage and stepwise C–C and CC coupling at the boron atom of a coordinated 1,5-cyclooctanediyldi(2-pyridyl)borato ligand with O2 as the sole oxidant.
Co-reporter:Andrei N. Vedernikov
ChemCatChem 2014 Volume 6( Issue 9) pp:2490-2492
Publication Date(Web):
DOI:10.1002/cctc.201402482
Co-reporter:Ina S. Dubinsky-Davidchik, Israel Goldberg, Arkadi Vigalok and Andrei N. Vedernikov
Chemical Communications 2013 vol. 49(Issue 33) pp:3446-3448
Publication Date(Web):08 Mar 2013
DOI:10.1039/C3CC41079J
Electrophilic fluorination of aryl α-naphthyl Pt(II) complexes leads to an unprecedented 1,3-migration of the aryl ligand to the β-position of the naphthyl group. The reaction proceeds via the initial oxidative addition of two fluoro ligands to the Pt center followed by C(sp2)–C(sp2) coupling and aryl migration.
Co-reporter:Williamson N. Oloo ; Peter Y. Zavalij
Organometallics 2013 Volume 32(Issue 19) pp:5601-5614
Publication Date(Web):October 3, 2013
DOI:10.1021/om400878u
A detailed mechanistic study of the di(2-pyridyl)ketone (dpk)-enabled oxidation with H2O2 in water of a series of monohydrocarbylpalladium(II) complexes derived from cyclopalladated 2-(3-R-benzoyl)pyridines (R = H, Me) and 2-(p-R′-phenyl)pyridines (R′ = H, Me, MeO, F) to produce corresponding PdIV monohydrocarbyl hydroxo complexes has been carried out, and the PdIV hydrocarbyls have been characterized in detail. The study involves kinetics, isotopic labeling experiments, and the DFT calculations. A reaction mechanism has been proposed for the oxidation of dpk-supported PdII complexes in water that includes elimination of water from the hydrated dpk ligand of the monohydrocarbylpalladium(II) species as the rate-limiting step. Subsequent reversible addition of H2O2 across the resulting ketone C═O bond leads to the formation of two diastereomeric hydroperoxoketals, one of which can rapidly produce a PdIV monohydrocarbyl and the second is unreactive in this type of transformation. All the monohydrocarbyl PdIV complexes undergo clean C–O reductive elimination to form the corresponding phenols or derived palladium(II) phenoxides. The kinetics of the C–O reductive elimination of the Pd(IV) monohydrocarbyls derived from cyclopalladated 2-(p-R-phenyl)pyridines was studied at 22 °C; the corresponding first-order rate constants were found to be only weakly dependent on the nature of the substituent R (H, Me, OMe, F). To account for these observations, a detailed DFT analysis of plausible C–O reductive elimination mechanisms in water was carried out. A direct elimination mechanism of six-coordinate complexes resulting from the oxidation above was proposed to be operational that involves an “early” C–O coupling transition state whose structure varies insignificantly among the substrates studied.
Co-reporter:Andrei N. Vedernikov
Accounts of Chemical Research 2012 Volume 45(Issue 6) pp:803
Publication Date(Web):November 16, 2011
DOI:10.1021/ar200191k
Atom economy and the use of “green” reagents in organic oxidation, including oxidation of hydrocarbons, remain challenges for organic synthesis. Solutions to this problem would lead to a more sustainable economy because of improved access to energy resources such as natural gas. Although natural gas is still abundant, about a third of methane extracted in distant oil fields currently cannot be used as a chemical feedstock because of a dearth of economically and ecologically viable methodologies for partial methane oxidation. Two readily available “atom-economical” “green” oxidants are dioxygen and hydrogen peroxide, but few methodologies have utilized these oxidants effectively in selective organic transformations. Hydrocarbon oxidation and C–H functionalization reactions rely on PdII and PtII complexes. These reagents have practical advantages because they can tolerate moisture and atmospheric oxygen. But this tolerance for atmospheric oxygen also makes it challenging to develop novel organometallic palladium and platinum-catalyzed C–H oxidation reactions utilizing O2 or H2O2.This Account focuses on these challenges: the development of M–C bond (M = PtII, PdII) functionalization and related selective hydrocarbon C–H oxidations with O2 or H2O2. Reactions discussed in this Account do not involve mediators, since the latter can impart low reaction selectivity and catalyst instability. As an efficient solution to the problem of direct M–C oxidation and functionalization with O2 and H2O2, this Account introduces the use of facially chelating semilabile ligands such as di(2-pyridyl)methanesulfonate and the hydrated form of di(2-pyridyl)ketone that enable selective and facile MII–C(spn) bond functionalization with O2 (M = Pt, n = 3; M = Pd, n = 3 (benzylic)) or H2O2 (M = Pd, n = 2). The reactions proceed efficiently in protic solvents such as water, methanol, or acetic acid. With the exception of benzylic PdII complexes, the organometallic substrates studied form isolable high-valent PtIV or PdIV intermediates as a result of an oxidant attack at the MII atom. The resulting high-valent MIV intermediates undergo C–O reductive elimination, leading to products in high yields. Guidelines for the synthesis of products containing other C–X bonds (X = OAc, Cl, Br) while using O2 or H2O2 as oxidants are also discussed. Although the MII–C bond functionalization reactions including high-valent intermediates are well understood, the mechanism for the aerobic functionalization of benzylic PdII complexes will require a more detailed exploration.Importantly, further optimization of the systems suitable for stoichiometric MII–C bond functionalization led to the development of catalytic reactions, including selective acetoxylation of benzylic C–H bonds with O2 as the oxidant and hydroxylation of aromatic C–H bonds with H2O2 in acetic acid solutions. Both reactions proceed efficiently with substrates that contain a directing heteroatom. This Account also describes catalytic methods for ethylene dioxygenation with H2O2 using MII complexes supported by facially chelating ligands.Mechanistic studies of these new oxidation reactions point to important ways to improve their substrate scope and to develop “green” CH functionalization chemistry.
Co-reporter:Ina S. Dubinsky-Davidchik ; Shay Potash ; Israel Goldberg ; Arkadi Vigalok
Journal of the American Chemical Society 2012 Volume 134(Issue 34) pp:14027-14032
Publication Date(Web):July 20, 2012
DOI:10.1021/ja3039272
A series of diphosphine Pt(II) aryl iodo complexes were reacted with XeF2 to cleanly produce the corresponding Pt(II) difluoro complexes and free iodoarenes. However, when aryl ligands bearing fluoro substituents in the ortho positions were used, the formation of the corresponding Pt(II) aryl fluoro complexes was observed in the reaction with XeF2. In the case of the Pt–C6F5 complex, the products of the fluoride-for-iodide exchange were the only products observed by means of 31P and 19F NMR spectroscopy. The experimental and theoretical studies suggest that the formation of iodine–fluorine bond may accompany this transformation. The plausible “I–F” species could be trapped by electron-richer organoplatinum complexes to give a Pt(IV) transient which subsequently eliminates the corresponding aryl iodide. Hence, in some cases a pathway involving an attack of XeF2 at the iodo ligand of Pt(II) aryl iodo complexes to generate I–F species can be operative in addition to or instead of the XeF2 attack at the metal center. Our DFT studies demonstrate that the electrophilic attacks of XeF2 at both sites, platinum and iodide, can be competitive.
Co-reporter:Shrinwantu Pal and Andrei N. Vedernikov
Dalton Transactions 2012 vol. 41(Issue 26) pp:8116-8122
Publication Date(Web):10 May 2012
DOI:10.1039/C2DT30905J
New dimethyldi(2-pyridyl)borato (dmdpb) platinum(II) complexes, (dmdpb)PtIIMe(SMe2) (1), (dmdpb)PtII(L)(SMe2)+, L = MeOH (2), MeCN (3), supported by dimethylsulfide ligand and featuring one (1) or no hydrocarbyls at the metal (2, 3) were prepared and their oxidation with hydrogen peroxide was studied. Both complex 1 bearing the formal charge of +1 on the metal and the methanol complex 2 capable of losing the proton of the methanol ligand to form the methoxide derivative 4 charged similarly to 1, are reactive towards H2O2. However, the cationic complex 3 with a formal charge of +2 on the metal does not react with H2O2. The oxidation of the monomethyl platinum(II) complex 1 leads to the B-to-Pt methyl transfer and formation of a robust dimethyl PtIV species 5 which does not undergo C–O reductive elimination up to 100 °C. By contrast, oxidation of 2 in methanol-d4 leads to quantitative formation of dimethyl ether-d3, CD3OCH3. It was presumed that the latter reaction involves the B-to-Pt methyl transfer and formation of a highly reactive cationic monomethyl PtIV species whose methyl group carbon atom can accept nucleophilic attack by the methanol-d4 solvent to form dimethyl ether-d3.
Co-reporter:Julia R. Khusnutdinova, Anthony S. Maiorana, Peter Y. Zavalij, Andrei N. Vedernikov
Inorganica Chimica Acta 2011 Volume 369(Issue 1) pp:274-283
Publication Date(Web):15 April 2011
DOI:10.1016/j.ica.2010.09.057
Chloro di(2-pyridyl)methanesulfonate ethylene platinum(II) complex was converted to corresponding N-alkylplatina(II)azetidines by reacting the former with primary amines, MeNH2 and tert-BuNH2, to produce 2-ammonioethyl chloro platinum(II) species and their subsequent cyclization in the presence of NaOH in methanol. The N-alkylplatina(II)azetidines are oxidized under air or the atmosphere of pure O2 to the corresponding N-alkylplatina(IV)azetidines in water or in methanol solution in the presence of one equivalent of a strong acid under ambient pressure at 22 °C. The resulting N-alkylplatina(IV)azetidines undergo C–O reductive elimination in acidic aqueous solutions to produce 2-(N-alkylamino)ethanols.Graphical abstractN-alkylplatina(II)azetidines (alkyl = Me, tert-Bu) were prepared from chloro di(2-pyridyl)methanesulfonate ethylene platinum(II) complex and corresponding alkylamine. These complexes react with O2 to form the corresponding N-alkylplatina(IV)azetidines in water at 20 °C; the latter complexes undergo C-O reductive elimination in acidic aqueous solutions to produce 2-(N-alkylamino)ethanols in high yield.Research highlights► Stoichiometric aminohydroxylation of coordinated ethylene in the presence of primary amines and dioxygen as a direct oxidant is achieved using di(2-pyridyl)methanesulfonate Pt(II) complexes in acidic aqueous solutions ► Di(2-pyridyl)methanesulfonate ligand enables aerobic oxidation of N-alkylplatina(II)azetidines to N-alkylplatina(IV)azetidines in acidic aqueous media. ► N-alkylplatina(IV)azetidines undergo C–O reductive elimination in acidic aqueous solutions to form 2-(N-alkylamino)ethanol-derived salts. ► C–N reductive elimination of N-alkylplatina(IV)azetidines is not observed in either aprotic or protic solvents.
Co-reporter:Julia R. Khusnutdinova, Peter Y. Zavalij, and Andrei N. Vedernikov
Organometallics 2011 Volume 30(Issue 12) pp:3392-3399
Publication Date(Web):June 1, 2011
DOI:10.1021/om2002766
Hydroxo olefin platinum(II) complex (dpms)PtII(OH)(olefin) (olefin = cis-cyclooctene) reacts in water, in methanol, and, much faster, in aprotic solvents, DMF, acetone, or CH2Cl2 at 20 °C to produce the corresponding η3-allylic complex. Allylic C–H bond deprotonation in (dpms)PtII(OH)(cis-cyclooctene) is reversible, leading to the selective H/D exchange of the olefin in D2O solutions. Attempted olefin-for-olefin ligand exchange in (dpms)PtII(OH)(C2H4) with olefin = cycloheptene or cyclopentene aimed at the preparation of other (dpms)PtII(OH)(olefin) complexes in water–organic solvent mixtures leads to corresponding η3-allylic derivatives via the intermediacy of (dpms)PtII(OH)(olefin) species. Consistent with mechanistic tests and results of DFT calculations, the PtII(OH) group is responsible for deprotonation of the allylic C–H bond of the coordinated olefin.
Co-reporter:Julia R. Khusnutdinova, Laura L. Newman, Andrei N. Vedernikov
Journal of Organometallic Chemistry 2011 696(25) pp: 3998-4006
Publication Date(Web):
DOI:10.1016/j.jorganchem.2011.06.016
Co-reporter:Williamson Oloo ; Peter Y. Zavalij ; Jing Zhang ; Eugene Khaskin
Journal of the American Chemical Society 2010 Volume 132(Issue 41) pp:14400-14402
Publication Date(Web):September 24, 2010
DOI:10.1021/ja107185w
Monohydrocarbyl palladium(IV) complexes bearing OH, OH2, Br, and Cl ligands at the metal and supported by facially chelating 1-hydroxy-1,1-bis(2-pyridyl)methoxide were readily prepared in water at 0 °C. These complexes reductively eliminate Ar−X (X = OH, Br, Cl) in water at room temperature in high yield, and the corresponding first-order rate constants kOH, kCl, and kBr are on the same order of magnitude.
Co-reporter:Anette Yahav-Levi, Israel Goldberg, Arkadi Vigalok and Andrei N. Vedernikov
Chemical Communications 2010 vol. 46(Issue 19) pp:3324-3326
Publication Date(Web):16 Apr 2010
DOI:10.1039/C001487G
A Pt(IV) complex bearing two aryl and two bromo ligands, which undergoes selective elimination of a bromoarene molecule has been prepared and fully-characterized. The mechanistic studies of this reaction are presented.
Co-reporter:Eugene Khaskin, Daniel L. Lew, Shrinwantu Pal and Andrei N. Vedernikov
Chemical Communications 2009 (Issue 41) pp:6270-6272
Publication Date(Web):03 Sep 2009
DOI:10.1039/B913319D
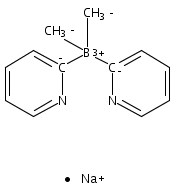
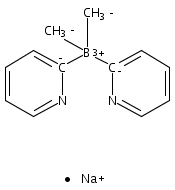
Unambiguous catalytic homogeneous alkane transfer dehydrogenation was observed with a group 10 metal complex catalyst, LPtII(cyclo-C6H10)H, supported by a lipophilic dimethyl-di(4-tert-butyl-2-pyridyl)borate anionic ligand and tert-butylethene as the sacrificial hydrogen acceptor.
Co-reporter:Jing Zhang, Eugene Khaskin, Nicholas P. Anderson, Peter Y. Zavalij and Andrei N. Vedernikov
Chemical Communications 2008 (Issue 31) pp:3625-3627
Publication Date(Web):16 Jun 2008
DOI:10.1039/B803156H
The ability of PdII complexes derived from 2,6-pyridinedicarboxylic acids to catalyze homogeneous regioselective aerobic oxidation of 5- and 6-substituted 8-methylquinolines in AcOH–Ac2O solution to produce corresponding 8-quinolylmethyl acetates in high yield was demonstrated; corresponding 8-quinoline carboxylic acids are minor reaction products.
Co-reporter:Eugene Khaskin;Peter Y. Zavalij Dr.;Andrei N. Vedernikov
Angewandte Chemie 2007 Volume 119(Issue 33) pp:
Publication Date(Web):19 JUL 2007
DOI:10.1002/ange.200701257
Protische Lösungsmittel als Reaktionsbeschleuniger: Die Übertragung einer Methylgruppe von Bor auf Platin erfolgt in den Titelverbindungen in Gegenwart eines Oxidationsmittels (O2 oder MeI) und eines protischen Solvens (Wasser oder ein Alkohol; siehe Schema). Wenn MeI eingesetzt wird, entsteht intermediär eine fünffach koordinierte Platin(IV)-Spezies mit einer agostischen Pt⋅⋅⋅CH-Wechselwirkung.
Co-reporter:Eugene Khaskin;Peter Y. Zavalij Dr.;Andrei N. Vedernikov
Angewandte Chemie International Edition 2007 Volume 46(Issue 33) pp:
Publication Date(Web):19 JUL 2007
DOI:10.1002/anie.200701257
A hydroxlic solvent promoted reaction: Boron-to-platinum methyl-group transfer occurs in the title complexes in the presence of an oxidant (O2 or MeI) and a protic solvent (water, alcohols; see scheme). With MeI as oxidant the reaction intermediate is shown to be a five-coordinate PtIV species containing a Pt⋅⋅⋅CH agostic interaction.
Co-reporter:S. Pal, P. Y. Zavalij and A. N. Vedernikov
Chemical Communications 2014 - vol. 50(Issue 40) pp:NaN5378-5378
Publication Date(Web):2013/11/11
DOI:10.1039/C3CC47445C
Dimethyl- and diphenylplatinum(II) fragments PtIIR2 (R = Me, Ph) enable facile and efficient oxidative C(sp3)–H bond cleavage and stepwise C–C and CC coupling at the boron atom of a coordinated 1,5-cyclooctanediyldi(2-pyridyl)borato ligand with O2 as the sole oxidant.
Co-reporter:Shrinwantu Pal and Andrei N. Vedernikov
Dalton Transactions 2012 - vol. 41(Issue 26) pp:NaN8122-8122
Publication Date(Web):2012/05/10
DOI:10.1039/C2DT30905J
New dimethyldi(2-pyridyl)borato (dmdpb) platinum(II) complexes, (dmdpb)PtIIMe(SMe2) (1), (dmdpb)PtII(L)(SMe2)+, L = MeOH (2), MeCN (3), supported by dimethylsulfide ligand and featuring one (1) or no hydrocarbyls at the metal (2, 3) were prepared and their oxidation with hydrogen peroxide was studied. Both complex 1 bearing the formal charge of +1 on the metal and the methanol complex 2 capable of losing the proton of the methanol ligand to form the methoxide derivative 4 charged similarly to 1, are reactive towards H2O2. However, the cationic complex 3 with a formal charge of +2 on the metal does not react with H2O2. The oxidation of the monomethyl platinum(II) complex 1 leads to the B-to-Pt methyl transfer and formation of a robust dimethyl PtIV species 5 which does not undergo C–O reductive elimination up to 100 °C. By contrast, oxidation of 2 in methanol-d4 leads to quantitative formation of dimethyl ether-d3, CD3OCH3. It was presumed that the latter reaction involves the B-to-Pt methyl transfer and formation of a highly reactive cationic monomethyl PtIV species whose methyl group carbon atom can accept nucleophilic attack by the methanol-d4 solvent to form dimethyl ether-d3.
Co-reporter:Eugene Khaskin, Daniel L. Lew, Shrinwantu Pal and Andrei N. Vedernikov
Chemical Communications 2009(Issue 41) pp:NaN6272-6272
Publication Date(Web):2009/09/03
DOI:10.1039/B913319D
Unambiguous catalytic homogeneous alkane transfer dehydrogenation was observed with a group 10 metal complex catalyst, LPtII(cyclo-C6H10)H, supported by a lipophilic dimethyl-di(4-tert-butyl-2-pyridyl)borate anionic ligand and tert-butylethene as the sacrificial hydrogen acceptor.
Co-reporter:Anette Yahav-Levi, Israel Goldberg, Arkadi Vigalok and Andrei N. Vedernikov
Chemical Communications 2010 - vol. 46(Issue 19) pp:NaN3326-3326
Publication Date(Web):2010/04/16
DOI:10.1039/C001487G
A Pt(IV) complex bearing two aryl and two bromo ligands, which undergoes selective elimination of a bromoarene molecule has been prepared and fully-characterized. The mechanistic studies of this reaction are presented.
Co-reporter:Ina S. Dubinsky-Davidchik, Israel Goldberg, Arkadi Vigalok and Andrei N. Vedernikov
Chemical Communications 2013 - vol. 49(Issue 33) pp:NaN3448-3448
Publication Date(Web):2013/03/08
DOI:10.1039/C3CC41079J
Electrophilic fluorination of aryl α-naphthyl Pt(II) complexes leads to an unprecedented 1,3-migration of the aryl ligand to the β-position of the naphthyl group. The reaction proceeds via the initial oxidative addition of two fluoro ligands to the Pt center followed by C(sp2)–C(sp2) coupling and aryl migration.
Co-reporter:Jing Zhang, Eugene Khaskin, Nicholas P. Anderson, Peter Y. Zavalij and Andrei N. Vedernikov
Chemical Communications 2008(Issue 31) pp:
Publication Date(Web):
DOI:10.1039/B803156H



![AZIRIDINE, 2-(2,2-DIMETHYLPROPYL)-1-[(4-METHYLPHENYL)SULFONYL]-](http://img.cochemist.com/ccimg/855800/855735-74-9.png)
![AZIRIDINE, 2-(2,2-DIMETHYLPROPYL)-1-[(4-METHYLPHENYL)SULFONYL]-](http://img.cochemist.com/ccimg/855800/855735-74-9_b.png)
![AZIRIDINE, 2,2,3-TRIMETHYL-1-[(4-METHYLPHENYL)SULFONYL]-](http://img.cochemist.com/ccimg/579500/579484-04-1.png)
![AZIRIDINE, 2,2,3-TRIMETHYL-1-[(4-METHYLPHENYL)SULFONYL]-](http://img.cochemist.com/ccimg/579500/579484-04-1_b.png)
![Aziridine, 2,3-dimethyl-1-[(4-methylphenyl)sulfonyl]-, (2R,3S)-rel-](http://img.cochemist.com/ccimg/579500/579483-97-9.png)
![Aziridine, 2,3-dimethyl-1-[(4-methylphenyl)sulfonyl]-, (2R,3S)-rel-](http://img.cochemist.com/ccimg/579500/579483-97-9_b.png)
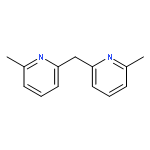
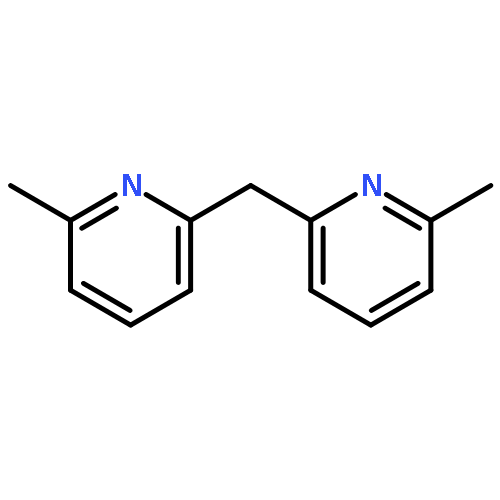


![Aziridine, 2-(1,1-dimethylethyl)-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/171000/170970-21-5.png)
![Aziridine, 2-(1,1-dimethylethyl)-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/171000/170970-21-5_b.png)
![9-Azabicyclo[6.1.0]nonane, 9-[(4-methylphenyl)sulfonyl]-, (1R,8S)-rel-](http://img.cochemist.com/ccimg/146100/146041-79-4.png)
![9-Azabicyclo[6.1.0]nonane, 9-[(4-methylphenyl)sulfonyl]-, (1R,8S)-rel-](http://img.cochemist.com/ccimg/146100/146041-79-4_b.png)


![Aziridine, 2-butyl-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/117000/116905-61-4.png)
![Aziridine, 2-butyl-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/117000/116905-61-4_b.png)


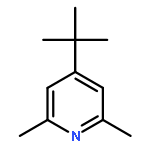
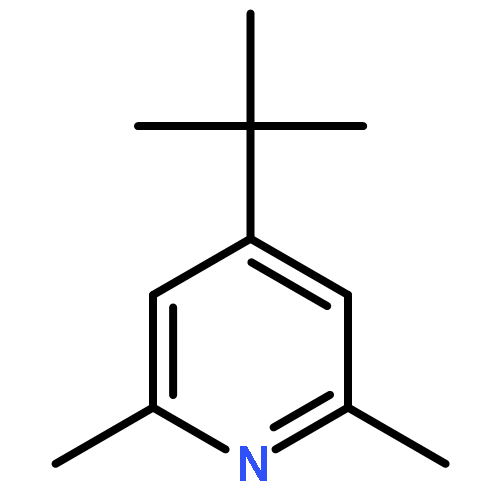
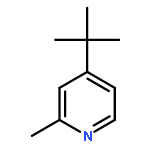
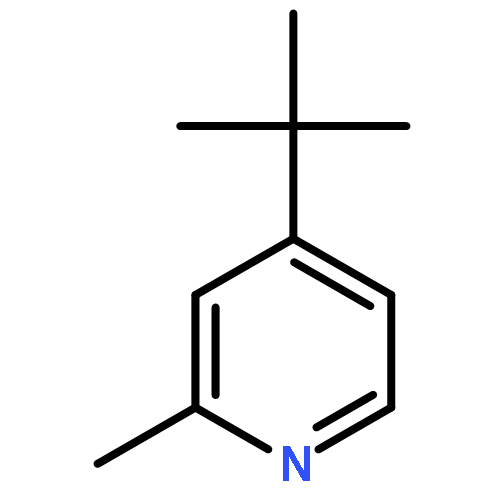





![6-Azabicyclo[3.1.0]hexane, 6-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/81100/81097-48-5.png)
![6-Azabicyclo[3.1.0]hexane, 6-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/81100/81097-48-5_b.png)
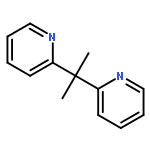






![Palladium,hexakis[m-(acetato-kO:kO')]tri-, cyclo](http://img.cochemist.com/ccimg/53200/53189-26-7.png)
![Palladium,hexakis[m-(acetato-kO:kO')]tri-, cyclo](http://img.cochemist.com/ccimg/53200/53189-26-7_b.png)
![Aziridine,1-[(4-methylphenyl)sulfonyl]-2-phenyl-](http://img.cochemist.com/ccimg/24400/24395-14-0.png)
![Aziridine,1-[(4-methylphenyl)sulfonyl]-2-phenyl-](http://img.cochemist.com/ccimg/24400/24395-14-0_b.png)
![4,7,13,16,21,24-Hexaoxa-1,10-diazabicyclo[8.8.8]hexacosane](http://img.cochemist.com/ccimg/24000/23978-09-8.png)
![4,7,13,16,21,24-Hexaoxa-1,10-diazabicyclo[8.8.8]hexacosane](http://img.cochemist.com/ccimg/24000/23978-09-8_b.png)