Co-reporter:Rebekka S. Klausen, C. Rose Kennedy, Alan M. Hyde, and Eric N. Jacobsen
Journal of the American Chemical Society September 6, 2017 Volume 139(Issue 35) pp:12299-12299
Publication Date(Web):August 8, 2017
DOI:10.1021/jacs.7b06811
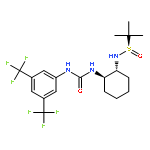
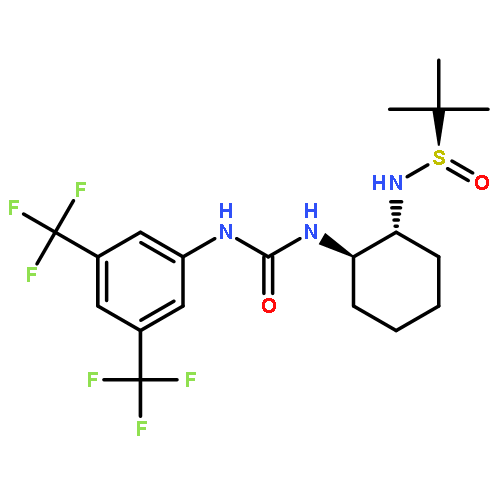
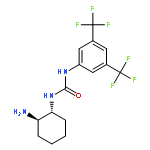
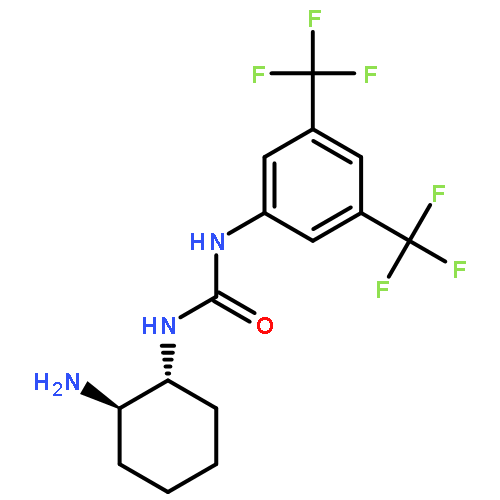
An investigation of the mechanism of benzoic acid/thiourea co-catalysis in the asymmetric Pictet–Spengler reaction is reported. Kinetic, computational, and structure–activity relationship studies provide evidence that rearomatization via deprotonation of the pentahydro-β-carbolinium ion intermediate by a chiral thiourea·carboxylate complex is both rate- and enantioselectivity-determining. The thiourea catalyst induces rate acceleration over the background reaction mediated by benzoic acid alone by stabilizing every intermediate and transition state leading up to and including the final selectivity-determining step. Distortion–interaction analyses of the transition structures for deprotonation predicted using density functional theory indicate that differential π–π and C–H···π interactions within a scaffold organized by multiple hydrogen bonds dictate stereoselectivity. The principles underlying rate acceleration and enantiocontrol described herein are expected to have general implications for the design of selective transformations involving deprotonation of high-energy intermediates.
Co-reporter:Steven M. Banik, Katrina M. Mennie, and Eric N. Jacobsen
Journal of the American Chemical Society July 12, 2017 Volume 139(Issue 27) pp:9152-9152
Publication Date(Web):June 16, 2017
DOI:10.1021/jacs.7b05160
Electronegative substituents arrayed in 1,3-relationships along saturated carbon frameworks can exert strong influence over molecular conformation due to dipole minimization effects. Simple and general methods for incorporation of such functional group relationships could thus provide a valuable tool for modulating molecular shape. Here, we describe a general strategy for the 1,3-oxidation of cyclopropanes using aryl iodine(I–III) catalysis, with emphasis on 1,3-difluorination reactions. These reactions make use of practical, commercially available reagents and can engage a variety of substituted cyclopropane substrates. Analysis of crystal and solution structures of several of the products reveal the consistent effect of 1,3-difluorides in dictating molecular conformation. The generality of the 1,3-oxidation strategy is demonstrated through the catalytic oxidative ring-opening of cyclopropanes for the synthesis of 1,3-fluoroacetoxylated products, 1,3-diols, 1,3-amino alcohols, and 1,3-diamines.
Co-reporter:Kaid C. Harper;Nadine Kuhl;Richard Y. Liu;Eugene E. Kwan;Yongho Park
Science 2017 Volume 355(Issue 6321) pp:
Publication Date(Web):
DOI:10.1126/science.aal1875
A cyclic catalyst to pair up sugars
Linking sugar molecules together to make complex carbohydrates is a geometrical challenge. For a six-carbon sugar such as glucose, there are six different possible linkage sites and also two possible configurations in which to anchor the incipient bond. Park et al. developed a ring-shaped, dimeric catalyst that pairs sugars after one of them has been modified with a chloride. The thiourea-based catalyst appears to pull away the chloride while simultaneously activating the incoming second sugar. The resultant bond-forming process reliably inverts the initial C–Cl configuration.
Science, this issue p. 162
Co-reporter:Steven M. Banik;Jonathan William Medley
Science 2016 Vol 353(6294) pp:51-54
Publication Date(Web):01 Jul 2016
DOI:10.1126/science.aaf8078
Abstract
Difluoromethyl groups possess specific steric and electronic properties that invite their use as chemically inert surrogates of alcohols, thiols, and other polar functional groups important in a wide assortment of molecular recognition processes. We report here a method for the catalytic, asymmetric, migratory geminal difluorination of β-substituted styrenes to access a variety of products bearing difluoromethylated tertiary or quaternary stereocenters. The reaction uses commercially available reagents (m-chloroperbenzoic acid and hydrogen fluoride pyridine) and a simple chiral aryl iodide catalyst and is carried out readily on a gram scale. Substituent effects and temperature-dependent variations in enantioselectivity suggest that cation-π interactions play an important role in stereodifferentiation by the catalyst.
Co-reporter:Steven M. Banik; Jonathan William Medley
Journal of the American Chemical Society 2016 Volume 138(Issue 15) pp:5000-5003
Publication Date(Web):April 5, 2016
DOI:10.1021/jacs.6b02391
We describe a direct, catalytic approach to the 1,2-difluorination of alkenes. The method utilizes a nucleophilic fluoride source and an oxidant in conjunction with an aryl iodide catalyst and is applicable to alkenes with all types of substitution patterns. In general, the vicinal difluoride products are produced with high diastereoselectivities. The observed sense of stereoinduction implicates anchimeric assistance pathways in reactions of alkenes bearing neighboring Lewis basic functionality.
Co-reporter:Eugene E. Kwan, Yongho Park, Harrison A. Besser, Thayer L. Anderson, and Eric N. Jacobsen
Journal of the American Chemical Society 2016 Volume 139(Issue 1) pp:43-46
Publication Date(Web):December 22, 2016
DOI:10.1021/jacs.6b10621
Polarization transfer is demonstrated as a sensitive technique for the measurement of isotopic fractionation of protonated carbons at natural abundance. This method allows kinetic isotope effects (KIEs) to be determined with substantially less material or shorter acquisition time compared with traditional experiments. Computations quantitatively reproduce the KIEs in a Diels–Alder reaction and a catalytic glycosylation. The glycosylation is shown to occur by an effectively concerted mechanism.
Co-reporter:Yongho Park, Corinna S. Schindler, and Eric N. Jacobsen
Journal of the American Chemical Society 2016 Volume 138(Issue 45) pp:14848-14851
Publication Date(Web):October 27, 2016
DOI:10.1021/jacs.6b09736
An enantioselective, catalytic aza-Sakurai cyclization of chlorolactams has been developed as an efficient entry into indolizidine and quinolizidine frameworks. Structure–enantioselectivity relationship studies and mechanistic analysis point to a dual role of the catalyst wherein the thiourea moiety of the catalyst is engaged in both anion binding and Lewis base activation of a substrate.
Co-reporter:C. Rose Kennedy, Dan Lehnherr, Naomi S. Rajapaksa, David D. Ford, Yongho Park, and Eric N. Jacobsen
Journal of the American Chemical Society 2016 Volume 138(Issue 41) pp:13525-13528
Publication Date(Web):October 5, 2016
DOI:10.1021/jacs.6b09205
We describe the rational design of a linked, bis-thiourea catalyst with enhanced activity relative to monomeric analogues in a representative enantioselective anion-abstraction reaction. Mechanistic insights guide development of this linking strategy to favor substrate activation though the intramolecular cooperation of two thiourea subunits while avoiding nonproductive aggregation. The resulting catalyst platform overcomes many of the practical limitations that have plagued hydrogen-bond-donor catalysis and enables use of catalyst loadings as low as 0.05 mol %. Computational analyses of possible anion-binding modes provide detailed insight into the precise mechanism of anion-abstraction catalysis with this pseudo-dimeric thiourea.
Co-reporter:Eric M. Woerly, Steven M. Banik, and Eric N. Jacobsen
Journal of the American Chemical Society 2016 Volume 138(Issue 42) pp:13858-13861
Publication Date(Web):October 6, 2016
DOI:10.1021/jacs.6b09499
The enantioselective synthesis of 4-fluoroisochromanones via chiral aryl iodide-catalyzed fluorolactonization is reported. This methodology uses HF-pyridine as a nucleophilic fluoride source with a peracid stoichiometric oxidant and provides access to lactones containing fluorine-bearing stereogenic centers in high enantio- and diastereoselectivity. The regioselectivity observed in these lactonization reactions is complementary to that obtained with established asymmetric electrophilic fluorination protocols.
Co-reporter:David D. Ford; Dan Lehnherr; C. Rose Kennedy
Journal of the American Chemical Society 2016 Volume 138(Issue 25) pp:7860-7863
Publication Date(Web):June 8, 2016
DOI:10.1021/jacs.6b04686
Chiral, neutral H-bond donors have found widespread use as catalysts in enantioselective reactions involving ion-pair intermediates. Herein, a systematic mechanistic study of a prototypical anion-binding reaction, the thiourea-catalyzed enantioselective alkylation of α-chloroethers, is detailed. This study reveals that the catalyst resting state is an inactive dimeric aggregate that must dissociate and then reassemble to form a 2:1 catalyst–substrate complex in the rate-determining transition structure. Insight into this mode of catalyst cooperativity sheds light on the practical limitations that have plagued many of the H-bond donor-catalyzed reactions developed to date and suggests design strategies for new, highly efficient catalyst structures.
Co-reporter:David D. Ford, Dan Lehnherr, C. Rose Kennedy, and Eric N. Jacobsen
ACS Catalysis 2016 Volume 6(Issue 7) pp:4616
Publication Date(Web):June 10, 2016
DOI:10.1021/acscatal.6b01384
In this paper, we provide a detailed mechanistic characterization of the electrophile-activation step in a representative thiourea-catalyzed enantioselective reaction proposed to involve generation of ion-pair intermediates. Comparison of catalyst-promoted substrate epimerization with catalytic alkylation points to the participation of a common intermediate in both pathways and provides conclusive evidence for anion abstraction via an SN1-like pathway involving the cooperative action of two catalyst molecules.Keywords: anion-abstraction catalysis; anion-binding catalysis; ion-pairing catalysis; organocatalysis; reaction mechanism
Co-reporter:Dan Lehnherr, David D. Ford, Andrew J. Bendelsmith, C. Rose Kennedy, and Eric N. Jacobsen
Organic Letters 2016 Volume 18(Issue 13) pp:3214-3217
Publication Date(Web):June 13, 2016
DOI:10.1021/acs.orglett.6b01435
While aryl pyrrolidinoamido-thioureas derived from α-amino acids are effective catalysts in a number of asymmetric transformations, they exist as mixtures of slowly interconverting amide rotamers. Herein, the compromising role of amide bond isomerism is analyzed experimentally and computationally. A modified catalyst structure that exists almost exclusively as a single amide rotamer is introduced. This modification is shown to result in improved reactivity and enantioselectivity by minimizing competing reaction pathways.
Co-reporter:Eric N. Jacobsen
Advanced Synthesis & Catalysis 2016 Volume 358( Issue 1) pp:
Publication Date(Web):
DOI:10.1002/adsc.201600013
Co-reporter:C. Rose Kennedy, Jennifer A. Guidera, and Eric N. Jacobsen
ACS Central Science 2016 Volume 2(Issue 6) pp:416
Publication Date(Web):June 14, 2016
DOI:10.1021/acscentsci.6b00125
Sigmatropic rearrangements number among the most powerful complexity-building transformations in organic synthesis but have remained largely insensitive to enantioselective catalysis due to the diffuse nature of their transition structures. Here, we describe a synergistic ion-binding strategy for asymmetric catalysis of anionic sigmatropic rearrangements. This approach is demonstrated with the enantioselective [2,3]-Wittig rearrangement of α-allyloxy carbonyl compounds to afford highly enantioenriched homoallylic alcohol products. Chiral thiourea catalysts are shown to engage reactive anions and their countercations through a cooperative set of attractive, noncovalent interactions. Catalyst structure–reactivity–selectivity relationship studies and computational analyses provide insight into catalyst–substrate interactions responsible for enantioinduction and allude to the potential generality of this catalytic strategy.
Co-reporter:Michael R. Witten and Eric N. Jacobsen
Organic Letters 2015 Volume 17(Issue 11) pp:2772-2775
Publication Date(Web):May 8, 2015
DOI:10.1021/acs.orglett.5b01193
A new primary amine catalyst for the asymmetric α-hydroxylation and α-fluorination of α-branched aldehydes is described. The products of the title transformations are generated in excellent yields with high enantioselectivities. Both processes can be performed within short reaction times and on gram scale. The similarity in results obtained in both reactions, combined with computational evidence, implies a common basis for stereoinduction and the possibility of a general catalytic mechanism for α-functionalizations. Promising initial results in α-amination and α-chlorination reactions support this hypothesis.
Co-reporter:Eric N. Jacobsen
Advanced Synthesis & Catalysis 2015 Volume 357( Issue 10) pp:2173-2174
Publication Date(Web):
DOI:10.1002/adsc.201500536
Co-reporter:Masayuki Wasa ; Richard Y. Liu ; Stéphane P. Roche
Journal of the American Chemical Society 2014 Volume 136(Issue 37) pp:12872-12875
Publication Date(Web):September 1, 2014
DOI:10.1021/ja5075163
We report a scalable, one-pot Mannich route to enantioenriched α-amino esters by direct reaction of α-chloroglycine ester as a practical imino ester surrogate. The reaction is promoted by a chiral aminothiourea, which is proposed to operate cooperatively by generating an iminium ion by chloride abstraction and an enolate by deprotonation, followed by highly stereoselective C–C bond formation between both reactive intermediates associated non-covalently within the catalyst framework.
Co-reporter:Charles S. Yeung ; Robert E. Ziegler ; John A. Porco ; Jr.
Journal of the American Chemical Society 2014 Volume 136(Issue 39) pp:13614-13617
Publication Date(Web):September 12, 2014
DOI:10.1021/ja508523g
We report the development of a catalytic method for the enantioselective addition of indoles to pyrone-derived electrophiles. Arylpyrrolidino-derived thioureas catalyze the addition with high stereoselectivity in the presence of catalytic quantities of an achiral Brønsted acid. The indole–pyrone adducts feature a quaternary stereocenter and represent an unusual class of indolines bearing structural resemblance to the hybrid natural product pleiocarpamine.
Co-reporter:Yuan-Qing Fang ; Pamela M. Tadross
Journal of the American Chemical Society 2014 Volume 136(Issue 52) pp:17966-17968
Publication Date(Web):December 15, 2014
DOI:10.1021/ja5117638
Bifunctional phosphinothiourea catalysts have been developed successfully for the highly regio- and enantioselective γ-hydroamination of allenyl and propargyl esters with N-methoxy carbamate nucleophiles to yield α,β-unsaturated γ-amino acid ester products. In the case of propargyl ester substrates, the reaction proceeds through reversible phosphinothiourea-catalyzed isomerization to the corresponding allenyl ester. The high enantioselectivity of the process is attributed to a cooperative conjugate addition of a thiourea-bound carbamate anion to a vinyl phosphonium ion resulting from covalent activation of the allenyl ester substrate.
Co-reporter:Hu Zhang ; Song Lin
Journal of the American Chemical Society 2014 Volume 136(Issue 47) pp:16485-16488
Publication Date(Web):November 7, 2014
DOI:10.1021/ja510113s
Highly enantioselective selenocyclization reactions are promoted by the combination of a new chiral squaramide catalyst, a mineral acid, and an achiral Lewis base. Mechanistic studies reveal that the enantioselectivity originates from the dynamic kinetic resolution of seleniranium ions through anion-binding catalysis.
Co-reporter:Giulia Bergonzini, Corinna S. Schindler, Carl-Johan Wallentin, Eric N. Jacobsen and Corey R. J. Stephenson
Chemical Science 2014 vol. 5(Issue 1) pp:112-116
Publication Date(Web):23 Aug 2013
DOI:10.1039/C3SC52265B
The enantioselective oxidative C–H functionalization of tetrahydroisoquinoline derivatives is achieved through the merger of photoredox and asymmetric anion-binding catalysis. This combination of two distinct catalysis concepts introduces a potentially general approach to asymmetric transformations in oxidative photocatalysis.
Co-reporter:Michael R. Witten ;Dr. Eric N. Jacobsen
Angewandte Chemie 2014 Volume 126( Issue 23) pp:6022-6026
Publication Date(Web):
DOI:10.1002/ange.201402834
Abstract
Highly enantioselective intermolecular [5+2] cycloadditions of pyrylium ion intermediates with electron-rich alkenes are promoted by a dual catalyst system composed of an achiral thiourea and a chiral primary aminothiourea. The observed enantioselectivity is highly dependent on the substitution pattern of the 5π component, and the basis for this effect is analyzed using experimental and computational evidence. The resultant 8-oxabicyclo[3.2.1]octane derivatives possess a scaffold common in natural products and medicinally active compounds and are also versatile chiral building blocks for further manipulations. Several stereoselective complexity-generating transformations of the 8-oxabicyclooctane products are presented.
Co-reporter:Michael R. Witten ;Dr. Eric N. Jacobsen
Angewandte Chemie International Edition 2014 Volume 53( Issue 23) pp:5912-5916
Publication Date(Web):
DOI:10.1002/anie.201402834
Abstract
Highly enantioselective intermolecular [5+2] cycloadditions of pyrylium ion intermediates with electron-rich alkenes are promoted by a dual catalyst system composed of an achiral thiourea and a chiral primary aminothiourea. The observed enantioselectivity is highly dependent on the substitution pattern of the 5π component, and the basis for this effect is analyzed using experimental and computational evidence. The resultant 8-oxabicyclo[3.2.1]octane derivatives possess a scaffold common in natural products and medicinally active compounds and are also versatile chiral building blocks for further manipulations. Several stereoselective complexity-generating transformations of the 8-oxabicyclooctane products are presented.
Co-reporter:David E. White, Pamela M. Tadross, Zhe Lu, Eric N. Jacobsen
Tetrahedron 2014 70(27–28) pp: 4165-4180
Publication Date(Web):
DOI:10.1016/j.tet.2014.03.043
Co-reporter:Mathieu P. Lalonde ; Meredeth A. McGowan ; Naomi S. Rajapaksa
Journal of the American Chemical Society 2013 Volume 135(Issue 5) pp:1891-1894
Publication Date(Web):January 15, 2013
DOI:10.1021/ja310718f
A highly enantio- and diastereoselective synthesis of indolo- and benzoquinolizidine compounds has been developed through the formal aza-Diels–Alder reaction of enones with cyclic imines. This transformation is catalyzed by a new bifunctional primary aminothiourea that achieves simultaneous activation of both the enone and imine reaction components.
Co-reporter:Adam R. Brown ; Christopher Uyeda ; Carolyn A. Brotherton
Journal of the American Chemical Society 2013 Volume 135(Issue 18) pp:6747-6749
Publication Date(Web):April 18, 2013
DOI:10.1021/ja402893z
Catalysis of Cope-type rearrangements of bis-homoallylic hydroxylamines is demonstrated using chiral thiourea derivatives. This formal intramolecular hydroamination reaction provides access to highly enantioenriched α-substituted pyrrolidine products and represents a complementary approach to metal-catalyzed methods.
Co-reporter:David D. Ford ; Lars P. C. Nielsen ; Stephan J. Zuend ; Charles B. Musgrave
Journal of the American Chemical Society 2013 Volume 135(Issue 41) pp:15595-15608
Publication Date(Web):September 16, 2013
DOI:10.1021/ja408027p
In the (salen)Co(III)-catalyzed hydrolytic kinetic resolution (HKR) of terminal epoxides, the rate- and stereoselectivity-determining epoxide ring-opening step occurs by a cooperative bimetallic mechanism with one Co(III) complex acting as a Lewis acid and another serving to deliver the hydroxide nucleophile. In this paper, we analyze the basis for the extraordinarily high stereoselectivity and broad substrate scope observed in the HKR. We demonstrate that the stereochemistry of each of the two (salen)Co(III) complexes in the rate-determining transition structure is important for productive catalysis: a measurable rate of hydrolysis occurs only if the absolute stereochemistry of each of these (salen)Co(III) complexes is the same. Experimental and computational studies provide strong evidence that stereochemical communication in the HKR is mediated by the stepped conformation of the salen ligand, and not the shape of the chiral diamine backbone of the ligand. A detailed computational analysis reveals that the epoxide binds the Lewis acidic Co(III) complex in a well-defined geometry imposed by stereoelectronic rather than steric effects. This insight serves as the basis of a complete stereochemical and transition structure model that sheds light on the reasons for the broad substrate generality of the HKR.
Co-reporter:Cheyenne S. Brindle, Charles S. Yeung and Eric N. Jacobsen
Chemical Science 2013 vol. 4(Issue 5) pp:2100-2104
Publication Date(Web):14 Mar 2013
DOI:10.1039/C3SC50410G
Highly enantioselective vicinal iodoamination of olefins is accomplished through the iodocyclization of alkenyl trichloroacetimidates catalysed by a new chiral Schiff-base urea derivative. The resulting products are converted readily to a variety of polyfunctional amine-containing chiral building blocks.
Co-reporter:Naomi S. Rajapaksa and Eric N. Jacobsen
Organic Letters 2013 Volume 15(Issue 16) pp:4238-4241
Publication Date(Web):August 8, 2013
DOI:10.1021/ol401968m
Highly enantio- and diastereoselective transannular ketone–ene reactions are catalyzed by a new chromium(III) triflate tridentate Schiff base complex. Electronically unactivated keto-olefins undergo heteroene reactions at ambient temperature to afford enantioenriched bicyclic alcohols, common structural motifs in natural products. The kinetic resolution of a configurationally stable planar-chiral cyclodecenone is also described.
Co-reporter:James A. Birrell and Eric N. Jacobsen
Organic Letters 2013 Volume 15(Issue 12) pp:2895-2897
Publication Date(Web):June 6, 2013
DOI:10.1021/ol401013s
A highly enantioselective addition of phenyl carbamate to meso-epoxides has been developed to efficiently generate protected trans-1,2-amino alcohols. This transformation is promoted by an oligomeric (salen)Co–OTf catalyst and has been used to prepare two useful 2-aminocycloalkanol hydrochlorides in enantiopure form on a multigram scale from commercially available starting materials.
Co-reporter:Naomi S. Rajapaksa, Meredeth A. McGowan, Matthew Rienzo, and Eric N. Jacobsen
Organic Letters 2013 Volume 15(Issue 3) pp:706-709
Publication Date(Web):January 18, 2013
DOI:10.1021/ol400046n
A catalytic, enantioselective synthesis of (+)-reserpine is reported. The route features a highly diastereoselective, chiral catalyst-controlled formal aza-Diels–Alder reaction between a 6-methoxytryptamine-derived dihydro-β-carboline and an enantioenriched α-substituted enone to form a key tetracyclic intermediate. This approach addresses the challenge of setting the C3 stereogenic center by using catalyst control. Elaboration of the tetracycle to (+)-reserpine includes an intramolecular aldol cyclization and a highly diastereoselective hydrogenation of a sterically hindered enoate.
Co-reporter:Dr. Katrien Brak ; Eric N. Jacobsen
Angewandte Chemie International Edition 2013 Volume 52( Issue 2) pp:534-561
Publication Date(Web):
DOI:10.1002/anie.201205449
Abstract
Charged intermediates and reagents are ubiquitous in organic transformations. The interaction of these ionic species with chiral neutral, anionic, or cationic small molecules has emerged as a powerful strategy for catalytic, enantioselective synthesis. This review describes developments in the burgeoning field of asymmetric ion-pairing catalysis with an emphasis on the insights that have been gleaned into the structural and mechanistic features that contribute to high asymmetric induction.
Co-reporter:Dr. Katrien Brak ; Eric N. Jacobsen
Angewandte Chemie 2013 Volume 125( Issue 2) pp:558-588
Publication Date(Web):
DOI:10.1002/ange.201205449
Abstract
Geladene Intermediate und Reagentien sind in organischen Transformationen ubiquitär. Die Wechselwirkungen dieser ionischen Spezies mit chiralen neutralen, anionischen oder kationischen kleinen Molekülen haben sich zu einer leistungsfähigen Strategie in der katalytischen enantioselektiven Synthese entwickelt. Dieser Aufsatz beschreibt die Entwicklungen in dem aufstrebenden Gebiet der asymmetrischen Ionenpaarkatalyse mit dem Schwerpunkt auf strukturellen und mechanistischen Eigenschaften, die zu der hohen asymmetrischen Induktion dieser Reaktionen beitragen.
Co-reporter:Lars P. C. Nielsen, Stephan J. Zuend, David D. Ford, and Eric N. Jacobsen
The Journal of Organic Chemistry 2012 Volume 77(Issue 5) pp:2486-2495
Publication Date(Web):February 1, 2012
DOI:10.1021/jo300181f
The (salen)Co(III)-catalyzed hydrolytic kinetic resolution (HKR) of terminal epoxides is a bimetallic process with a rate controlled by partitioning between a nucleophilic (salen)Co–OH catalyst and a Lewis acidic (salen)Co–X catalyst. The commonly used (salen)Co–OAc and (salen)Co–Cl precatalysts undergo complete and irreversible counterion addition to epoxide during the course of the epoxide hydrolysis reaction, resulting in quantitative formation of weakly Lewis acidic (salen)Co–OH and severely diminished reaction rates in the late stages of HKR reactions. In contrast, (salen)Co–OTs maintains high reactivity over the entire course of HKR reactions. We describe here an investigation of catalyst partitioning with different (salen)Co–X precatalysts and demonstrate that counterion addition to epoxide is reversible in the case of the (salen)Co–OTs. This reversible counterion addition results in stable partitioning between nucleophilic and Lewis acidic catalyst species, allowing highly efficient catalysis throughout the course of the HKR reaction.
Co-reporter:James A. Birrell ; Jean-Nicolas Desrosiers
Journal of the American Chemical Society 2011 Volume 133(Issue 35) pp:13872-13875
Publication Date(Web):August 1, 2011
DOI:10.1021/ja205602j
A highly enantioselective acylation of silyl ketene acetals with acyl fluorides has been developed to generate useful α,α-disubstituted butyrolactone products. This transformation is promoted by a new thiourea catalyst and 4-pyrrolidinopyridine and represents the first example of enantioselective thiourea anion-binding catalysis with fluoride.
Co-reporter:Noah Z. Burns ; Michael R. Witten
Journal of the American Chemical Society 2011 Volume 133(Issue 37) pp:14578-14581
Publication Date(Web):August 17, 2011
DOI:10.1021/ja206997e
A new method for effecting catalytic enantioselective intramolecular [5 + 2] cycloadditions based on oxidopyrylium intermediates is reported. The dual catalyst system consists of a chiral primary aminothiourea and a second achiral thiourea. Experimental evidence points to a new type of cooperative catalysis with each species being necessary to generate a reactive pyrylium ion pair that undergoes subsequent cycloaddition with high enantioselectivity.
Co-reporter:Christopher Uyeda
Journal of the American Chemical Society 2011 Volume 133(Issue 13) pp:5062-5075
Publication Date(Web):March 10, 2011
DOI:10.1021/ja110842s
The mechanism by which chiral arylpyrrole-substituted guanidinium ions promote the Claisen rearrangement of O-allyl α-ketoesters and induce enantioselectivity was investigated by experimental and computational methods. In addition to stabilization of the developing negative charge on the oxallyl fragment of the rearrangement transition state by hydrogen-bond donation, evidence was obtained for a secondary attractive interaction between the π-system of a catalyst aromatic substituent and the cationic allyl fragment. Across a series of substituted arylpyrrole derivatives, enantioselectivity was observed to vary predictably according to this proposal. This mechanistic analysis led to the development of a new p-dimethylaminophenyl-substituted catalyst, which afforded improvements in enantioselectivity relative to the parent phenyl catalyst for a representative set of substrates.
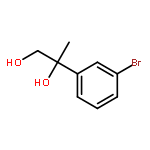
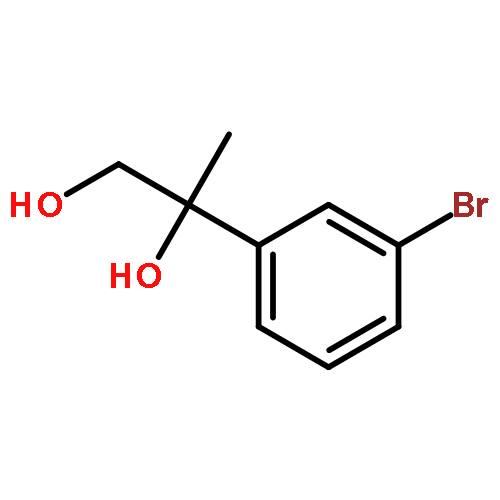
Co-reporter:Yunmi Lee, Rebekka S. Klausen, and Eric N. Jacobsen
Organic Letters 2011 Volume 13(Issue 20) pp:5564-5567
Publication Date(Web):September 15, 2011
DOI:10.1021/ol202300t
A one-pot condensation of isotryptamines and aldehydes that affords enantiomerically enriched 4-substituted tetrahydro-γ-carbolines is reported. The reaction is induced by a chiral thiourea/benzoic acid dual catalyst system. Purification of the N-Boc-protected products by trituration or crystallization provides the optically pure tetrahydro-γ-carboline derivatives in a scalable and highly practical procedure.
Co-reporter:Elizabeth M. Beck, Alan M. Hyde, and Eric N. Jacobsen
Organic Letters 2011 Volume 13(Issue 16) pp:4260-4263
Publication Date(Web):July 25, 2011
DOI:10.1021/ol201608a
The application of chiral sulfinamides and achiral sulfonic acids as a cocatalyst system for enantioselective protonation reactions is described. Structurally simple, easily accessible sulfinamides were found to induce moderate-to-high ee’s in the formation of 2-aryl-substituted cycloalkanones from the corresponding trimethylsilyl enol ethers.
Co-reporter:Adam R. Brown ; Wen-Hsin Kuo
Journal of the American Chemical Society 2010 Volume 132(Issue 27) pp:9286-9288
Publication Date(Web):June 22, 2010
DOI:10.1021/ja103618r
Primary aminothiourea derivatives are shown to catalyze enantioselective alkylation of α-arylpriopionaldehdyes with diarylbromomethane. Evidence for a stepwise, SN1 mechanism in the substitution reaction induced by anion binding to the catalyst is provided by catalyst structure−activity studies, kinetic isotope effects, linear free-energy relationship studies, and competition experiments.
Co-reporter:Robert R. Knowles ; Song Lin
Journal of the American Chemical Society 2010 Volume 132(Issue 14) pp:5030-5032
Publication Date(Web):April 7, 2010
DOI:10.1021/ja101256v
A new thiourea catalyst is reported for the enantioselective cationic polycyclization of hydroxylactams. Both the yield and enantioselectivity of this transformation were found to vary strongly with the identity of a single aromatic residue on a common catalyst framework, with more expansive and polarizable arenes proving optimal. Evidence is presented for a mechanism in which stabilizing cation−π interactions are a principal determinant of enantioselectivity.
Co-reporter:Dr. Gemma E. Veitch ;Dr. Eric N. Jacobsen
Angewandte Chemie 2010 Volume 122( Issue 40) pp:7490-7493
Publication Date(Web):
DOI:10.1002/ange.201003681
Co-reporter:Stephan J. Zuend;Hao Xu;Ye Tao;Matthew G. Woll
Science 2010 Volume 327(Issue 5968) pp:986-990
Publication Date(Web):19 Feb 2010
DOI:10.1126/science.1182826
Co-reporter:Eric N. Jacobsen;David W. C. MacMillan
PNAS 2010 Volume 107 (Issue 48 ) pp:20618-20619
Publication Date(Web):2010-11-30
DOI:10.1073/pnas.1016087107
The use of small-molecule organic catalysts in organic synthesis has flourished over the past decade. Examples of defining
concepts and cutting-edge results are provided in the papers in this Special Feature.
Co-reporter:Robert R. Knowles
PNAS 2010 Volume 107 (Issue 48 ) pp:20678-20685
Publication Date(Web):2010-11-30
DOI:10.1073/pnas.1006402107
Catalysis by neutral, organic, small molecules capable of binding and activating substrates solely via noncovalent interactions—particularly
H-bonding—has emerged as an important approach in organocatalysis. The mechanisms by which such small molecule catalysts induce
high enantioselectivity may be quite different from those used by catalysts that rely on covalent interactions with substrates.
Attractive noncovalent interactions are weaker, less distance dependent, less directional, and more affected by entropy than
covalent interactions. However, the conformational constraint required for high stereoinduction may be achieved, in principle,
if multiple noncovalent attractive interactions are operating in concert. This perspective will outline some recent efforts
to elucidate the cooperative mechanisms responsible for stereoinduction in highly enantioselective reactions promoted by noncovalent
catalysts.
Co-reporter:MeredethA. McGowan;ChristianP. Stevenson;MatthewA. Schiffler ;EricN. Jacobsen Dr.
Angewandte Chemie 2010 Volume 122( Issue 35) pp:6283-6286
Publication Date(Web):
DOI:10.1002/ange.201002177
Co-reporter:Christopher Uyeda;Andreas R. Rötheli ;Dr. Eric N. Jacobsen
Angewandte Chemie 2010 Volume 122( Issue 50) pp:9947-9950
Publication Date(Web):
DOI:10.1002/ange.201005183
Co-reporter:MeredethA. McGowan;ChristianP. Stevenson;MatthewA. Schiffler ;EricN. Jacobsen Dr.
Angewandte Chemie International Edition 2010 Volume 49( Issue 35) pp:6147-6150
Publication Date(Web):
DOI:10.1002/anie.201002177
Co-reporter:Dr. Gemma E. Veitch ;Dr. Eric N. Jacobsen
Angewandte Chemie International Edition 2010 Volume 49( Issue 40) pp:7332-7335
Publication Date(Web):
DOI:10.1002/anie.201003681
Co-reporter:Christopher Uyeda;Andreas R. Rötheli ;Dr. Eric N. Jacobsen
Angewandte Chemie International Edition 2010 Volume 49( Issue 50) pp:9753-9756
Publication Date(Web):
DOI:10.1002/anie.201005183
Co-reporter:Stephan J. Zuend
Journal of the American Chemical Society 2009 Volume 131(Issue 42) pp:15358-15374
Publication Date(Web):September 24, 2009
DOI:10.1021/ja9058958
An experimental and computational investigation of amido-thiourea promoted imine hydrocyanation has revealed a new and unexpected mechanism of catalysis. Rather than direct activation of the imine by the thiourea, as had been proposed previously in related systems, the data are consistent with a mechanism involving catalyst-promoted proton transfer from hydrogen isocyanide to imine to generate diastereomeric iminium/cyanide ion pairs that are bound to catalyst through multiple noncovalent interactions; these ion pairs collapse to form the enantiomeric α-aminonitrile products. This mechanistic proposal is supported by the observation of a statistically significant correlation between experimental and calculated enantioselectivities induced by eight different catalysts (P ≪ 0.01). The computed models reveal a basis for enantioselectivity that involves multiple stabilizing and destabilizing interactions between substrate and catalyst, including thiourea-cyanide and amide-iminium interactions.
Co-reporter:EmilyA. Peterson Dr. ;EricN. Jacobsen Dr.
Angewandte Chemie International Edition 2009 Volume 48( Issue 34) pp:6328-6331
Publication Date(Web):
DOI:10.1002/anie.200902420
Co-reporter:EmilyA. Peterson Dr. ;EricN. Jacobsen Dr.
Angewandte Chemie 2009 Volume 121( Issue 34) pp:6446-6449
Publication Date(Web):
DOI:10.1002/ange.200902420
Co-reporter:Stephan J. Zuend,
Matthew P. Coughlin,
Mathieu P. Lalonde
&
Eric N. Jacobsen
Nature 2009 461(7266) pp:968
Publication Date(Web):2009-10-15
DOI:10.1038/nature08484
Efficient methods for the synthesis of enantioenriched -amino acids — the building blocks of proteins — have been developed, but it remains a challenge to obtain non-natural amino acids. A new catalytic asymmetric method is now reported for the syntheses of highly enantiomerically enriched non-natural amino acids using a simple and robust chiral amido-thiourea catalyst. The method also uses a safer source of cyanide.
Co-reporter:Thomas Belser;EricN. Jacobsen
Advanced Synthesis & Catalysis 2008 Volume 350( Issue 7-8) pp:967-971
Publication Date(Web):
DOI:10.1002/adsc.200800028
Abstract
Chiral salen ligands were incorporated into self-assembled thiolate monolayers (SAMs) on gold colloids. Treatment of the immobilized ligand with Co(OAc)2⋅4 H2O yielded the corresponding [(salen)Co(II)] complex, and aerobic oxidation in the presence of triflic acid afforded the catalytically active [(salen)Co(III)] complex. Functionalized gold colloids with a diameter of 3.4 nm, coated with a mixed monolayer of n-octanethiolates and thiolates with chiral [(salen)Co(III)] end groups were studied as catalysts in the hydrolytic kinetic resolution (HKR) of hexene-1-oxide. Extremely high selectivitiy and significant rate acceleration relative to homogeneous monomeric catalysts were observed. Recovery of the immobilized catalyst was accomplished by simple filtration, and catalyst reoxidation and repeated recycling (seven times) was possible with no loss of reactivity or enantioselectivity.
Co-reporter:MelissaL. Grachan;MatthewT. Tudge Dr. ;Eric.N. Jacobsen
Angewandte Chemie 2008 Volume 120( Issue 8) pp:1491-1494
Publication Date(Web):
DOI:10.1002/ange.200704439
Co-reporter:Clément Mazet Dr. ;EricN. Jacobsen
Angewandte Chemie 2008 Volume 120( Issue 9) pp:1786-1789
Publication Date(Web):
DOI:10.1002/ange.200704461
Co-reporter:MelissaL. Grachan;MatthewT. Tudge Dr. ;Eric.N. Jacobsen
Angewandte Chemie International Edition 2008 Volume 47( Issue 8) pp:1469-1472
Publication Date(Web):
DOI:10.1002/anie.200704439
Co-reporter:Clément Mazet Dr. ;EricN. Jacobsen
Angewandte Chemie International Edition 2008 Volume 47( Issue 9) pp:1762-1765
Publication Date(Web):
DOI:10.1002/anie.200704461
Co-reporter:Emily P. Balskus
Science 2007 Volume 317(Issue 5845) pp:1736-1740
Publication Date(Web):21 Sep 2007
DOI:10.1126/science.1146939
Abstract
Transannular chemical reactions are unparalleled in their ability to generate high degrees of stereochemical and architectural complexity in a single transformation. However, the successful application of this approach in synthesis depends on the ability to predict and control the outcome of the transannular reaction. Use of a chiral catalyst in this context represents an attractive, yet unused, strategy. This report describes a catalytic, asymmetric transannnular Diels-Alder (TADA) reaction that affords polycyclic products in high enantiomeric excess. This catalyst system can also alter the inherent diastereoselectivity of cyclizations with substrates containing chiral centers. Additionally, the catalytic enantioselective TADA has been used as the key step in a total synthesis of the sesquiterpene 11,12-diacetoxydrimane; this route may provide a general approach to the polycyclic carbon framework shared by many terpene natural products.
Co-reporter:Abigail G. Doyle;Eric N. Jacobsen
Angewandte Chemie 2007 Volume 119(Issue 20) pp:
Publication Date(Web):3 APR 2007
DOI:10.1002/ange.200604901
„Entmischt“: Eine dynamische Mischung acyclischer Isomere von Tributylzinnenolaten liefert in einer {Cr(salen)}-katalysierten Alkylierung in hoher Ausbeute und Enantioselektivität Methylketone mit einem quartären Stereozentrum in α-Stellung zur Carbonylgruppe (salen=N,N′-Bis(salicyliden)ethylendiamin-Dianion). Als Mechanismus wird eine Halogenidaktivierung durch einen kationischen Metallkomplex vorgeschlagen.
Co-reporter:Kian L. Tan Dr.;Eric N. Jacobsen
Angewandte Chemie 2007 Volume 119(Issue 8) pp:
Publication Date(Web):9 JAN 2007
DOI:10.1002/ange.200603354
In einer neuen Rolle finden sich chirale Harnstoffkatalysatoren bei der asymmetrischen Allylierung von Acylhydrazonen mit Allylindiumreagentien (siehe Schema; Bz=Benzoyl). Die besten Ergebnisse lieferte der difunktionelle Katalysator 1 mit Lewis-basischer Sulfinamideinheit. Das Beispiel zeigt die erste hoch enantioselektive Addition eines Organometallreagens mithilfe eines chiralen Harnstoffkatalysators.
Co-reporter:Abigail G. Doyle;Eric N. Jacobsen
Angewandte Chemie International Edition 2007 Volume 46(Issue 20) pp:
Publication Date(Web):3 APR 2007
DOI:10.1002/anie.200604901
Straighten out the mixture: A dynamic mixture of acyclic isomers of tributyltin enolates undergoes a {Cr(salen)}-catalyzed alkylation reaction to generate methyl ketones that contain α-carbonyl quaternary stereocenters in high yield and enantioselectivity (salen=N,N′-bis(salicylidene)ethylenediamine dianion). A mechanism is proposed which involves halide activation by a cationic metal complex.
Co-reporter:Kian L. Tan Dr.;Eric N. Jacobsen
Angewandte Chemie International Edition 2007 Volume 46(Issue 8) pp:
Publication Date(Web):9 JAN 2007
DOI:10.1002/anie.200603354
Urea-ka! A new role for urea-based chiral catalysts has been uncovered in the asymmetric allylation of acylhydrazones with allylindium reagents (see scheme; Bz: benzoyl). The best results were obtained using the bifunctional catalyst 1, which bears a Lewis basic sulfinamide. This example represents the first highly enantioselective addition of an organometallic reagent catalyzed by a chiral urea catalyst.
Co-reporter:Mathieu P. Lalonde;Yonggang Chen Dr. Dr.
Angewandte Chemie International Edition 2006 Volume 45(Issue 38) pp:
Publication Date(Web):28 AUG 2006
DOI:10.1002/anie.200602221
Dual activation: The bifunctional primary amine thiourea catalyst 1 promotes the highly enantioselective direct conjugate addition of α-branched aldehydes to nitroalkenes (see scheme). Cooperative activation of both the nucleophile and electrophile allows the use of mild reaction conditions and provides access to a wide variety of adducts with vicinal quaternary and tertiary stereogenic centers (>90 % ee).
Co-reporter:Mark S. Taylor,Eric N. Jacobsen
Angewandte Chemie International Edition 2006 45(10) pp:1520-1543
Publication Date(Web):
DOI:10.1002/anie.200503132
Co-reporter:Mathieu P. Lalonde;Yonggang Chen Dr. Dr.
Angewandte Chemie 2006 Volume 118(Issue 38) pp:
Publication Date(Web):28 AUG 2006
DOI:10.1002/ange.200602221
Doppelte Aktivierung: Der difunktionelle Katalysator 1 – ein primäres Amin und zugleich ein Thioharnstoffderivat – ermöglicht die hoch enantioselektive direkte konjugierte Addition von α-verzweigten Aldehyden an Nitroalkene (siehe Schema) durch kooperative Aktivierung von Nucleophil und Elektrophil. Unter milden Reaktionsbedingungen ist eine Vielzahl an Addukten mit benachbarten quartären und tertiären Stereozentren zugänglich (>90 % ee).
Co-reporter:Mark S. Taylor
Angewandte Chemie 2006 Volume 118(Issue 10) pp:
Publication Date(Web):21 FEB 2006
DOI:10.1002/ange.200503132
Wasserstoffbrücken bestimmen die Struktur eines großen Teils der uns umgebenden Materie. Die ungewöhnlichen Eigenschaften von flüssigem Wasser, die Faltung von Proteinen zu stabilen dreidimensionalen Strukturen, die DNA-Basenpaarung und die Bindung von Liganden an Rezeptoren beruhen auf dieser nichtkovalenten Wechselwirkung. Neben ihrer strukturgebenden Rolle haben Wasserstoffbrücken auch entscheidende Funktionen bei katalytischen Prozessen. Enzyme beschleunigen viele biochemische Reaktionen, indem sie die Elektronendichte eines Elektrophils durch Bildung einer Wasserstoffbrücke verringern und es so für den nucleophilen Angriff aktivieren. In der organischen Synthese hat man das Potenzial dieser Strategie erkannt, und niedermolekulare Verbindungen, insbesondere chirale Wasserstoffbrückendonoren, wurden als Katalysatoren für enantioselektive Synthesen entwickelt. Dieser Aufsatz zeigt, welche strukturellen und mechanistischen Merkmale in wasserstoffbrückenvermittelten Katalyseprozessen zu hohen Enantioselektivitäten führen.
Co-reporter:Eric N. Jacobsen;Andreas Pfaltz;Masakatsu Shibasaki
Advanced Synthesis & Catalysis 2005 Volume 347(Issue 11-13) pp:
Publication Date(Web):19 OCT 2005
DOI:10.1002/adsc.200505313
Co-reporter:Izzat T. Raheem;Eric N. Jacobsen
Advanced Synthesis & Catalysis 2005 Volume 347(Issue 11-13) pp:
Publication Date(Web):19 OCT 2005
DOI:10.1002/adsc.200505230
We report the discovery of asymmetric aza-Baylis–Hillman (ABH) reactions of N-p-nitrobenzenesulfonylimines with methyl acrylate catalyzed by chiral thiourea derivatives. A series of aromatic imines was found to undergo coupling with methyl acrylate with unprecedented levels of enantioselectivity (87–99% ee), albeit only in modest (25–49%) yields. A DABCO-acrylate-imine adduct was isolated as a key intermediate in the ABH reaction. We provide a mechanistic analysis based on the identity of this intermediate as well as kinetic investigations and isotope studies, and propose a rationale for the observed limitations in yield. Synthetic applications of the ABH products are also described.
Co-reporter:Mark S. Taylor, Norihito Tokunaga,Eric N. Jacobsen
Angewandte Chemie International Edition 2005 44(41) pp:6700-6704
Publication Date(Web):
DOI:10.1002/anie.200502277
Co-reporter:Alessandro A. Boezio, Elizabeth R. Jarvo, Brian M. Lawrence,Eric N. Jacobsen
Angewandte Chemie International Edition 2005 44(37) pp:6046-6050
Publication Date(Web):
DOI:10.1002/anie.200502178
Co-reporter:Elizabeth R. Jarvo, Brian M. Lawrence,Eric N. Jacobsen
Angewandte Chemie International Edition 2005 44(37) pp:6043-6046
Publication Date(Web):
DOI:10.1002/anie.200502176
Co-reporter:Mark Gelman and
Angewandte Chemie International Edition 2005 Volume 44(Issue 16) pp:
Publication Date(Web):10 MAR 2005
DOI:10.1002/anie.200463058
New nucleophiles in the arena of asymmetric catalysis, fused and nonfused N-heterocycles undergo conjugate addition to unsaturated imides and enones in the presence of a chiral [(salen)AlIII] complex with generally high enantioselectivities (see scheme; salen=N,N′-bis(salicylaldehydo)ethylenediamine).
Co-reporter:Tehshik P. Yoon
Angewandte Chemie International Edition 2005 Volume 44(Issue 3) pp:
Publication Date(Web):29 DEC 2004
DOI:10.1002/anie.200461814
Approaching “privileged” status? A new and highly effective enantioselective catalyst for the nitro-Mannich reaction was identified by modification of thiourea-based Strecker catalysts (see scheme). Products are obtained in high enantiomeric purity and up to 16:1 diastereoselectivity.
Co-reporter:Tehshik P. Yoon
Angewandte Chemie 2005 Volume 117(Issue 45) pp:
Publication Date(Web):16 NOV 2005
DOI:10.1002/ange.200590150
Co-reporter:Mark S. Taylor;Norihito Tokunaga Dr.
Angewandte Chemie 2005 Volume 117(Issue 41) pp:
Publication Date(Web):21 SEP 2005
DOI:10.1002/ange.200502277
Preiswerte aromatische Substanzen sind die Substrate für hoch enantioselektive acylierende Mannich-Reaktionen, die durch den chiralen H-Brücken-Donor-Thioharnstoff 1 katalysiert werden. Auf diese Art sind nützliche 1-substituierte Dihydroisochinoline zugänglich (siehe Schema; TrocCl=(2,2,2-Trichlorethyl)chlorformiat, TBS=Tributylsilyl), die in enantiomerenangereicherte 1-substituierte Tetrahydroisochinoline überführt werden können.
Co-reporter:Elizabeth R. Jarvo Dr.;Brian M. Lawrence Dr.
Angewandte Chemie 2005 Volume 117(Issue 37) pp:
Publication Date(Web):31 AUG 2005
DOI:10.1002/ange.200502176
Naturstoffe dienen als Vorbilder für die asymmetrische Katalyse in Syntheseansätzen für Colombiasin A und verwandten komplexen Zielverbindungen. Die Entdeckung hoch selektiver Diels-Alder-Reaktionen von Chinonen mit dem neuen, monomeren Schiff-Base-Chrom(III)-Komplex 1 als Katalysator war ein wichtiger Schritt in dem Unterfangen (siehe Schema; TES=Triethylsilyl, DBU=1,8-Diazabicyclo[5.4.0]undec-7-en).
Co-reporter:Alessro A. Boezio Dr.;Elizabeth R. Jarvo Dr.;Brian M. Lawrence Dr.
Angewandte Chemie 2005 Volume 117(Issue 37) pp:
Publication Date(Web):18 AUG 2005
DOI:10.1002/ange.200502178
Eine maßgeschneiderte asymmetrische katalytische Reaktion wurde für den zentralen Chinon-Diels-Alder-Schritt in den Totalsynthesen der Titelverbindungen eingesetzt (siehe Schema für die Synthese von Colombiasin A).
Co-reporter:Mark Gelman and
Angewandte Chemie 2005 Volume 117(Issue 16) pp:
Publication Date(Web):10 MAR 2005
DOI:10.1002/ange.200463058
Ins Rampenlicht der asymmetrischen Katalyse treten kondensierte und nichtkondensierte N-Heterocyclen als neue Nucleophile für die konjugierte Addition an ungesättigte Imide und Enone. In Gegenwart eines chiralen (Salen)aluminium(III)-Komplexes werden überwiegend hohe Enantioselektivititäten erzielt (siehe Schema). Salen=N,N′-Bis (salicylaldehydo)ethylendiamin).
Co-reporter:Tehshik P. Yoon,Eric N. Jacobsen
Angewandte Chemie International Edition 2005 44(45) pp:7327
Publication Date(Web):
DOI:10.1002/anie.200590151
Co-reporter:Eric N. Jacobsen
Advanced Synthesis & Catalysis 2004 Volume 346(Issue 2-3) pp:
Publication Date(Web):29 MAR 2004
DOI:10.1002/adsc.200404009
Co-reporter:Sang Kyun Kim
Angewandte Chemie International Edition 2004 Volume 43(Issue 30) pp:
Publication Date(Web):20 JUL 2004
DOI:10.1002/anie.200460369
If life deals you a lemon …: Low selectivity in the [Co(salen)]-catalyzed kinetic resolution of terminal epoxides with N-Boc sulfonamides is parlayed into an efficient one-pot synthesis of enantiopure amino alcohol derivatives. This provides the foundation for a general route to the title compounds (see scheme; Boc=tert-butoxycarbonyl; Ns=2-nitrobenzenesulfonyl).
Co-reporter:Sang Kyun Kim
Angewandte Chemie 2004 Volume 116(Issue 30) pp:
Publication Date(Web):20 JUL 2004
DOI:10.1002/ange.200460369
Mach's beste draus: Nutze die geringe Selektivität der [Co(salen)]-katalysierten kinetischen Enantiomerentrennung terminaler Epoxide mit N-Boc-Sulfonamiden für eine effiziente Eintopfsynthese enantiomerenreiner Aminoalkoholderivate und gelange so zu einem allgemeinen Weg zu den Titelverbindungen (siehe Schema; Boc=tert-Butoxycarbonyl; Ns=2-Nitrobenzolsulfonyl).
Co-reporter:Mark S. Taylor
PNAS 2004 Volume 101 (Issue 15 ) pp:5368-5373
Publication Date(Web):2004-04-13
DOI:10.1073/pnas.0307893101
This article describes three distinct strategies by which stereochemically complex molecules are synthesized and the ways
asymmetric catalysis can impact on all three. The development of general methods to prepare synthetically useful building
blocks leads to an expanded “chiral pool” of potential starting materials for asymmetric synthesis. The possibility of discovering
new reactions to access new types of building blocks is particularly attractive and serves to help define the frontiers of
the field. Asymmetric catalysis can also be applied to diastereoselective synthesis such that the stereochemistry of the catalyst,
and not that of the substrate, determines the relative configuration of the product. Finally, in reactions where multiple
stereocenters are generated simultaneously or in tandem, catalyst and substrate control can operate in a complementary manner
to achieve one of many possible stereochemical outcomes selectively.
Co-reporter:David E. White, Eric N. Jacobsen
Tetrahedron: Asymmetry 2003 Volume 14(Issue 22) pp:3633-3638
Publication Date(Web):14 November 2003
DOI:10.1016/j.tetasy.2003.09.024
The solvent-free hydrolytic kinetic resolution of terminal epoxides catalyzed by a new oligomeric (salen)Co complex 2 is described. Extremely low loadings of catalyst were used to provide all epoxides examined in good yields and >99% ee under ambient conditions within 24 h.Graphic(S)-EpichlorohydrinC3H5ClO>99% ee (by GC analysis)[α]31D=+32.9 (c 0.998, MeOH)Source of chirality: catalytic hydrolytic kinetic resolution(R)-Propylene oxideC3H6O>99% ee (by HPLC analysis of the 2-napthalenethiol terminal addition derivative)[α]31D=+13.8 (neat)Source of chirality: catalytic hydrolytic kinetic resolution(R)-Styrene oxideC8H8O>99% ee (by HPLC analysis)[α]31D=−23.7 (c 1.01, CHCl3)Source of chirality: catalytic hydrolytic kinetic resolution(S)-Methyl glycidateC4H6O3>99% ee (by GC analysis)[α]31D=−9.8 (c 1.01, MeOH)Source of chirality: catalytic hydrolytic kinetic resolution(S)-Allyl glycidyl etherC6H10O2>99% ee (by GC analysis)[α]31D=+11.6 (c 1.00, EtOH)Source of chirality: catalytic hydrolytic kinetic resolution
Co-reporter:Rebecca T. Ruck
Angewandte Chemie International Edition 2003 Volume 42(Issue 39) pp:
Publication Date(Web):23 SEP 2003
DOI:10.1002/anie.200351591
A concerted ene reaction between silyl enol ethers and simple aldehydes is catalyzed by chiral (Schiff base) CrIII complexes with high stereoselectivity (see scheme). This method leads directly to enantioenriched β-hydroxy silyl enol ether derivatives, which are known to be useful intermediates for the stereocontrolled synthesis of substituted polyol frameworks.
Co-reporter:Rebecca T. Ruck
Angewandte Chemie 2003 Volume 115(Issue 39) pp:
Publication Date(Web):23 SEP 2003
DOI:10.1002/ange.200351591
Eine konzertierte En-Reaktion zwischen Silylenolethern und einfachen Aldehyden wird von chiralen (Schiff-Base-)CrIII-Komplexen mit hoher Stereoselektivität katalysiert (siehe Schema). Auf diesem Weg werden direkt enantioangereicherte β-Hydroxysilylenolether-Derivative erhalten, die bekanntlich nützliche Intermediate für die stereokontrollierte Synthese von substituierten Polyolgerüsten sind.
Co-reporter:Eric N. Jacobsen
Advanced Synthesis & Catalysis 2002 Volume 344(Issue 1) pp:
Publication Date(Web):28 FEB 2002
DOI:10.1002/1615-4169(200201)344:1<1::AID-ADSC1>3.0.CO;2-A
Co-reporter:Steven N. Goodman;Eric N. Jacobsen
Advanced Synthesis & Catalysis 2002 Volume 344(Issue 9) pp:
Publication Date(Web):28 OCT 2002
DOI:10.1002/1615-4169(200210)344:9<953::AID-ADSC953>3.0.CO;2-A
We report an improved synthesis of α,β-unsaturated imides, a class of compounds that has been identified as broadly useful in (salen)aluminum-catalyzed asymmetric conjugate addition reactions. An efficient, scaleable procedure for the synthesis of phosphonate imide reagent 4 is described, as well as a DBU-mediated Horner WadsworthEmmons reaction that affords the target compounds in high yield and (E)-selectivity and with good functional group tolerance.
Co-reporter:Karl Gademann Dr.;David E. Chavez Dr.
Angewandte Chemie International Edition 2002 Volume 41(Issue 16) pp:
Publication Date(Web):21 AUG 2002
DOI:10.1002/1521-3773(20020816)41:16<3059::AID-ANIE3059>3.0.CO;2-I
Straightforward access to useful synthetic intermediates is provided by this new method. Simple, α,β-unsaturated aldehydes are excellent substrates in the hetero-Diels–Alder reaction with inverse electron demand, catalyzed by CrIII–Schiff base complexes (see scheme; R1, R2=alkyl or aryl) in the presence of 4-Å molecular sieves and no solvent. The resulting dihydropyrans are obtained in high enantio- (89–98 % ee) and diastereoselectivity (>95 % de) and yield (40–95 %).
Co-reporter:Joseph M. Ready
Angewandte Chemie International Edition 2002 Volume 41(Issue 8) pp:
Publication Date(Web):16 APR 2002
DOI:10.1002/1521-3773(20020415)41:8<1374::AID-ANIE1374>3.0.CO;2-8
Prepared by a high yield, chromatography-free route, “oligosalen” exists as a mixture of dimer, trimer, and tetramer (see picture). Derived catalyst systems display remarkable activity and selectivity in the asymmetric ring-opening of epoxides, with turnover numbers exceeding 100 000 in some cases.
Co-reporter:Steven N. Goodman Dr.
Angewandte Chemie 2002 Volume 114(Issue 24) pp:
Publication Date(Web):12 DEC 2002
DOI:10.1002/ange.200290021
Den CO-Druck niedrig halten – dies ist wichtig bei einer neuen Methode für die Carbonylierung terminaler Epoxide. Hierbei werden enantiomerenreine β-Hydroxymorpholinylamide direkt gebildet [Gl. (1)]; diese Synthesebausteine lassen sich in einer Reihe von Acyltransferreaktionen einsetzen. Ein präparativ besonders nützliches Beispiel ist die kurze Synthese von δ-Hydroxy-β-oxoestern, Schlüsselintermediaten für die Herstellung von HMG-CoA-Reduktase-Inhibitoren aus der Klasse der Statine.
Co-reporter:Karl Gademann Dr.;David E. Chavez Dr.
Angewandte Chemie 2002 Volume 114(Issue 16) pp:
Publication Date(Web):21 AUG 2002
DOI:10.1002/1521-3757(20020816)114:16<3185::AID-ANGE3185>3.0.CO;2-S
Unkomplizierter Zugang zu präparativ nützlichen Zwischenprodukten: Einfache α,β-ungesättigte Aldehyde sind für die asymmetrische Hetero-Diels-Alder-Reaktion mit inversem Elektronenbedarf ausgezeichnete Substrate, wenn die Reaktion durch einen CrIII-Schiff-Basen-Komplex katalysiert wird und ohne Lösungsmittel in Gegenwart eines 4-Å-Molekularsiebs abläuft (siehe Schema; R1, R2=Alkyl oder Aryl). Dihydropyrane wurden mit guten bis sehr guten Ausbeuten (40–95 %) sowie hohen Enantio- (89–98 % ee) und Diasteroselektivitäten (>95 % de) hergestellt.
Co-reporter:Joseph M. Ready
Angewandte Chemie 2002 Volume 114(Issue 8) pp:
Publication Date(Web):16 APR 2002
DOI:10.1002/1521-3757(20020415)114:8<1432::AID-ANGE1432>3.0.CO;2-6
Ohne Chromatographie-Schritte liefert die Synthese von „Oligosalen“ eine Mischung aus Dimeren, Trimeren und Tetrameren in hoher Ausbeute (siehe Bild). Katalysatorsysteme auf der Basis dieses Oligosalens zeigen eine bemerkenswerte Aktivität und Selektivität bei der asymmetrischen Ringöffnung von Epoxiden mit zum Teil mehr als 100 000 Katalysezyklen.
Co-reporter:Steven N. Goodman Dr.
Angewandte Chemie International Edition 2002 Volume 41(Issue 24) pp:
Publication Date(Web):12 DEC 2002
DOI:10.1002/anie.200290022
Keeping the CO pressure down is a crucial, practical feature of the new methodology for the carbonylation of terminal epoxides. β-Hydroxy morpholinyl amides are generated directly in optically pure form [Eq. (1)], and these building blocks can be applied in a variety of acyl transfer reactions. A useful illustration is provided in the concise synthesis of δ-hydroxy-β-ketoesters, key intermediates for the preparation of the statin class of HMG-CoA reductase inhibitors.
Co-reporter:Julius T. Su;Petr Vachal;Eric N. Jacobsen
Advanced Synthesis & Catalysis 2001 Volume 343(Issue 2) pp:
Publication Date(Web):5 MAR 2001
DOI:10.1002/1615-4169(20010226)343:2<197::AID-ADSC197>3.0.CO;2-8
Co-reporter:John M. Keith;Jay F. Larrow;Eric N. Jacobsen
Advanced Synthesis & Catalysis 2001 Volume 343(Issue 1) pp:
Publication Date(Web):6 FEB 2001
DOI:10.1002/1615-4169(20010129)343:1<5::AID-ADSC5>3.0.CO;2-I
This review provides a critical analysis of catalytic kinetic resolution reactions from a practical perspective, asking the question of when, if ever, is kinetic resolution the best option for the synthesis of an optically active target. A series of crucial conditions are identified, and it is postulated that if all of them are met, then indeed kinetic resolution can be highly practical. A variety of catalytic kinetic resolution processes are evaluated in the context of these criteria, with particular emphasis on catalyst availability, substrate scope, availability of the racemic substrate and of alternative methods for accessing enantiopure substrate or product, and key experimental considerations. It is found that several catalytic systems have been developed that offer almost unbeatable methods for the preparation of useful chiral building blocks.
Co-reporter:David E. Chavez
Angewandte Chemie International Edition 2001 Volume 40(Issue 19) pp:
Publication Date(Web):2 OCT 2001
DOI:10.1002/1521-3773(20011001)40:19<3667::AID-ANIE3667>3.0.CO;2-6
The most selective protein phosphatase inhibitor identified to date, fostriecin was synthesized in a highly convergent manner by a chiral building block approach (see picture). Three of the four stereocenters were introduced by using catalytic methods, including novel and practical applications of asymmetric hetero-Diels–Alder (HDA) and hydrolytic kinetic resolution reactions.
Co-reporter:David E. Chavez
Angewandte Chemie 2001 Volume 113(Issue 19) pp:
Publication Date(Web):2 OCT 2001
DOI:10.1002/1521-3757(20011001)113:19<3779::AID-ANGE3779>3.0.CO;2-C
Der bisher selektivste Proteinphosphatase-Inhibitor, Fostriecin, wurde auf hochkonvergente Weise aus chiralen Bausteinen synthetisiert (siehe Bild). Drei der vier Stereozentren wurden mit katalytischen Methoden eingeführt, wobei neue und praktische Varianten der asymmetrischen Hetero-Diels-Alder(HDA)-Reaktion und der hydrolytischen kinetischen Racematspaltung angewendet wurden.
Co-reporter:Rolf Breinbauer Dr.;Eric N. Jacobsen
Angewandte Chemie 2000 Volume 112(Issue 20) pp:
Publication Date(Web):13 OCT 2000
DOI:10.1002/1521-3757(20001016)112:20<3750::AID-ANGE3750>3.0.CO;2-T
Co-reporter:Matthew S. Sigman Dr.;Petr Vachal
Angewandte Chemie 2000 Volume 112(Issue 7) pp:
Publication Date(Web):4 APR 2000
DOI:10.1002/(SICI)1521-3757(20000403)112:7<1336::AID-ANGE1336>3.0.CO;2-Z
Co-reporter:Matthew B. Francis
Angewandte Chemie 1999 Volume 111(Issue 7) pp:
Publication Date(Web):26 MAR 1999
DOI:10.1002/(SICI)1521-3757(19990401)111:7<987::AID-ANGE987>3.0.CO;2-9
Neue Leitstrukturen für Olefinepoxidierungskatalysatoren wurden ausgehend von einer kombinatorischen Bibliothek aus 5760 harzgebundenen Metall-Ligand-Komplexen (siehe mikroskopische Aufnahme) erhalten. Bei Verwendung von H2O2 als Oxidationsmittel lieferten Eisenkomplexe glatt das Epoxidierungsprodukt. Parallel hergestellte Bibliotheken wurden verwendet, um die für eine hohe katalytische Aktivität der Komplexe erforderlichen Ligandeneigenschaften zu bestimmen und auch enantioselektive Katalysatoren aufzufinden (siehe Gleichung).
Co-reporter:Alexer G. Dossetter;Timothy F. Jamison
Angewandte Chemie International Edition 1999 Volume 38(Issue 16) pp:
Publication Date(Web):6 AUG 1999
DOI:10.1002/(SICI)1521-3773(19990816)38:16<2398::AID-ANIE2398>3.0.CO;2-E
Even moderately nucleophilic dienes react with simple aldehydes in the presence of a new CrIII catalyst in a hetero-Diels–Alder reaction [Eq. (1)]. Tetrahydropyranyl products with up to three stereogenic centers are generated in near-perfect diastereoselectivities and with greater than 90 % ee (99 % ee for the example shown). TBAF=tetrabutylammonium fluoride; TBS=tert-butyldimethylsilyl; TES=triethylsilyl.
Co-reporter:Michael H. Wu;Karl B. Hansen
Angewandte Chemie International Edition 1999 Volume 38(Issue 13‐14) pp:
Publication Date(Web):12 JUL 1999
DOI:10.1002/(SICI)1521-3773(19990712)38:13/14<2012::AID-ANIE2012>3.0.CO;2-H
The intramolecular cyclization of epoxy alcohols was catalyzed with excellent regio- and enantiocontrol by a [CoIII(salen)] complex. High endo selectivity was observed for the enantioselective cyclization of terminal epoxy alcohols [Eq. (a)], while the reaction of meso substrates produced novel cyclic and bicyclic ethers in good yields and high enantiopurity. TBME=tert-butyl methyl ether.
Co-reporter:Matthew B. Francis
Angewandte Chemie International Edition 1999 Volume 38(Issue 7) pp:
Publication Date(Web):26 MAR 1999
DOI:10.1002/(SICI)1521-3773(19990401)38:7<937::AID-ANIE937>3.0.CO;2-O
New lead structures for olefin oxidation catalysts have been obtained from a combinatorial library of 5760 metal–ligand complexes (see the microscopy picture). Iron complexes led to clean epoxide product formation using H2O2 as the terminal oxidant. Parallel libraries were used to determine ligand features important for high catalytic activity and to identify enantioselective catalyst structures (see the Equation).
Co-reporter:Alexer G. Dossetter;Timothy F. Jamison;Eric N. Jacobsen
Angewandte Chemie 1999 Volume 111(Issue 16) pp:
Publication Date(Web):6 AUG 1999
DOI:10.1002/(SICI)1521-3757(19990816)111:16<2549::AID-ANGE2549>3.0.CO;2-H
Selbst schwach nucleophile Diene reagieren mit einfachen Aldehyden unter Verwendung eines neuen CrIII-Katalysators in einer Hetero-Diels-Alder-Reaktion [Gl. (1)]. Dabei werden Tetrahydropyrane, die bis zu drei stereogene Zentren enthalten, mit nahezu perfekter Diastereoselektivität und >90 % ee (99 % ee für das gezeigte Beispiel) erhalten. MS = Molekularsieb; TBAF = Tetrabutylammoniumfluorid; TBS = tert-Butyldimethylsilyl; TES = Triethylsilyl.
Co-reporter:Michael H. Wu;Karl B. Hansen
Angewandte Chemie 1999 Volume 111(Issue 13‐14) pp:
Publication Date(Web):12 JUL 1999
DOI:10.1002/(SICI)1521-3757(19990712)111:13/14<2167::AID-ANGE2167>3.0.CO;2-F
Mit exzellenter Regio-und Enantiokontrolle katalysieren [(salen)CoIII]-Komplexe die intramolekulare Cyclisierung von Epoxyalkoholen. Terminale Epoxyalkohole werden mit hoher endo-Selektivität enantioselektiv cyclisiert [Gl. (a)]. Die Reaktion von meso-Substraten führte in guten Ausbeuten zu neuartigen cyclischen und bicyclischen Ethern mit hoher Enantiomerenreinheit. TBME=tert-Butylmethylether.
Co-reporter:D. Allen Annis;Olivier Helluin
Angewandte Chemie 1998 Volume 110(Issue 13‐14) pp:
Publication Date(Web):12 MAR 1999
DOI:10.1002/(SICI)1521-3757(19980703)110:13/14<2010::AID-ANGE2010>3.0.CO;2-3
Unterschiedliche biologische Aktivitäten weisen die diastereomeren, aus chiralen Aminoalkoholen synthetisierten cyclischen Peptide wie 1 (nur ein Diastereomer gezeigt) mit der universalen Zellerkennungssequenz Arg-Gly-Asp aufgrund verschiedener Konformationen auf. Die erforderlichen Aminoalkohole wurden aus polymergebunden Epoxiden durch enantioselektive Ringöffnung erhalten, was den Nutzen enantioselektiver Katalysereaktionen für die kombinatorische Synthese unterstreicht.
Co-reporter:D. Allen Annis;Olivier Helluin
Angewandte Chemie International Edition 1998 Volume 37(Issue 13‐14) pp:
Publication Date(Web):17 DEC 1998
DOI:10.1002/(SICI)1521-3773(19980803)37:13/14<1907::AID-ANIE1907>3.0.CO;2-A
Dramatic differences in biological activity as a result of different conformations are exhibited by diastereomeric cyclic peptides such as 1 (only one diastereoisomer is shown) containing the universal cell-recognition sequence Arg-Gly-Asp, which were synthesized on amino alcohol templates. The amino alcohols were prepared by enantioselective ring opening of polymer-bound epoxides, a reaction that exemplifies the usefulness of enantioselective catalysis in combinatorial synthesis.
Co-reporter:Nathaniel S. Finney;Paul J. Pospisil;Sukbok Chang;Michael Palucki;Reed G. Konsler;Karl B. Hansen; Eric N. Jacobsen
Angewandte Chemie 1997 Volume 109(Issue 16) pp:
Publication Date(Web):31 JAN 2006
DOI:10.1002/ange.19971091614
Co-reporter:Bridget D. Brandes, Eric N. Jacobsen
Tetrahedron: Asymmetry 1997 Volume 8(Issue 23) pp:3927-3933
Publication Date(Web):11 December 1997
DOI:10.1016/S0957-4166(97)00568-5
3-Chlorostyrene oxide was prepared in >99% ee employing the (salen)Co-catalyzed hydrolytic kinetic resolution (HKR) reaction. The HKR was performed successfully on both racemic and enantiomerically enriched epoxides, the latter was obtained via (salen)Mn-catalyzed asymmetric epoxidation reactions.Graphic
Co-reporter:Paul J. Pospisil;Douglas H. Carsten; Eric N. Jacobsen
Chemistry - A European Journal 1996 Volume 2(Issue 8) pp:
Publication Date(Web):21 FEB 2006
DOI:10.1002/chem.19960020812
The relationship between catalyst structure and enantioselectivity in the asymmetric epoxidation of unfunctionalized olefins by a series of chiral Mn(sa1en) complexes (1 – 10) was examined. The X-ray structures of 5-coordinate complexes 5, 8, of 6-coordinate 9 ([6,6′= -tBu; 4,4 = -tBu]+ClO4−), and 10 (6,6′= -tBu; 4,4′=-Br) were determined. Catalysts 1 – 9 were derived from (R,R)-1, 2 diaminocyclohexane and catalyst 10 from (S,S)-1,2-diphenylethylenediamine. Catalysts 1–9 differ in the stereoelectronic substitution of the ortho (6,6) and para (4,4) positions of the salicylidene moiety. A comparison between structures 5, 8, and 9 reveals that the ligand geometry around the metal center and the chiral diimine backbone remains remarkably constant in both five- and six-coordinate cyclohexanediamine-derived complexes; in contrast, the salicylidene regions of the complexes display a wide range of conformations. The asymmetric epoxidation of indene and 6-cyano-2,2-dimethylchromene with NaOCl catalyzed by complexes 1 – 10 was effected. Systematically increasing the steric bulk on the ortho and then the para position in the order 1 (6,6′ = -H; 4,4′ = -H),2(6,6′ = -CH3; 4,4′ = -CH3),3(6,6′=-tBu;4,4′=-H),4(6,6′=-tBu; 4,4′ =-CH3), 5 (6,6′=-tBu; 4,4′=-tBu), and 6 (6,6′=-tBu; 4,4′= -trityl), and electronically modifying the para substituents in 7 (6,6 = -tBu; 4,4 =-OMe) and 8 (6.6′ = -tBu; 4,4′=-OTIPS) resulted in enhanced enantioselectivities of the desired epoxides. The conformational variations observed in the solid state are likely to reflect accessible solution conformations and may help explain the high levels of stereoinduction obtained with these catalysts in the asymmetric epoxidation of unfunctionalized olefins.
Co-reporter:Giulia Bergonzini, Corinna S. Schindler, Carl-Johan Wallentin, Eric N. Jacobsen and Corey R. J. Stephenson
Chemical Science (2010-Present) 2014 - vol. 5(Issue 1) pp:NaN116-116
Publication Date(Web):2013/08/23
DOI:10.1039/C3SC52265B
The enantioselective oxidative C–H functionalization of tetrahydroisoquinoline derivatives is achieved through the merger of photoredox and asymmetric anion-binding catalysis. This combination of two distinct catalysis concepts introduces a potentially general approach to asymmetric transformations in oxidative photocatalysis.
Co-reporter:Cheyenne S. Brindle, Charles S. Yeung and Eric N. Jacobsen
Chemical Science (2010-Present) 2013 - vol. 4(Issue 5) pp:NaN2104-2104
Publication Date(Web):2013/03/14
DOI:10.1039/C3SC50410G
Highly enantioselective vicinal iodoamination of olefins is accomplished through the iodocyclization of alkenyl trichloroacetimidates catalysed by a new chiral Schiff-base urea derivative. The resulting products are converted readily to a variety of polyfunctional amine-containing chiral building blocks.







![2H-Pyran, tetrahydro-2-[[(4Z)-5-iodo-4-penten-1-yl]oxy]-](http://img.cochemist.com/ccimg/924900/924889-40-7.png)
![2H-Pyran, tetrahydro-2-[[(4Z)-5-iodo-4-penten-1-yl]oxy]-](http://img.cochemist.com/ccimg/924900/924889-40-7_b.png)
![Silane, [(1-methoxy-2-methyl-1-propenyl)oxy]tris(1-methylethyl)-](http://img.cochemist.com/ccimg/868100/868070-20-6.png)
![Silane, [(1-methoxy-2-methyl-1-propenyl)oxy]tris(1-methylethyl)-](http://img.cochemist.com/ccimg/868100/868070-20-6_b.png)



![TERT-BUTYL 3-METHYLIDENE-8-AZABICYCLO[3.2.1]OCTANE-8-CARBOXYLATE](http://img.cochemist.com/ccimg/273300/273207-57-1.png)
![TERT-BUTYL 3-METHYLIDENE-8-AZABICYCLO[3.2.1]OCTANE-8-CARBOXYLATE](http://img.cochemist.com/ccimg/273300/273207-57-1_b.png)
![1H-Imidazole, 2,2'-[5-(1,1-dimethylethyl)-1,3-phenylene]bis[4,5-dihydro-](http://img.cochemist.com/ccimg/225600/225500-86-7.png)
![1H-Imidazole, 2,2'-[5-(1,1-dimethylethyl)-1,3-phenylene]bis[4,5-dihydro-](http://img.cochemist.com/ccimg/225600/225500-86-7_b.png)








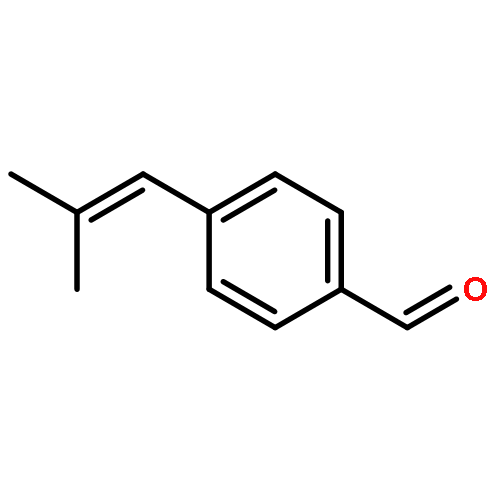


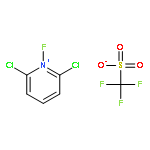
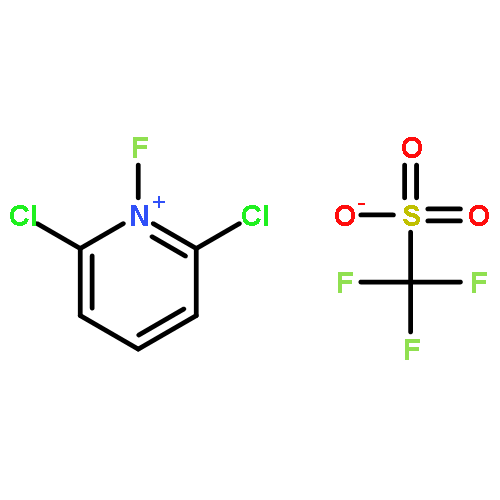


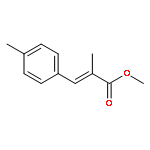

![2-Propen-1-ol, 3-[4-(trifluoromethyl)phenyl]-, (2E)-](/data/chemimg/115900/125617-18-7.png)
![2-Propen-1-ol, 3-[4-(trifluoromethyl)phenyl]-, (2E)-](/data/chemimg/115900/125617-18-7_b.png)



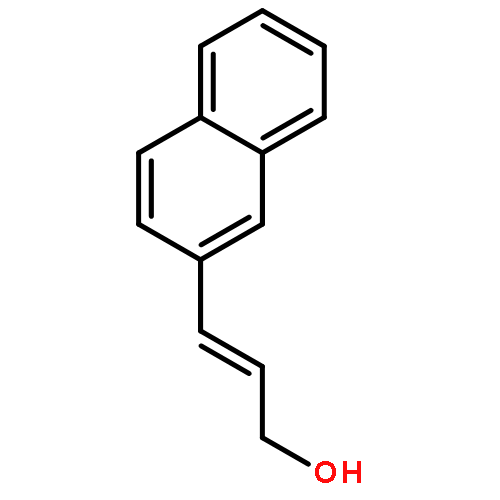






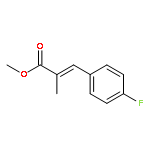
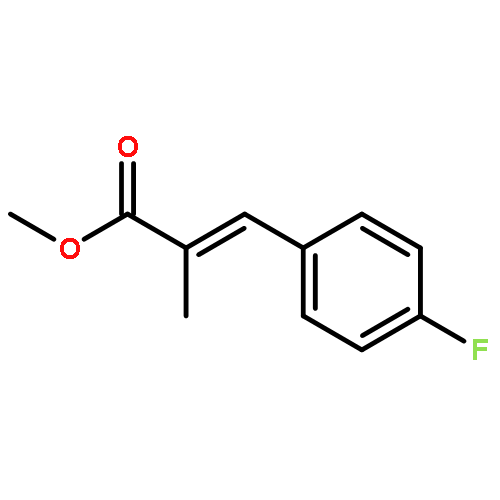


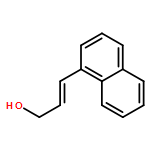




![Benzeneacetic acid, a-[(methylsulfonyl)oxy]-, methyl ester](http://img.cochemist.com/ccimg/82100/82027-15-4.png)
![Benzeneacetic acid, a-[(methylsulfonyl)oxy]-, methyl ester](http://img.cochemist.com/ccimg/82100/82027-15-4_b.png)


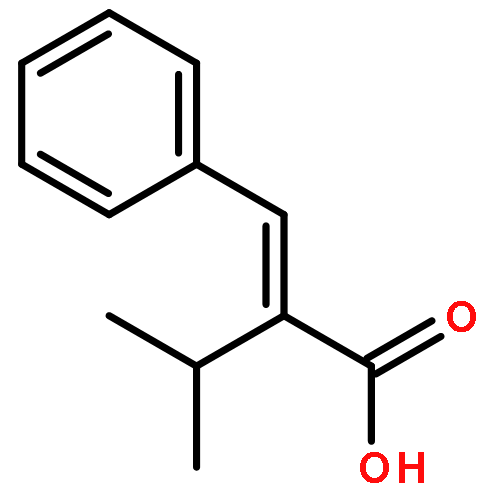






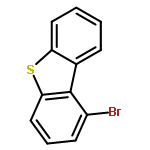
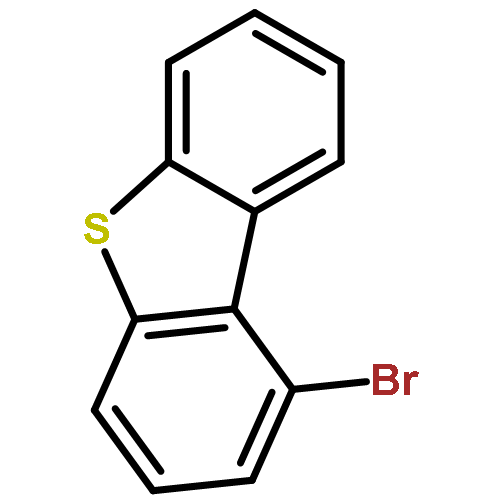
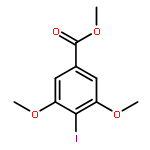
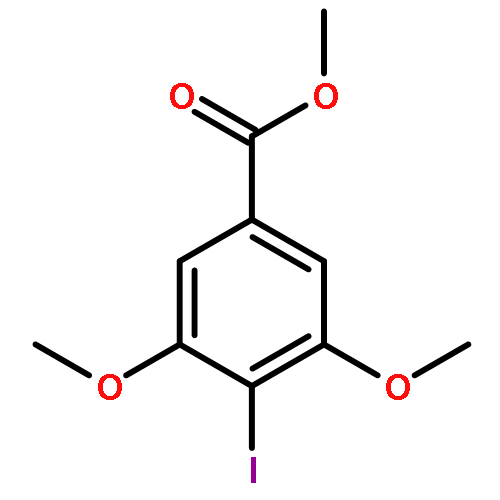



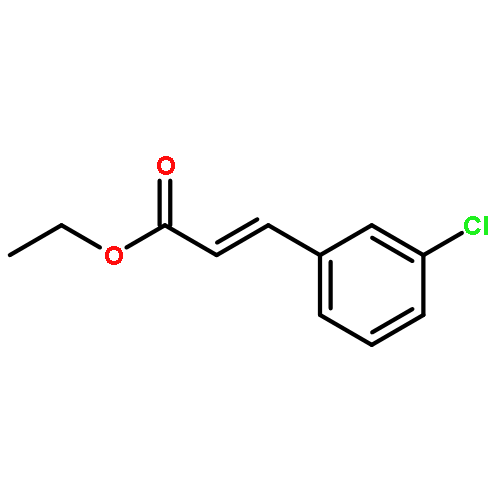
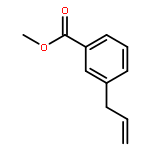
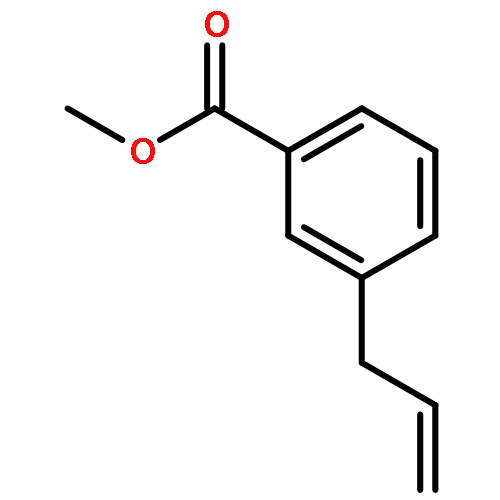
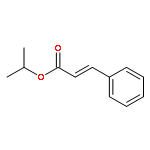

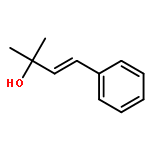



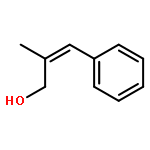
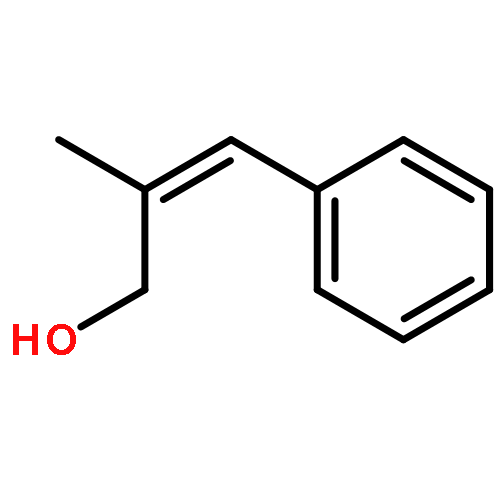
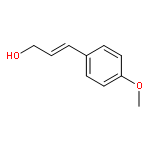
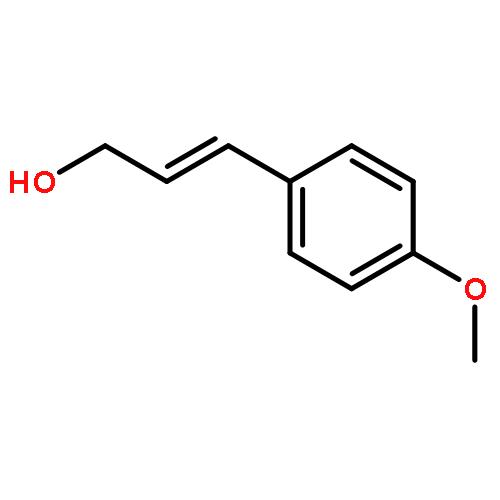


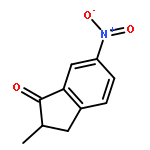
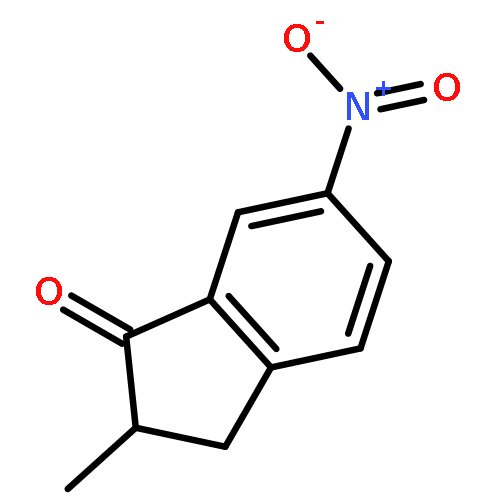





![[2-tert-butoxy-1-(tert-butoxycarbonyl)-2-oxoethylidene]diazenium](http://img.cochemist.com/ccimg/35300/35207-75-1.png)
![[2-tert-butoxy-1-(tert-butoxycarbonyl)-2-oxoethylidene]diazenium](http://img.cochemist.com/ccimg/35300/35207-75-1_b.png)





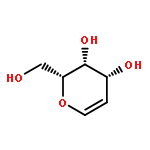
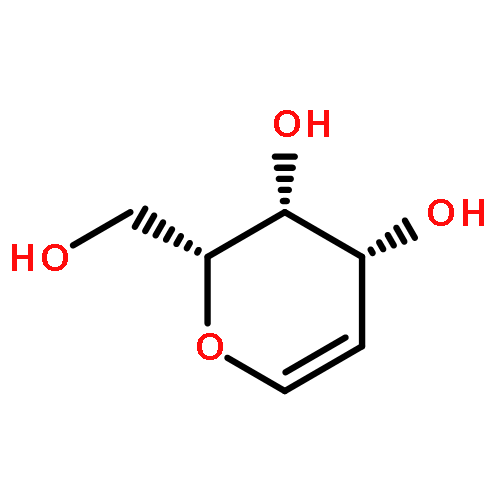
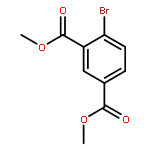
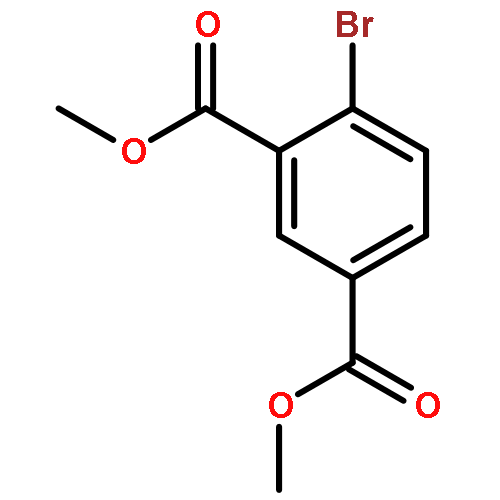









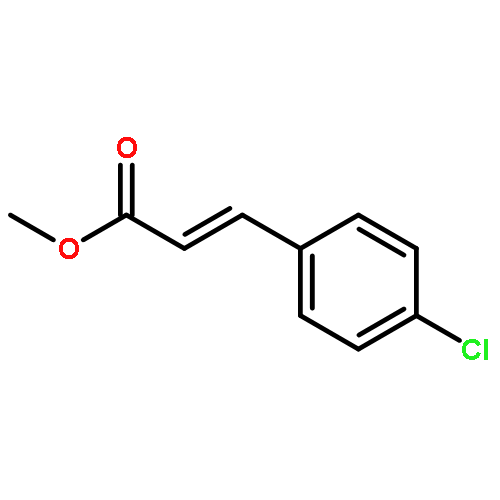
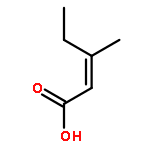
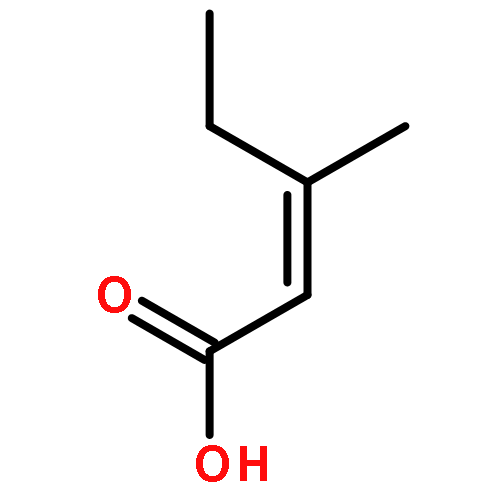
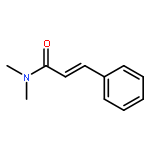




![4-CHLORO-6-METHYLTHIENO[3,2-D]PYRIMIDINE](http://img.cochemist.com/ccimg/7700/7622-21-1.png)
![4-CHLORO-6-METHYLTHIENO[3,2-D]PYRIMIDINE](http://img.cochemist.com/ccimg/7700/7622-21-1_b.png)
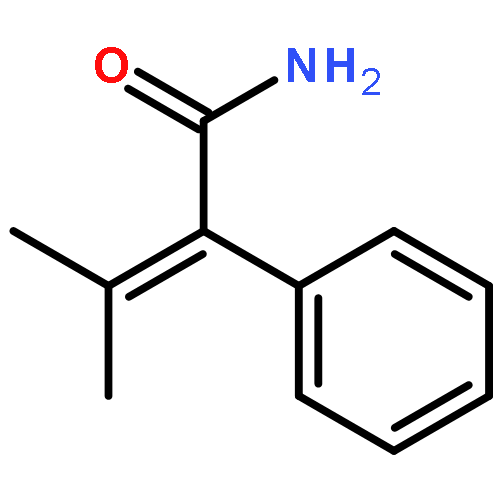
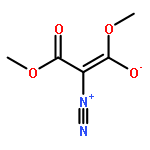
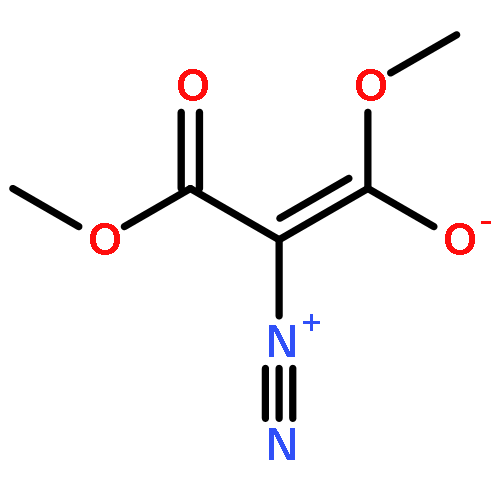
![2-[(4-methoxyphenyl)methyl]benzoic Acid](http://img.cochemist.com/ccimg/5400/5398-17-4.png)
![2-[(4-methoxyphenyl)methyl]benzoic Acid](http://img.cochemist.com/ccimg/5400/5398-17-4_b.png)


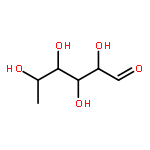
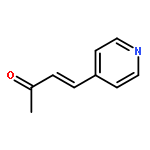
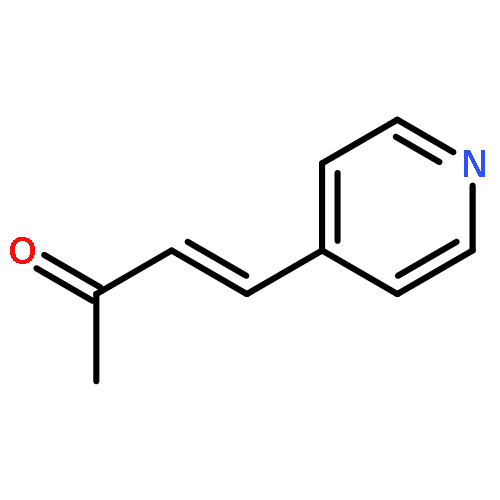
![PYRIDINE, 4-[(1E)-2-PHENYLETHENYL]-](http://img.cochemist.com/ccimg/5100/5097-93-8.png)
![PYRIDINE, 4-[(1E)-2-PHENYLETHENYL]-](http://img.cochemist.com/ccimg/5100/5097-93-8_b.png)



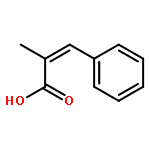
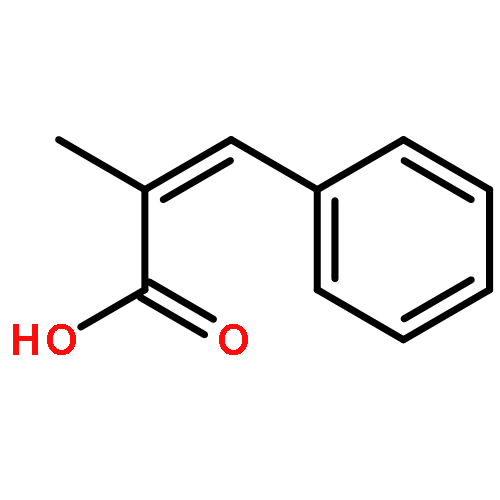
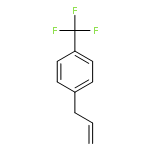
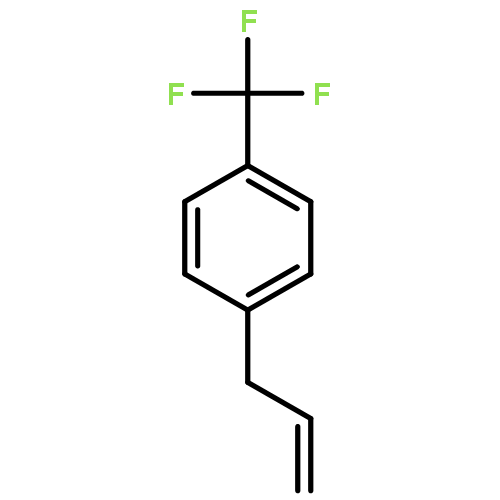










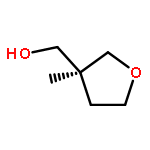
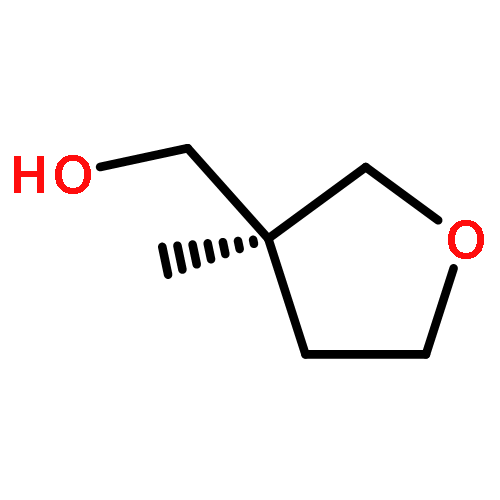
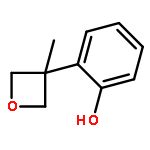
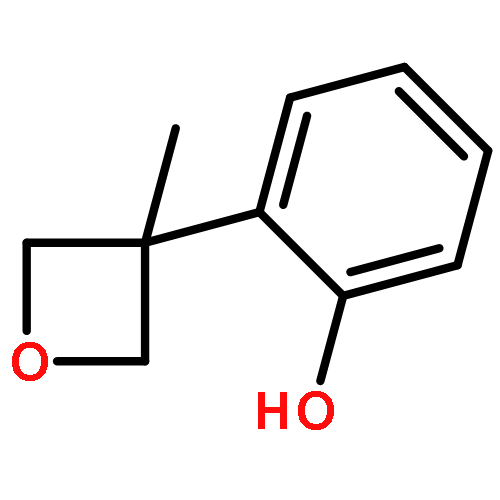














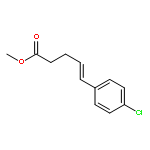
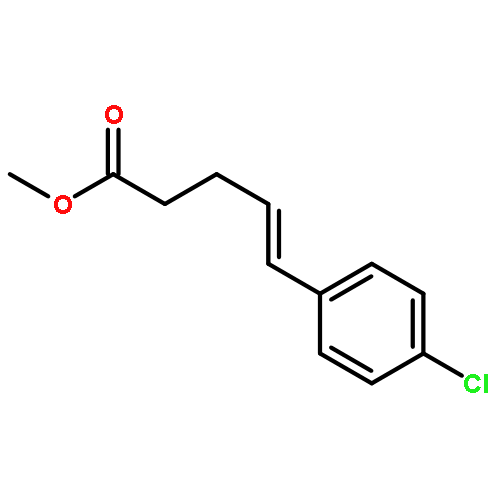
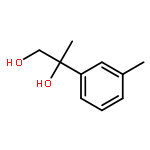
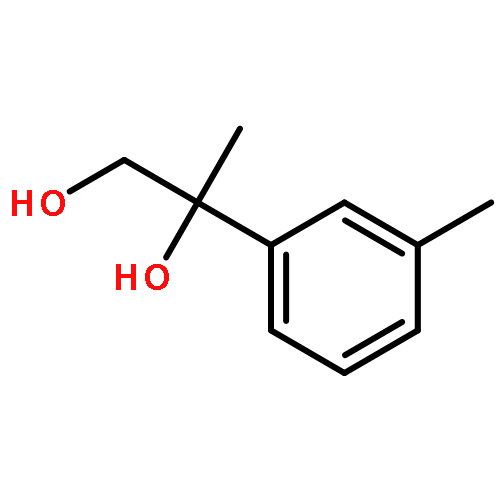


![Silane, trimethyl[[2-(4-methylphenyl)-1-cyclohexen-1-yl]oxy]-](http://img.cochemist.com/ccimg/851400/851332-01-9.png)
![Silane, trimethyl[[2-(4-methylphenyl)-1-cyclohexen-1-yl]oxy]-](http://img.cochemist.com/ccimg/851400/851332-01-9_b.png)
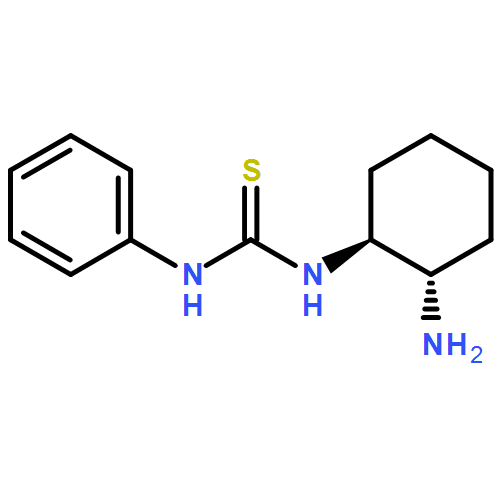
![UREA, N-[(1S,2S)-2-AMINOCYCLOHEXYL]-N'-[3,5-BIS(TRIFLUOROMETHYL)PHENYL]-](http://img.cochemist.com/ccimg/850100/850079-15-1.png)
![UREA, N-[(1S,2S)-2-AMINOCYCLOHEXYL]-N'-[3,5-BIS(TRIFLUOROMETHYL)PHENYL]-](http://img.cochemist.com/ccimg/850100/850079-15-1_b.png)
![CYCLOHEXANECARBONITRILE, 1-[(2E)-3-PHENYL-2-PROPENYL]-](http://img.cochemist.com/ccimg/850100/850011-75-5.png)
![CYCLOHEXANECARBONITRILE, 1-[(2E)-3-PHENYL-2-PROPENYL]-](http://img.cochemist.com/ccimg/850100/850011-75-5_b.png)
![Dinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin,4-hydroxy-2,6-bis[2,4,6-tris(1-methylethyl)phenyl]-, 4-oxide, (11bR)-](/data/chemimg/115800/791616-63-2.png)
![Dinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin,4-hydroxy-2,6-bis[2,4,6-tris(1-methylethyl)phenyl]-, 4-oxide, (11bR)-](/data/chemimg/115800/791616-63-2_b.png)
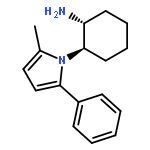
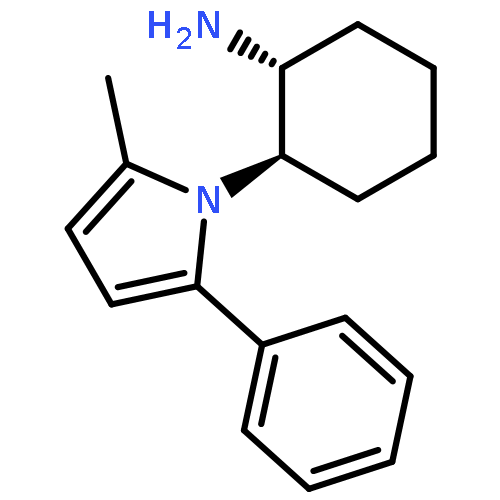
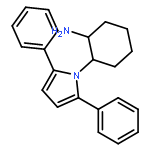
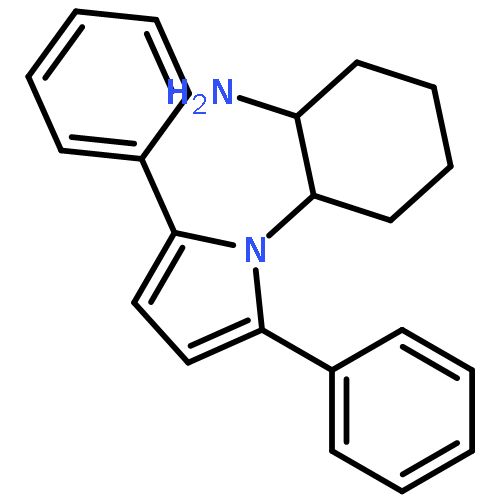
![2-Furanmethanol, 5-[[[(1,1-dimethylethyl)dimethylsilyl]oxy]methyl]-](http://img.cochemist.com/ccimg/753100/753004-76-1.png)
![2-Furanmethanol, 5-[[[(1,1-dimethylethyl)dimethylsilyl]oxy]methyl]-](http://img.cochemist.com/ccimg/753100/753004-76-1_b.png)
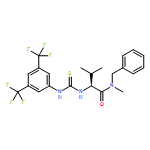
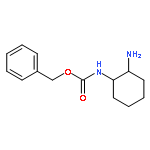
![(2S)-2-[[[[(1R,2R)-2-Aminocyclohexyl]amino]thioxomethyl]amino]-N-3,3-trimethyl-N-(phenylmethyl)butanamide, (S)-2-[[[[(1R,2R)-2-Aminocyclohexyl]amino]thioxomethyl]amino]-N-benzyl-N-3,3-trimethylbutanamide](/data/chemimg/1589000/479423-21-7.png)
![(2S)-2-[[[[(1R,2R)-2-Aminocyclohexyl]amino]thioxomethyl]amino]-N-3,3-trimethyl-N-(phenylmethyl)butanamide, (S)-2-[[[[(1R,2R)-2-Aminocyclohexyl]amino]thioxomethyl]amino]-N-benzyl-N-3,3-trimethylbutanamide](/data/chemimg/1589000/479423-21-7_b.png)


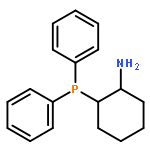

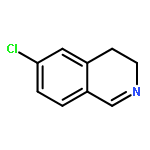
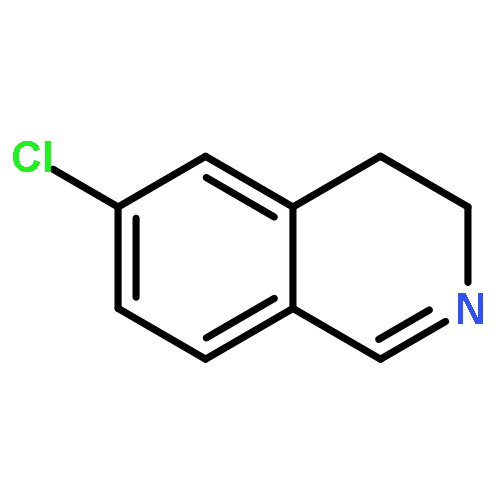






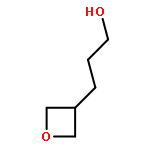
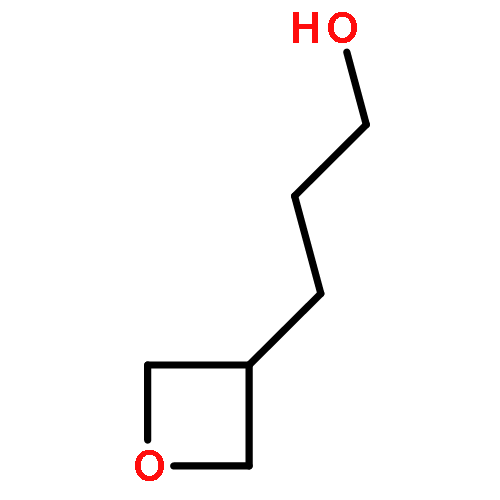
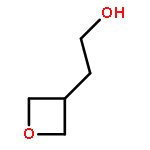
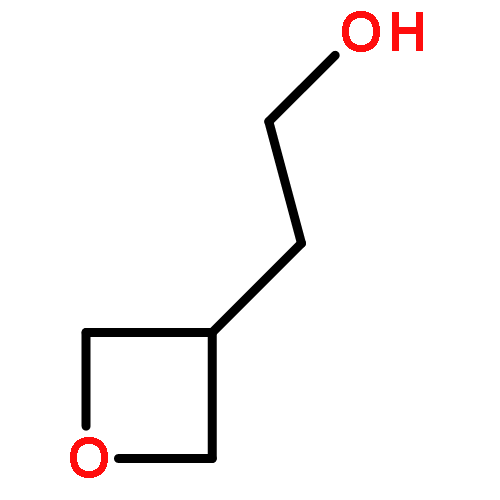
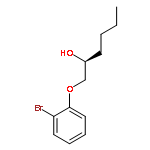
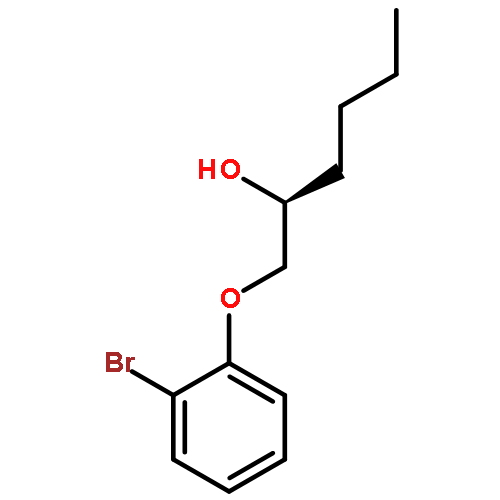
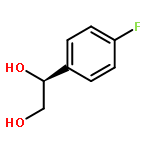
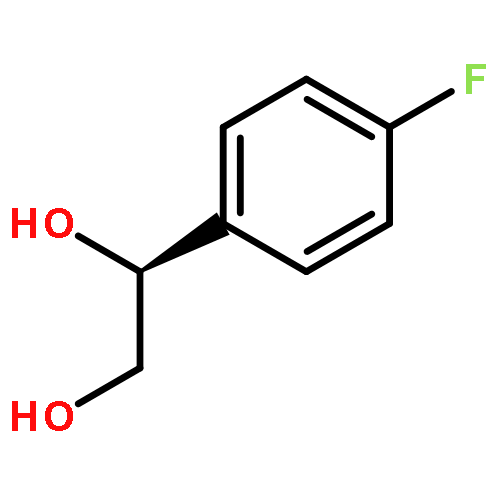


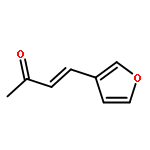
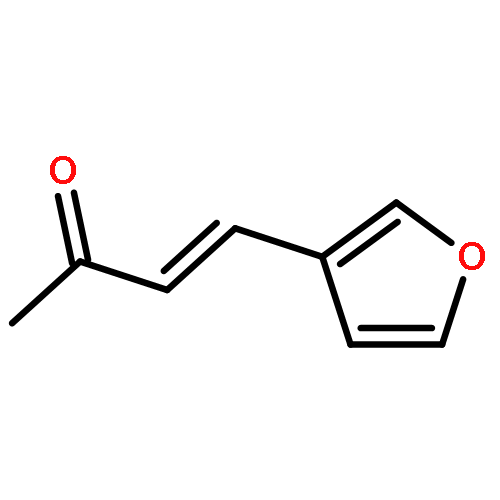


![Urea, N-[3,5-bis(trifluoromethyl)phenyl]-N'-cyclohexyl-](http://img.cochemist.com/ccimg/192100/192049-20-0.png)
![Urea, N-[3,5-bis(trifluoromethyl)phenyl]-N'-cyclohexyl-](http://img.cochemist.com/ccimg/192100/192049-20-0_b.png)
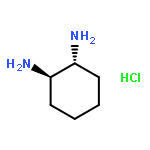

![6-Oxabicyclo[3.1.0]hexane-3-carboxylicacid,ethylester,(1alpha,3alpha,5alpha)-(9CI)](http://img.cochemist.com/ccimg/183100/183065-60-3.png)
![6-Oxabicyclo[3.1.0]hexane-3-carboxylicacid,ethylester,(1alpha,3alpha,5alpha)-(9CI)](http://img.cochemist.com/ccimg/183100/183065-60-3_b.png)

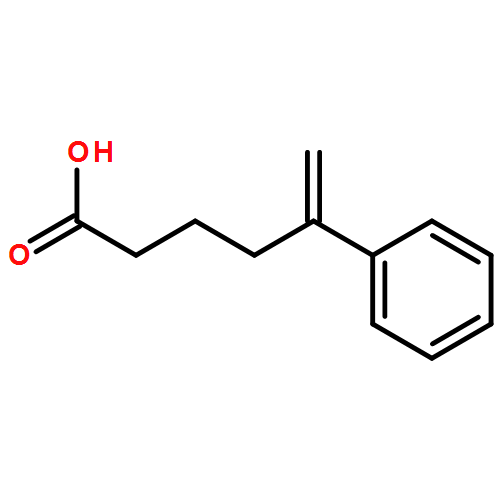
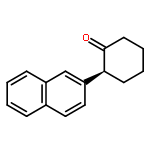
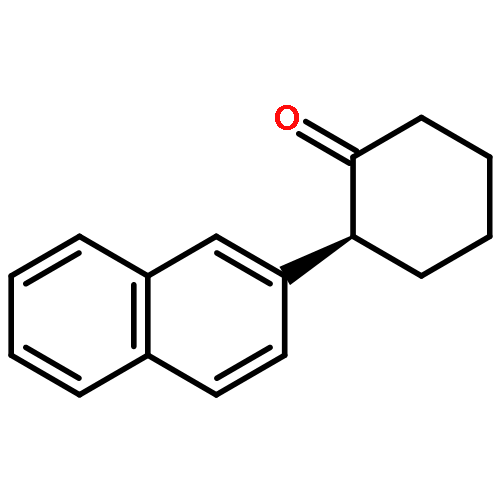
![Silane, trimethyl[[2-(2-naphthalenyl)-1-cyclohexen-1-yl]oxy]-](http://img.cochemist.com/ccimg/167500/167476-36-0.png)
![Silane, trimethyl[[2-(2-naphthalenyl)-1-cyclohexen-1-yl]oxy]-](http://img.cochemist.com/ccimg/167500/167476-36-0_b.png)
![3H-Pyrido[3,4-b]indole, 6-chloro-4,9-dihydro-](http://img.cochemist.com/ccimg/138600/138565-85-2.png)
![3H-Pyrido[3,4-b]indole, 6-chloro-4,9-dihydro-](http://img.cochemist.com/ccimg/138600/138565-85-2_b.png)



![3-Buten-2-one, 4-[4-(trifluoromethyl)phenyl]-, (3E)-](http://img.cochemist.com/ccimg/115700/115665-92-4.png)
![3-Buten-2-one, 4-[4-(trifluoromethyl)phenyl]-, (3E)-](http://img.cochemist.com/ccimg/115700/115665-92-4_b.png)

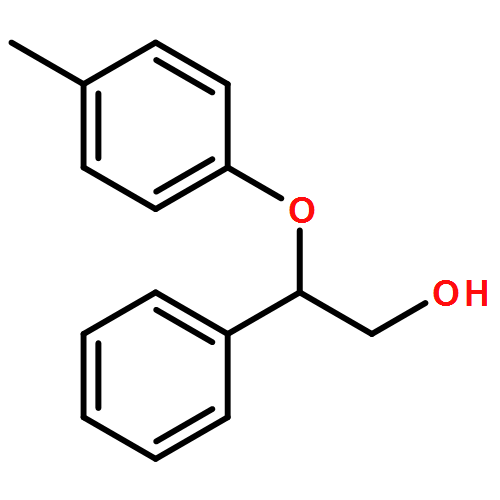
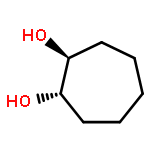
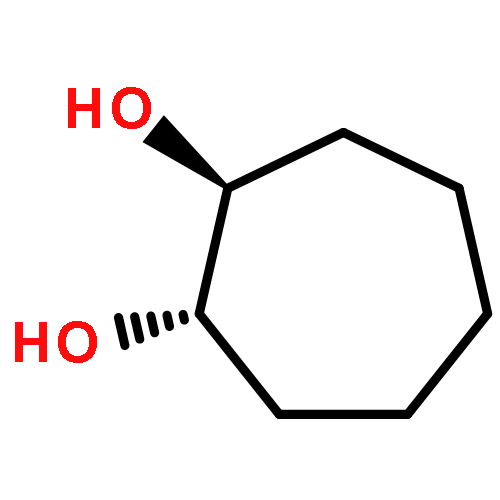


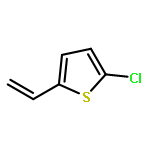


![Oxirane, [(2-propenyloxy)methyl]-, (2S)-](http://img.cochemist.com/ccimg/95100/95043-55-3.png)
![Oxirane, [(2-propenyloxy)methyl]-, (2S)-](http://img.cochemist.com/ccimg/95100/95043-55-3_b.png)
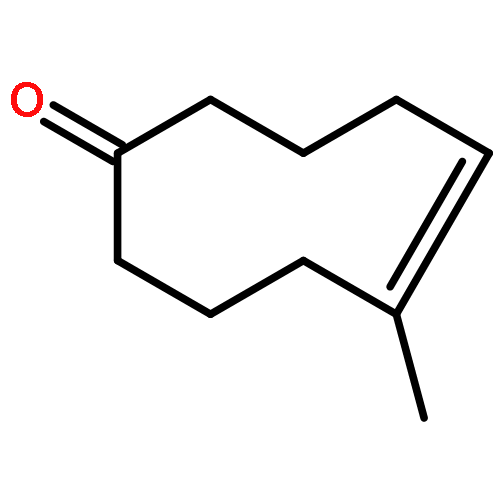
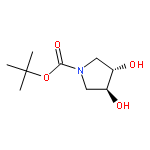
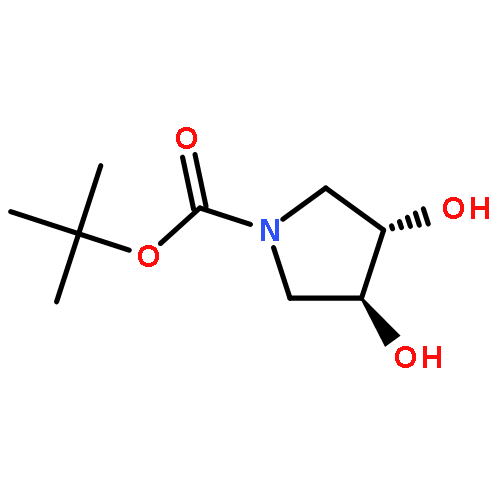
![Silane, [(4,5-dihydro-3-phenyl-2-furanyl)oxy]triethyl-](http://img.cochemist.com/ccimg/89600/89588-45-4.png)
![Silane, [(4,5-dihydro-3-phenyl-2-furanyl)oxy]triethyl-](http://img.cochemist.com/ccimg/89600/89588-45-4_b.png)
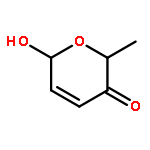
![[1,1':3',1''-Terphenyl]-2'-amine](http://img.cochemist.com/ccimg/87700/87666-57-7.png)
![[1,1':3',1''-Terphenyl]-2'-amine](http://img.cochemist.com/ccimg/87700/87666-57-7_b.png)





![tert-butyl{[(3E)-4-iodo-3-methylbut-3-en-1-yl]oxy}dimethylsilane](http://img.cochemist.com/ccimg/78600/78592-77-5.png)
![tert-butyl{[(3E)-4-iodo-3-methylbut-3-en-1-yl]oxy}dimethylsilane](http://img.cochemist.com/ccimg/78600/78592-77-5_b.png)
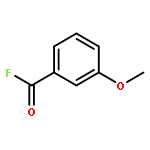
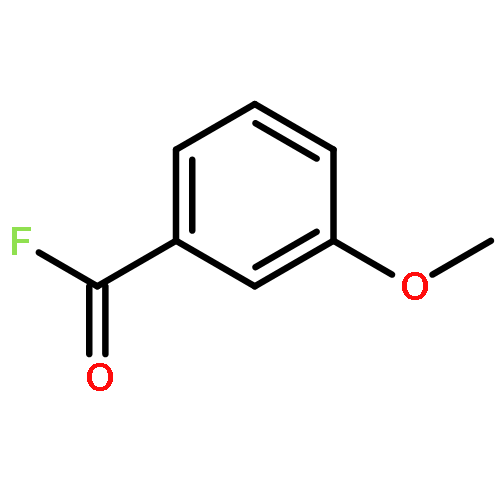
![5-{[4-(2-METHYLPHENYL)-1-PIPERAZINYL]METHYL}-2-THIOXO-2,3-DIHYDRO<WBR />-4(1H)-PYRIMIDINONE](http://img.cochemist.com/ccimg/77200/77132-99-1.png)
![5-{[4-(2-METHYLPHENYL)-1-PIPERAZINYL]METHYL}-2-THIOXO-2,3-DIHYDRO<WBR />-4(1H)-PYRIMIDINONE](http://img.cochemist.com/ccimg/77200/77132-99-1_b.png)
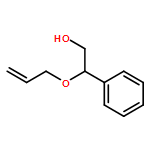
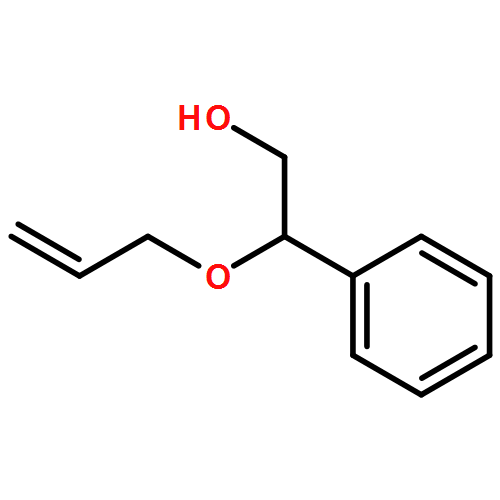
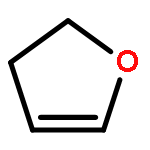
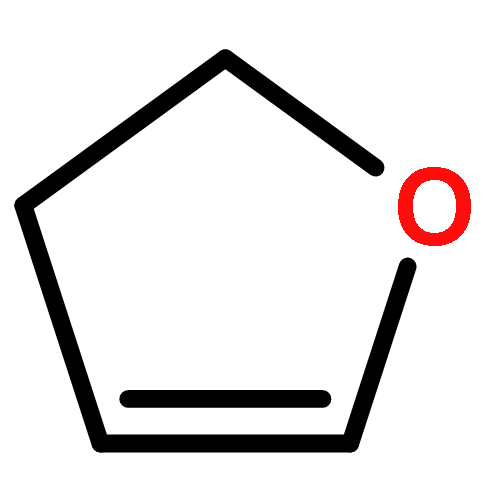


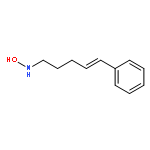
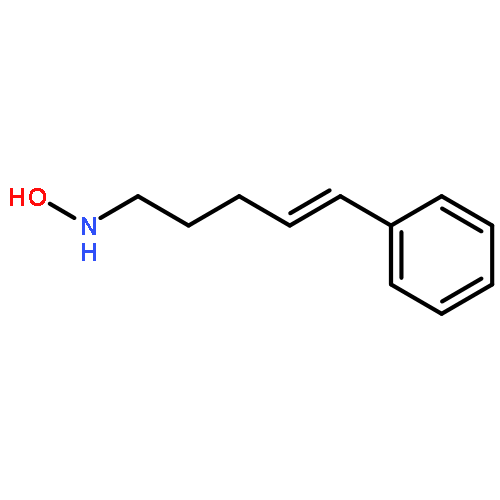




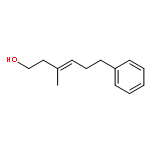
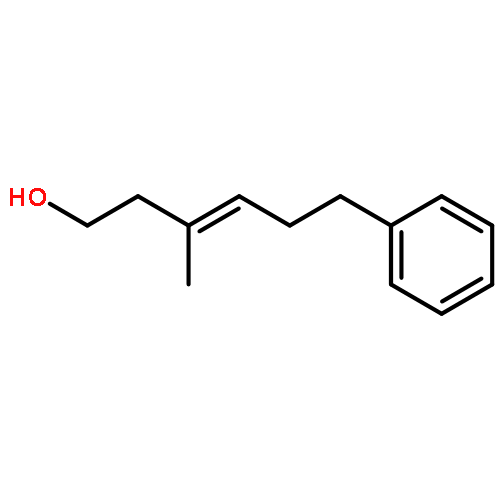
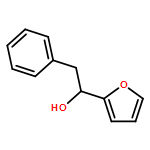
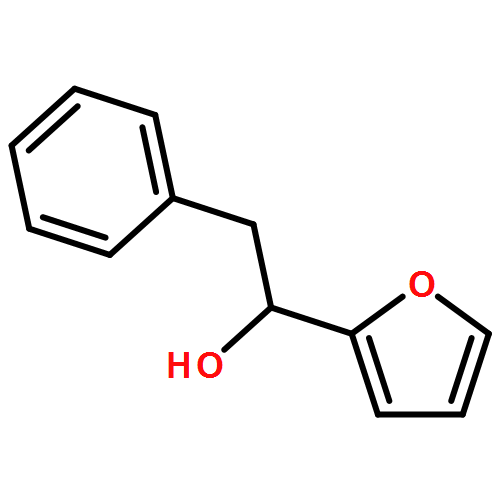
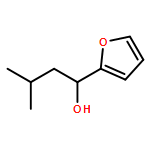
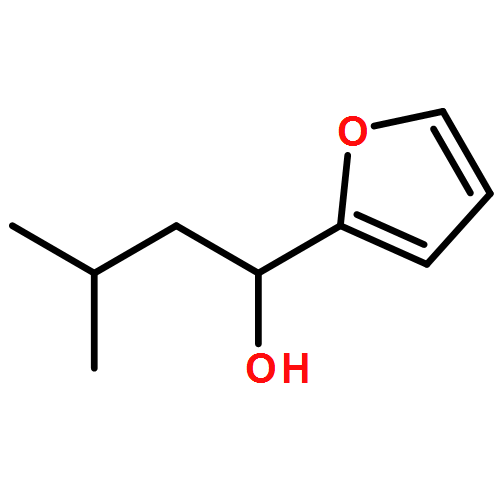

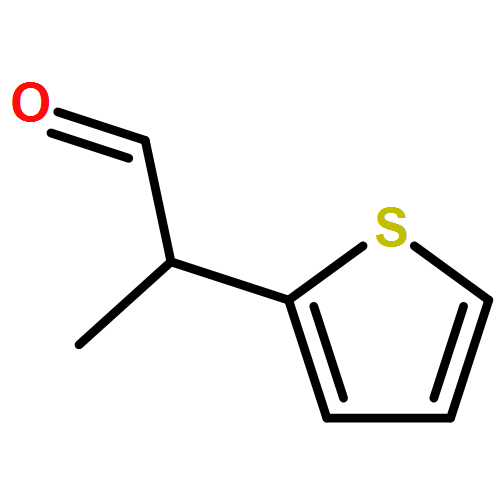
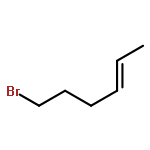

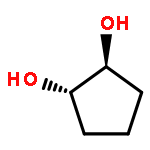



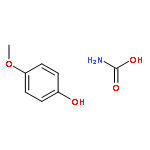
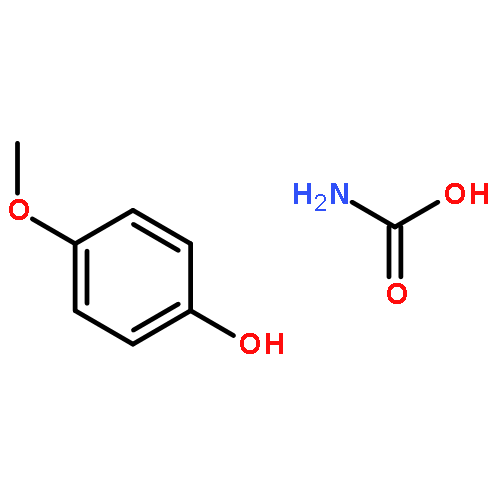
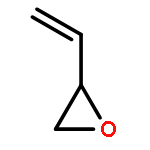
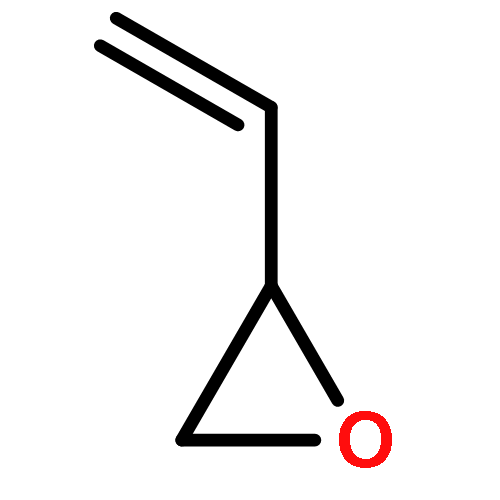
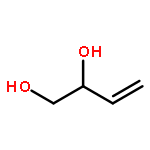
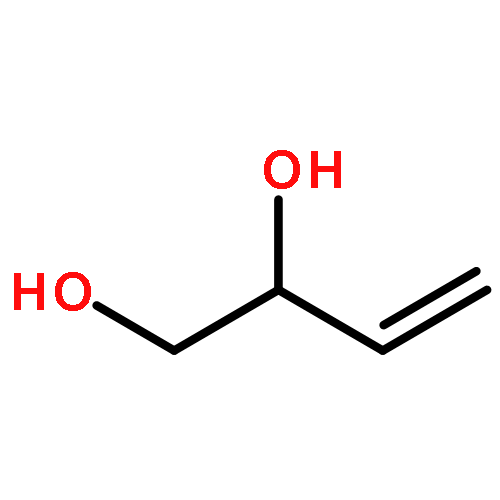
![1,4-Dioxaspiro[4.5]decan-8-ol, 7-(1-methylethenyl)-, trans-](http://img.cochemist.com/ccimg/61600/61543-81-5.png)
![1,4-Dioxaspiro[4.5]decan-8-ol, 7-(1-methylethenyl)-, trans-](http://img.cochemist.com/ccimg/61600/61543-81-5_b.png)
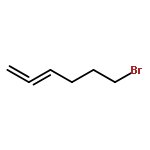










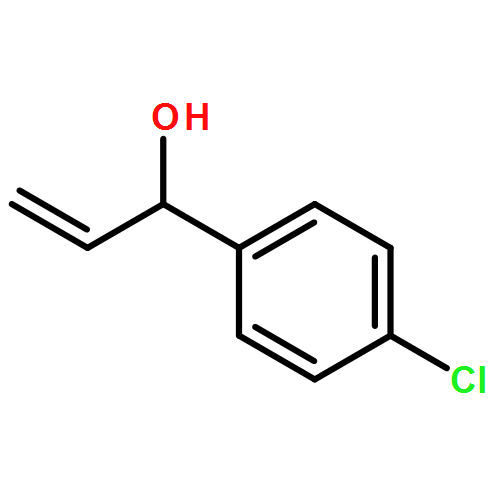
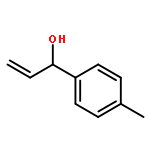
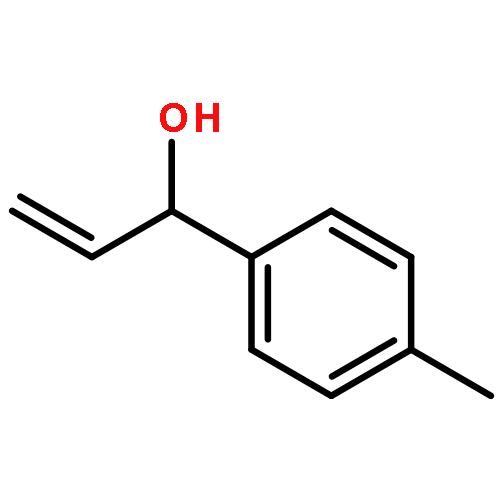


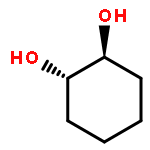


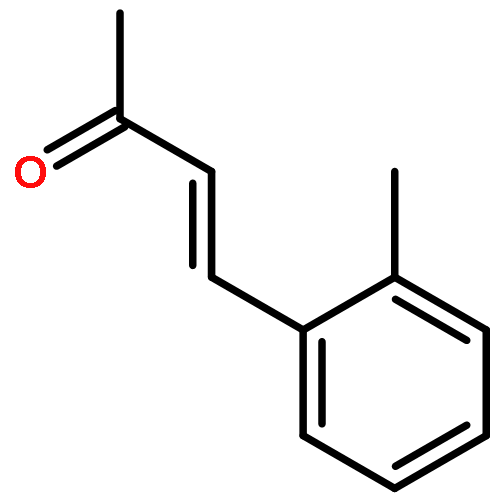
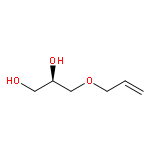

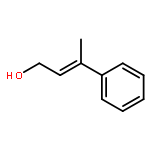
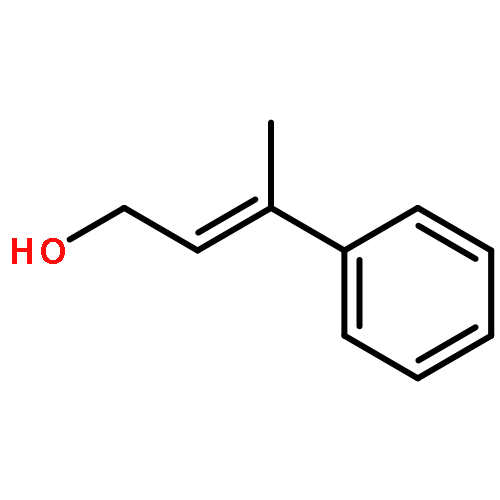

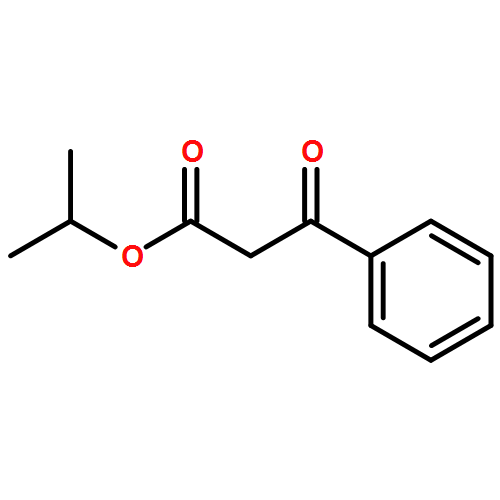
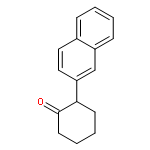
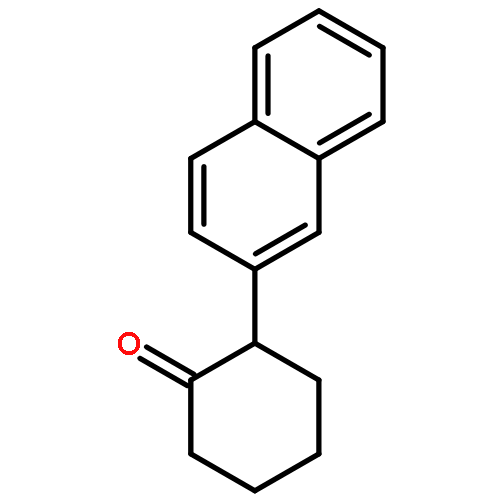






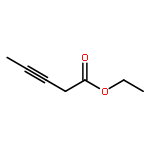























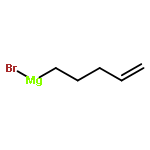
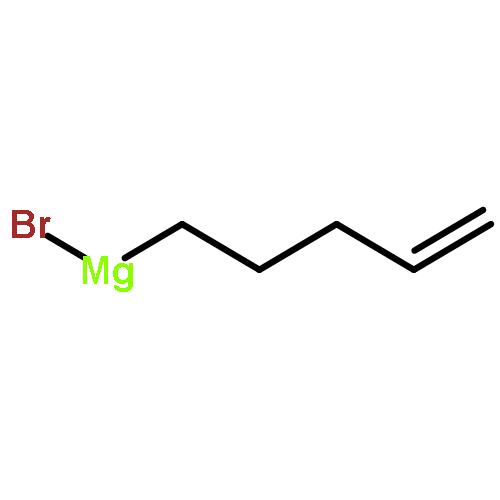
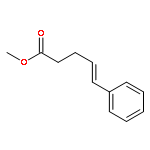



![7-METHOXY-4,9-DIHYDRO-3H-PYRIDO[3,4-B]INDOLE](http://img.cochemist.com/ccimg/31700/31652-37-6.png)
![7-METHOXY-4,9-DIHYDRO-3H-PYRIDO[3,4-B]INDOLE](http://img.cochemist.com/ccimg/31700/31652-37-6_b.png)


![4-[4-(DIMETHYLAMINO)PHENYL]BUT-3-EN-2-ONE](http://img.cochemist.com/ccimg/30700/30625-58-2.png)
![4-[4-(DIMETHYLAMINO)PHENYL]BUT-3-EN-2-ONE](http://img.cochemist.com/ccimg/30700/30625-58-2_b.png)








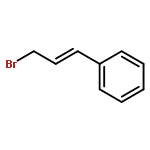
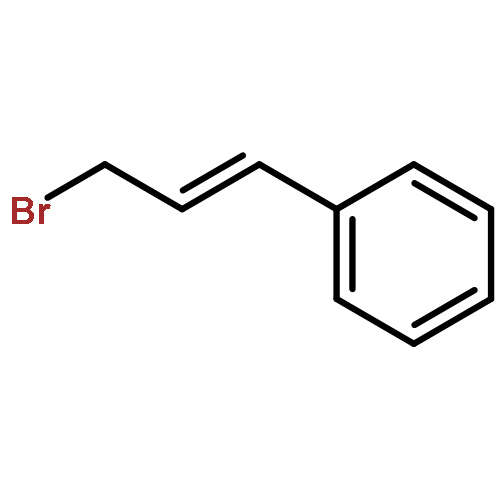
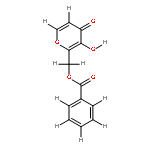
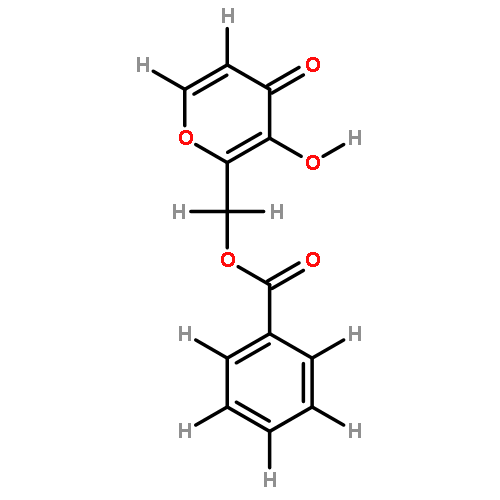
![2H-Pyran-3(6H)-one,4-(benzoyloxy)-2-[(benzoyloxy)methyl]-6-methoxy-, (2R,6S)-](http://img.cochemist.com/ccimg/25600/25552-06-1.png)
![2H-Pyran-3(6H)-one,4-(benzoyloxy)-2-[(benzoyloxy)methyl]-6-methoxy-, (2R,6S)-](http://img.cochemist.com/ccimg/25600/25552-06-1_b.png)


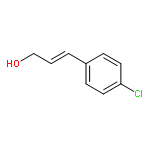


![(Z)-cis-9-oxabicyclo[6.1.0]non-4-ene](http://img.cochemist.com/ccimg/19800/19740-90-0.png)
![(Z)-cis-9-oxabicyclo[6.1.0]non-4-ene](http://img.cochemist.com/ccimg/19800/19740-90-0_b.png)






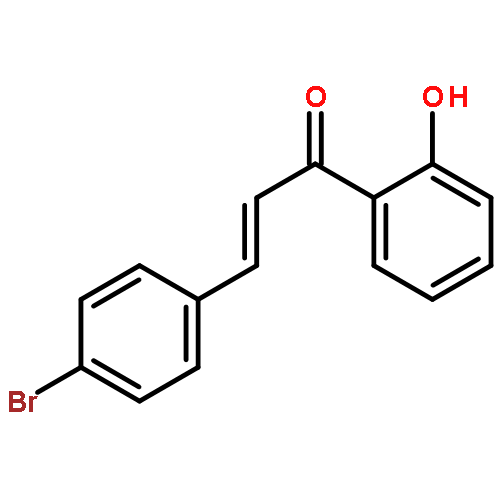
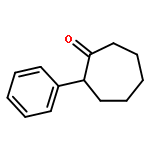









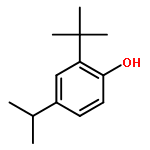
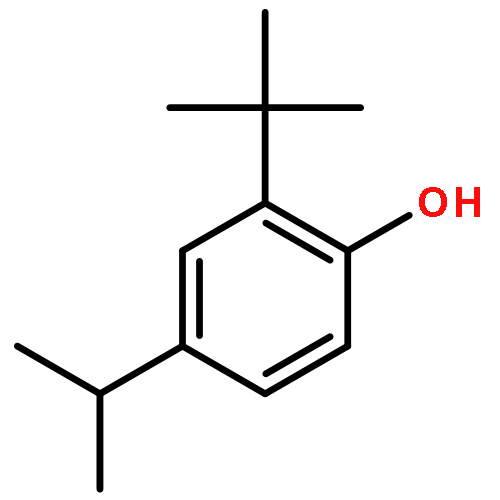
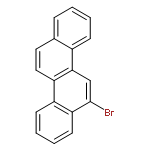
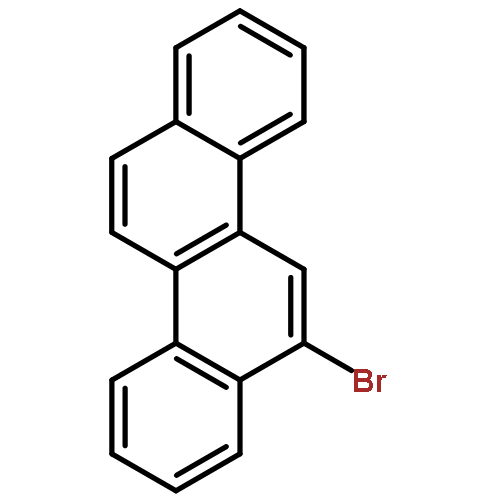
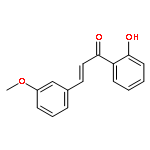
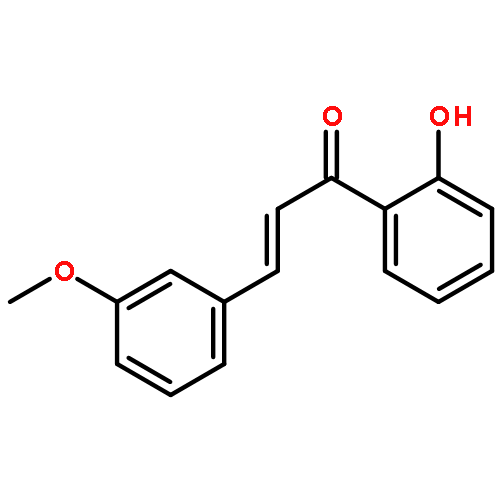
![[1]Benzopyrano[4,3-b]indole, 6,11-dihydro-](http://img.cochemist.com/ccimg/6800/6722-06-1.png)
![[1]Benzopyrano[4,3-b]indole, 6,11-dihydro-](http://img.cochemist.com/ccimg/6800/6722-06-1_b.png)
![Formamide, N-[2-(1H-indol-3-yl)ethyl]-](http://img.cochemist.com/ccimg/6600/6502-82-5.png)
![Formamide, N-[2-(1H-indol-3-yl)ethyl]-](http://img.cochemist.com/ccimg/6600/6502-82-5_b.png)
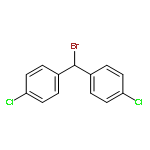
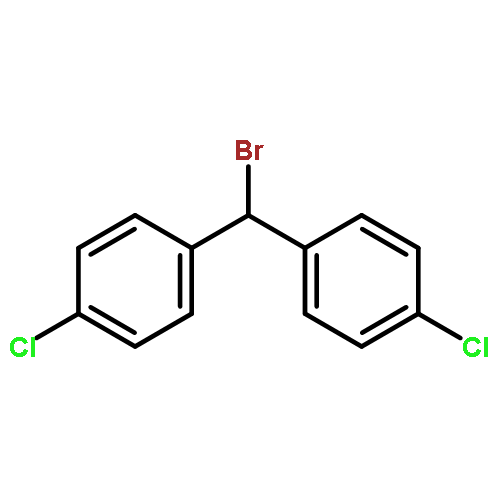

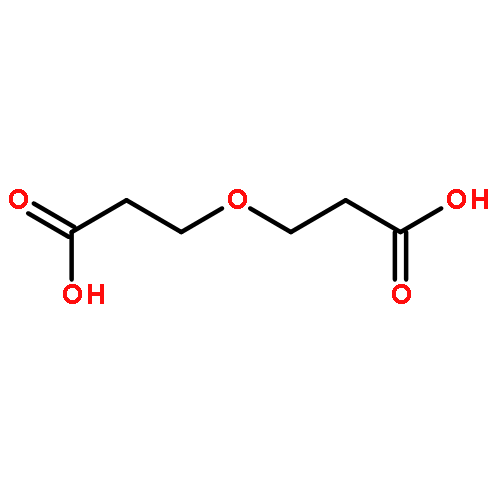






![4,9-dihydro-3h-pyrido[3,4-b]indole](http://img.cochemist.com/ccimg/4900/4894-26-2.png)
![4,9-dihydro-3h-pyrido[3,4-b]indole](http://img.cochemist.com/ccimg/4900/4894-26-2_b.png)



![1,3-BIS[3,5-BIS(TRIFLUOROMETHYL)PHENYL]UREA](http://img.cochemist.com/ccimg/3900/3824-74-6.png)
![1,3-BIS[3,5-BIS(TRIFLUOROMETHYL)PHENYL]UREA](http://img.cochemist.com/ccimg/3900/3824-74-6_b.png)
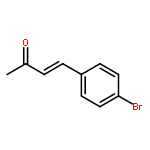
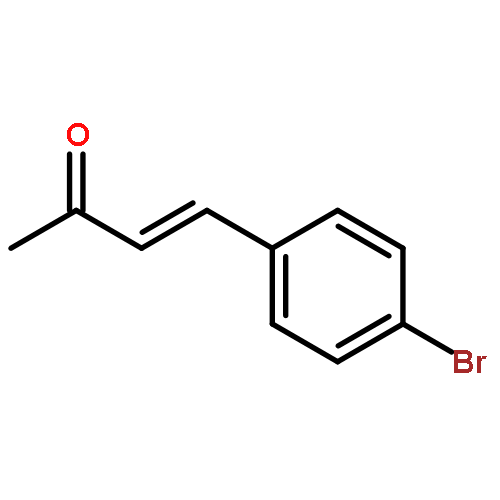
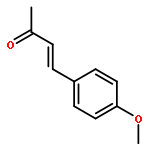
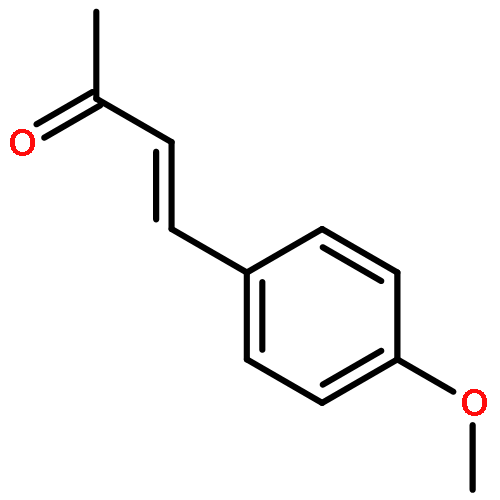


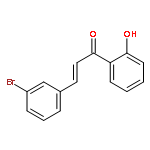
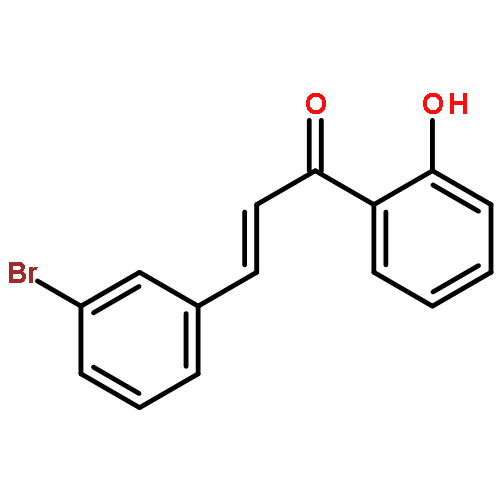




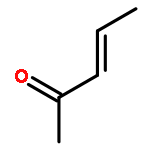
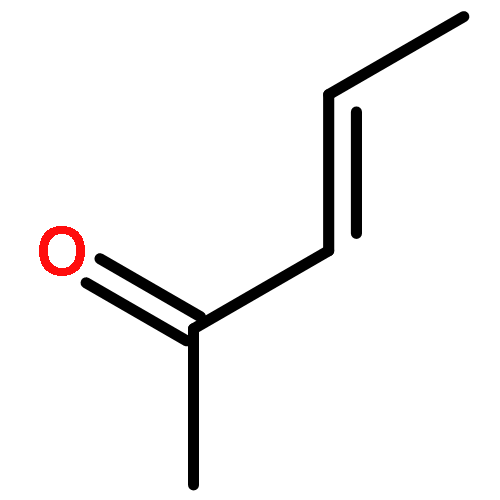






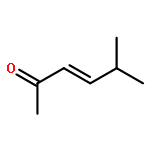
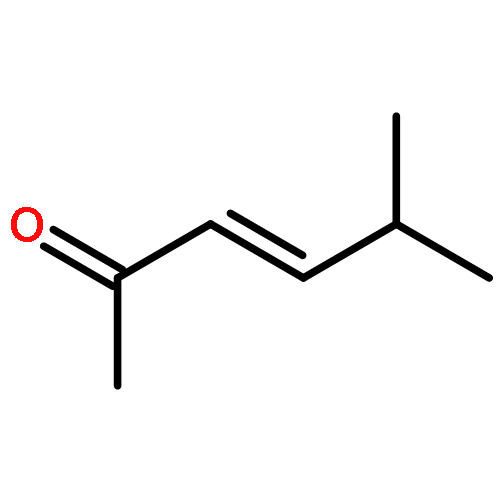


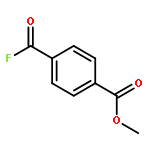
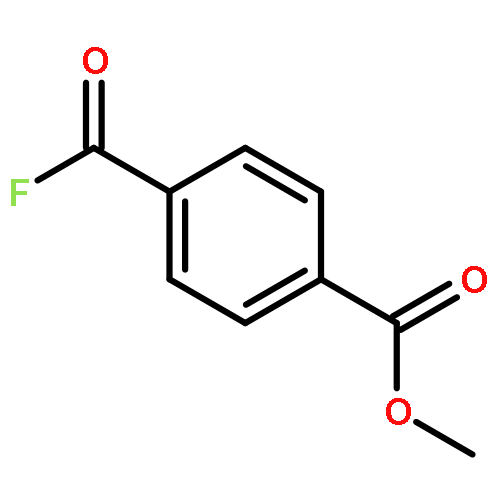





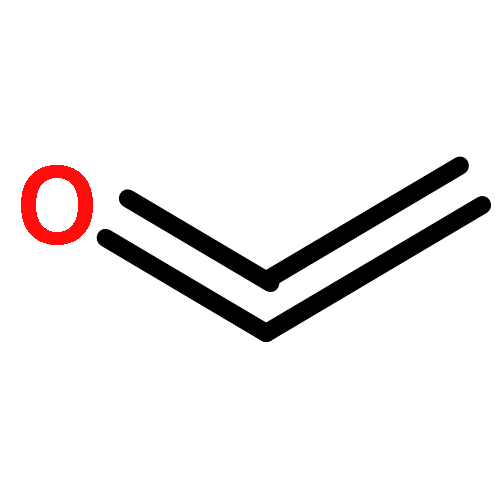
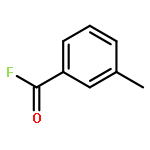
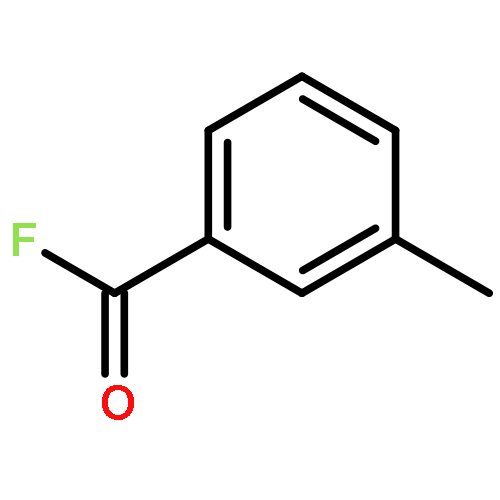


![5H-Pyrido[4,3-b]indole](http://img.cochemist.com/ccimg/300/244-69-9.png)
![5H-Pyrido[4,3-b]indole](http://img.cochemist.com/ccimg/300/244-69-9_b.png)


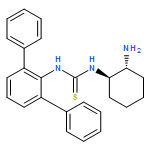

![3-Cyclobutene-1,2-dione, 3-[[3,5-bis(trifluoromethyl)phenyl]amino]-4-methoxy-](/data/chemimg/144700/1223105-85-8.png)
![3-Cyclobutene-1,2-dione, 3-[[3,5-bis(trifluoromethyl)phenyl]amino]-4-methoxy-](/data/chemimg/144700/1223105-85-8_b.png)




![Thiourea,N-[(1R,2R)-2-aminocyclohexyl]-N'-[3,5-bis(trifluoromethyl)phenyl]-](/data/chemimg/103900/860994-58-7.png)
![Thiourea,N-[(1R,2R)-2-aminocyclohexyl]-N'-[3,5-bis(trifluoromethyl)phenyl]-](/data/chemimg/103900/860994-58-7_b.png)
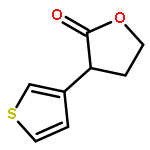
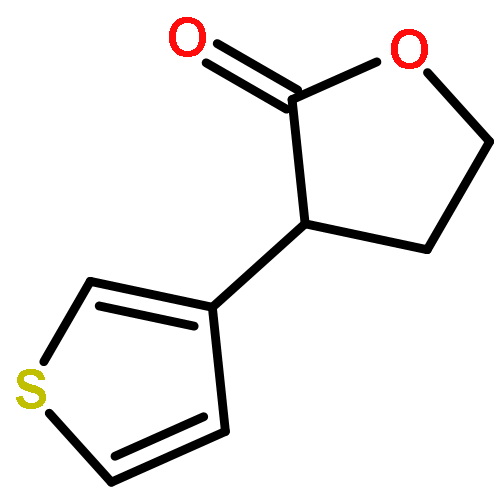
![SILANE, [[4,5-DIHYDRO-3-(3-THIENYL)-2-FURANYL]OXY]TRIMETHYL-](http://img.cochemist.com/ccimg/525000/524937-48-2.png)
![SILANE, [[4,5-DIHYDRO-3-(3-THIENYL)-2-FURANYL]OXY]TRIMETHYL-](http://img.cochemist.com/ccimg/525000/524937-48-2_b.png)


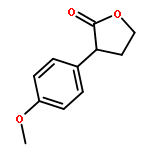
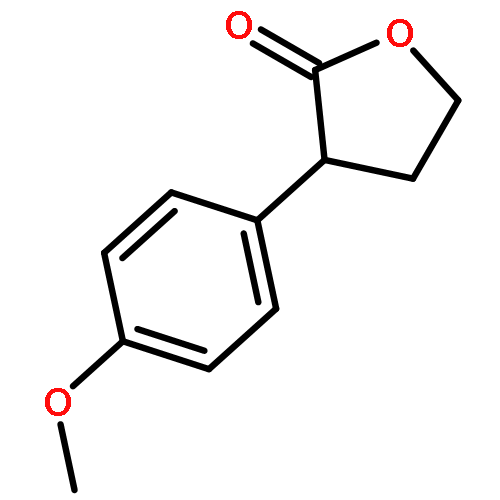
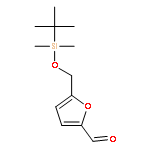
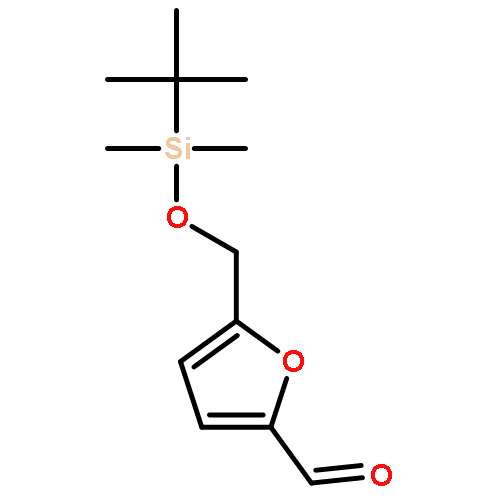
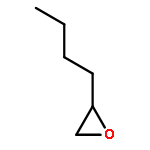
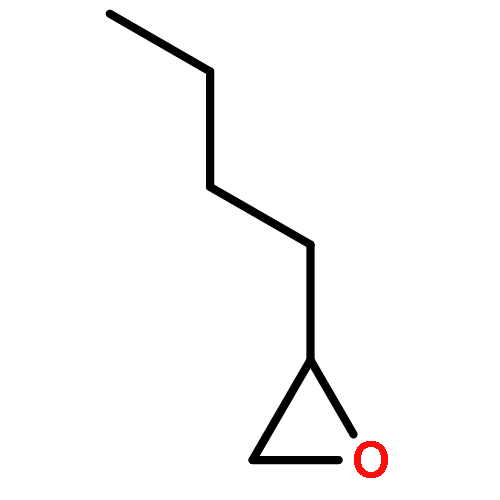
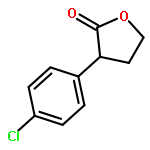
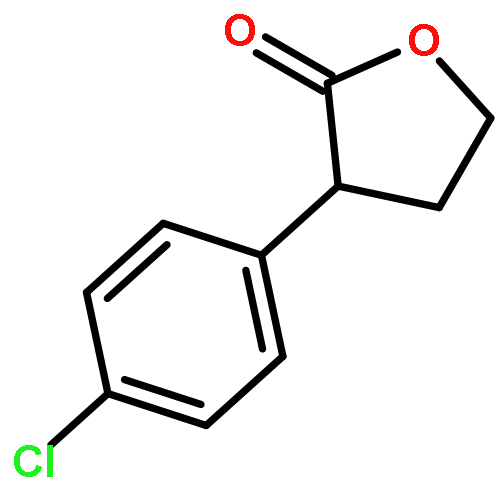



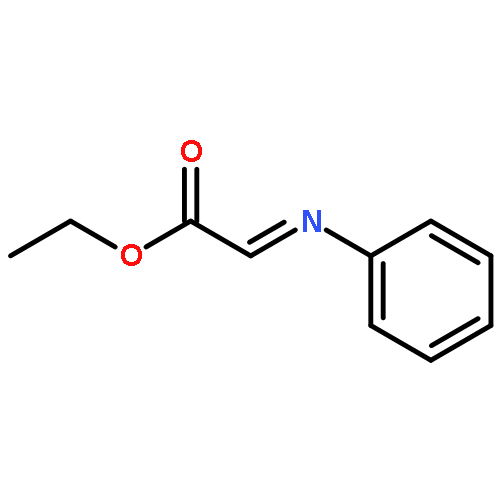
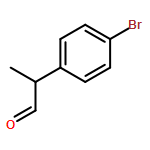



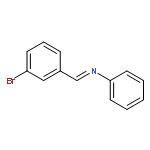
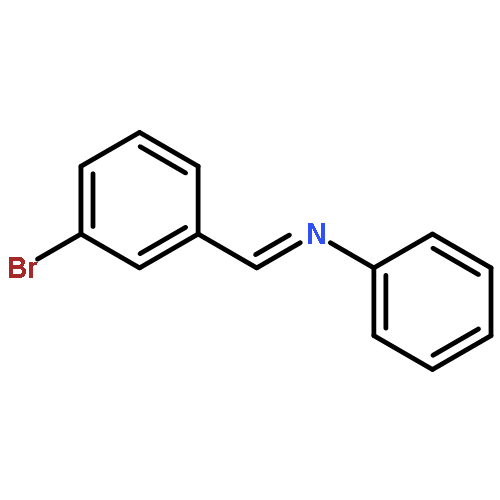
![Benzenamine, N-[(3-methylphenyl)methylene]-(E)-](http://img.cochemist.com/ccimg/7000/6906-25-8.png)
![Benzenamine, N-[(3-methylphenyl)methylene]-(E)-](http://img.cochemist.com/ccimg/7000/6906-25-8_b.png)


![7-oxabicyclo[4.1.0]hept-3-ene](http://img.cochemist.com/ccimg/6300/6253-27-6.png)
![7-oxabicyclo[4.1.0]hept-3-ene](http://img.cochemist.com/ccimg/6300/6253-27-6_b.png)
![BENZENAMINE, N-[(3-CHLOROPHENYL)METHYLENE]-](http://img.cochemist.com/ccimg/5900/5877-57-6.png)
![BENZENAMINE, N-[(3-CHLOROPHENYL)METHYLENE]-](http://img.cochemist.com/ccimg/5900/5877-57-6_b.png)
![Benzenamine, N-[(4-bromophenyl)methylene]-](http://img.cochemist.com/ccimg/5900/5877-51-0.png)
![Benzenamine, N-[(4-bromophenyl)methylene]-](http://img.cochemist.com/ccimg/5900/5877-51-0_b.png)


![Benzoic acid, 4-[(phenylmethylene)amino]-, methyl ester](http://img.cochemist.com/ccimg/4200/4112-09-8.png)
![Benzoic acid, 4-[(phenylmethylene)amino]-, methyl ester](http://img.cochemist.com/ccimg/4200/4112-09-8_b.png)
![Naphth[2,3-b]oxirene,1a,2,7,7a-tetrahydro-](http://img.cochemist.com/ccimg/2500/2461-35-0.png)
![Naphth[2,3-b]oxirene,1a,2,7,7a-tetrahydro-](http://img.cochemist.com/ccimg/2500/2461-35-0_b.png)
![Benzenamine,N-[(4-chlorophenyl)methylene]-](http://img.cochemist.com/ccimg/2400/2362-79-0.png)
![Benzenamine,N-[(4-chlorophenyl)methylene]-](http://img.cochemist.com/ccimg/2400/2362-79-0_b.png)
![Benzenamine,N-[(4-methylphenyl)methylene]-](http://img.cochemist.com/ccimg/2400/2362-77-8.png)
![Benzenamine,N-[(4-methylphenyl)methylene]-](http://img.cochemist.com/ccimg/2400/2362-77-8_b.png)
![1,3-Bis[3,5-bis(trifluoromethyl)phenyl]thiourea](/data/chemimg/315700/1060-92-0.png)
![1,3-Bis[3,5-bis(trifluoromethyl)phenyl]thiourea](/data/chemimg/315700/1060-92-0_b.png)