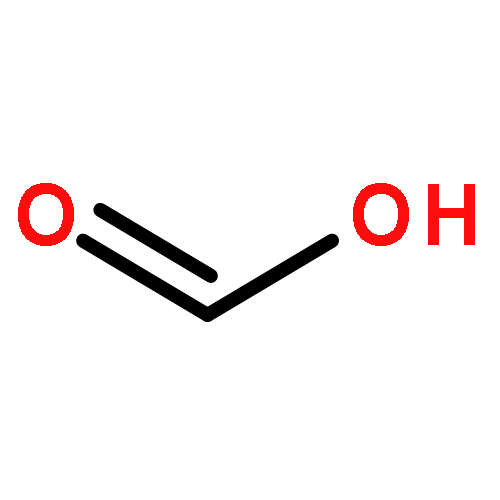
Co-reporter:Shuo Wang;Junyi Liu
Journal of Materials Chemistry A 2017 vol. 5(Issue 32) pp:16936-16943
Publication Date(Web):2017/08/15
DOI:10.1039/C7TA04941B
Layered Li2MnO3 as a cathode material is well known for its high lithium storage capacity. However, its poor cycling performance restricts its applications due to surface oxygen release and structural degradation. So it is highly desirable to search for new structures of Li2MnO3 with longer cycle life. In this work using pressure as an effective method in synthesis and global structure search, we find two metastable phases of Li2MnO3 that belong to the space group of C2/C and RC, respectively. The former one has the following features: a narrow band gap of 0.17 eV, a high open-circuit voltage of >4.4 V, a better structural stability against oxygen release (long cycle-life), a higher reversible capacity than the conventional layered Li2MnO3 phase, appropriate three-dimensional migration energy barriers of 0.45–1.14 eV and enhanced ionic mobility when more vacancies were introduced, while the latter one is metallic with an average open-circuit voltage of 4 V, comparative delithiation ability, and lower migration barriers of 0.10–0.69 eV. Both of them exhibit some merits over the conventional layered phase when used as cathode materials.
Co-reporter:Hong Fang;Shuo Wang;Junyi Liu;Puru Jena
Journal of Materials Chemistry A 2017 vol. 5(Issue 26) pp:13373-13381
Publication Date(Web):2017/07/04
DOI:10.1039/C7TA01648D
The development of cheap, durable, and safe inorganic solid electrolytes with superionic conductivity is the key for the next generation of all-solid metal-ion batteries. The recent discovery of antiperovskites with composition Li3OA (A = halogen) shows promise in this regard. Here, we demonstrate the potential of a new class of antiperovskites where halogens are replaced by BH4 superhalogens. In addition to maintaining the high ionic conductivity of Li3OA, Li3O(BH4) is lightweight, mechanically flexible, thermodynamically more stable, and electronically more insulating than Li3OA. By mixing BH4 with Cl to make Li3O(BH4)0.5Cl0.5, we further show that the conductivity will be doubled. The Li+-ion conductivity of the new materials is of the order of 10−4 to over 10−3 S cm−1 at room temperature and will be well above 10−2 S cm−1 at higher temperatures below the melting point. The conduction mechanism of the material is revealed by identifying the relationship between the orientational symmetry of the BH4− rotors and the potential surface felt by the lithium ions.
Co-reporter:Junyi Liu;Shuo Wang
PNAS 2017 Volume 114 (Issue 4 ) pp:651-656
Publication Date(Web):2017-01-24
DOI:10.1073/pnas.1618051114
Topological state of matter and lithium batteries are currently two hot topics in science and technology. Here we combine
these two by exploring the possibility of using all-carbon-based porous topological semimetal for lithium battery anode material.
Based on density-functional theory and the cluster-expansion method, we find that the recently identified topological semimetal
bco-C16 is a promising anode material with higher specific capacity (Li-C4) than that of the commonly used graphite anode (Li-C6), and Li ions in bco-C16 exhibit a remarkable one-dimensional (1D) migration feature, and the ion diffusion channels are robust against the compressive
and tensile strains during charging/discharging. Moreover, the energy barrier decreases with increasing Li insertion and can
reach 0.019 eV at high Li ion concentration; the average voltage is as low as 0.23 V, and the volume change during the operation
is comparable to that of graphite. These intriguing theoretical findings would stimulate experimental work on topological
carbon materials.
Co-reporter:Haoming ShenYawei Li, Qiang Sun
The Journal of Physical Chemistry C 2017 Volume 121(Issue 7) pp:
Publication Date(Web):February 3, 2017
DOI:10.1021/acs.jpcc.7b00317
Due to the high surface ratio and dispersed metal sites, organometallic sheets provide a very special platform for catalysis. Here we investigate the CO2 electroreduction performance of expanded phthalocyanine sheets with different transition metal dimers using density functional theory. We have determined Mn dimer to be the best active center, and the reaction path CO2 → COOH* → CO* → CHO* → CH2O* → CH3O* → CH3OH is identified as the preferable one with the overpotential of 0.84 eV. Electronic structures analyses show that σ-bonding−π-backbonding mode exists when COOH* adsorbed on Mn2-Pc, which is different from the bonding mode on Mn-Pc counterpart. Our study indicates that the introduction of metal dimer in porous covalent organic frameworks provides a new strategy for the design of catalytic materials for CO2 electroreduction.
Co-reporter:Guizhi Zhu, Yawei Li, Haiyan Zhu, Haibin Su, Siew Hwa Chan, and Qiang Sun
ACS Catalysis 2016 Volume 6(Issue 9) pp:6294
Publication Date(Web):August 11, 2016
DOI:10.1021/acscatal.6b02020
The selective electrocatalytic conversion of CO2 into useful products is a major challenge in facilitating a closed carbon cycle. Here, on the basis of first-principles calculations combined with computational hydrogen electrode model, we report a curvature-dependent selectivity of CO2 reduction on cobalt–porphyrin nanotubes which are thermodynamically stable, displaying tunable geometric and electronic properties with tube radius. We have found that CO production is preferred on nanotubes with larger diameter, and the predicted current density from microkinetics is larger than that on Au, the best metal catalyst for CO production from CO2 electroreduction. In contrast, highly curved nanotubes with small radii tend to further catalyze CO reduction to CH4 gas and the overpotential is much lower in comparison with the cases on Cu surfaces. The selectivity and the feasibility of synthesis make cobalt–porphyrin nanotubes very promising for CO2 conversion.Keywords: CO2 reduction; cobalt−porphyrin nanotubes; density functional theory; electrocatalysis; microkinetic modeling
Co-reporter:Yu Chen, Qiang Sun and Puru Jena
Journal of Materials Chemistry A 2016 vol. 4(Issue 26) pp:6353-6361
Publication Date(Web):07 Jun 2016
DOI:10.1039/C6TC01138A
Complementing the group of two-dimensional (2D) binary phosphorene analogues, we carried out first-principles calculations for α-SiTe and β-SiTe monolayers which are, respectively, black-phosphorene-like and blue-phosphorene-like. We show that both the SiTe monolayers are dynamically, thermally and mechanically stable, although α-SiTe with significant elastic anisotropy is energetically more favorable than β-SiTe. Both monolayers exhibit superior mechanical flexibility and are indirect-gap semiconductors with band gaps of 0.57 and 2.36 eV, respectively. What is even more important is that the α-SiTe monolayer can be tuned from an indirect band gap semiconductor to a direct band gap semiconductor and eventually to a metal when biaxial strains are applied, showing a high degree of flexibility in band engineering which is absent in non-silicon based analogues.
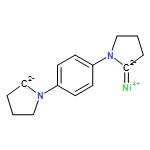
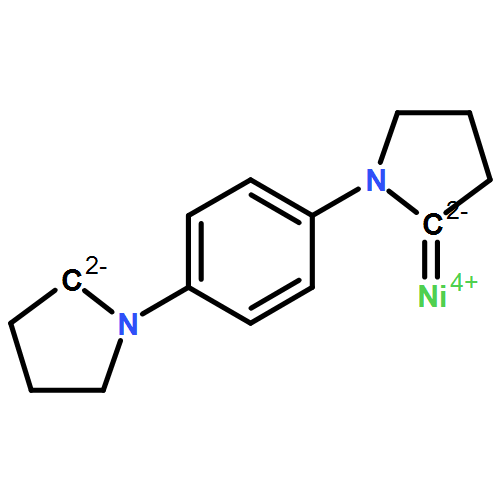
Co-reporter:Guizhi Zhu, Junyi Liu, Qiang Sun and Puru Jena
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 27) pp:17868-17874
Publication Date(Web):07 Jun 2016
DOI:10.1039/C6CP01908K
Motivated by the recent synthesis of bi-coordinated transition metal–organic complexes [Samuel, et al., Chem. Sci., 2015, 6, 3148], we have studied the structure and magnetic properties of a series of bi-coordinated transition metal based nanorings by folding quasi-1D chains. Among the cyclic alkyl(amino)carbine (CAAC) based quasi-1D chains (TM-CAAC, TM = Cr, Mn, Fe, Co, Ni), only Cr-CAAC is found to be ferromagnetic. First-principles calculations combined with Heisenberg–Dirac–van Vleck models were performed to understand the onset of robust ferromagnetism in Cr-based systems. With increasing size, the infrared intensity increases and the exchange energy oscillates. In particular, when the number, n, of TM-CAAC units is even and larger than 3, the magnetic coupling in nanorings is stronger than that in quasi-1D chains. The band gap changes very slowly with size. More importantly, compared with the highly coordinated Cr single molecular magnets, the low coordination of Cr ions enhances magnetic moment and stabilizes ferromagnetic coupling.
Co-reporter:Junyi Liu, Qiang Sun, Yoshiyuki Kawazoe and Puru Jena
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 13) pp:8777-8784
Publication Date(Web):30 Sep 2015
DOI:10.1039/C5CP04835D
In addition to spintronics another motivation for exploring ferromagnetic two-dimensional materials is for biomedical applications such as magnetic labeling and hyperthermia treatment of tumors. Unfortunately, the widely studied Mn-containing monolayer is not biocompatible, although it is ferromagnetic. Here using first principles calculations combined with Monte Carlo simulations based on the Ising model, we systematically study a class of 2D ferromagnetic monolayers CrX3 (X = Cl, Br, I). The feasibility of exfoliation from their layered bulk phase is confirmed by the small cleavage energy and high in-plane stiffness. Spin-polarized calculations, combined with self-consistently determined Hubbard U that accounts for strong correlation energy, demonstrate that CrX3 (X = Cl, Br, I) monolayers are ferromagnetic and that Cr is trivalent and carries a magnetic moment of 3 μB; the resulting Cr3+ ions are biocompatible. The corresponding Curie temperatures for CrCl3, CrBr3 and CrI3 are found to be 66, 86, and 107 K, respectively, which can be increased to 323, 314, and 293 K by hole doping. The biocompatibility and ferromagnetism render these Cr-containing trihalide monolayers unique for applications.
Co-reporter:Guizhi Zhu, Qiang Sun
Computational Materials Science 2016 Volume 112(Part B) pp:492-502
Publication Date(Web):1 February 2016
DOI:10.1016/j.commatsci.2015.07.020
The unique geometry and novel properties of graphene have tremendously motivated scientists to explore other monolayer materials, especially those with separately distributed and exposed metal ions for magnetism, hydrogen storage, CO2 capture and catalysis. The recent successful synthesis of 2D organometallic sheets has opened a new pathway to design and fabricate such desirable 2D materials going beyond graphene and other inorganic sheets. This article briefly reviews the recent advances in computational studies of 2D organometallic sheets based on density functional theory, quantum chemistry modeling and Monte Carlo simulation focusing on stability, magnetic coupling, magnetism tuning, hydrogen storage, CO2 capture and catalysis. Future research directions in this field are also discussed.
Co-reporter:Jingbo Wang, Yoshiyuki Kawazoe, Qiang Sun, Siewhwa Chan, Haibin Su
Surface Science 2016 Volume 645() pp:30-40
Publication Date(Web):March 2016
DOI:10.1016/j.susc.2015.10.035
•The selectivity of Cu, Co and Ni catalysts for syngas conversion is elucidated.•The trend on activity of catalyst helps to understand the selectivity.•Brønsted–Evans–Polanyi relation is observed for C–O bond breaking reactions.•Kinetic effect affects the barrier height when special transition state exists.Typical Fischer–Tropsch catalysts display different selectivity and activity in catalyzing CO hydrogenation to diverse products. In this work, the preferable routes for CH3OH formation on Cu, chain growth on Co and CH4 formation on Ni are identified guided by the comprehensive reaction network that is mapped out by density function theory calculations. The difference in selectivity among catalysts is controlled delicately by several reactions, including CH3O + H ↔ CH3OH, CH3 + H ↔ CH4 and CH2 + CO ↔ CH2CO. The equilibrium shifts of CH2O + H ↔ CH3O and CH2 + H ↔ CH3 also make an impact on selectivity. The distinct selectivity can be understood further with the activity of catalysts. Our results show that the ability of surface to absorb species increases in the order Cu < Ni < Co. Generally, Cu catalyzes the association reaction better than Co and Ni, while Co facilitates the dissociation reaction. Two key factors, thermodynamic effect and kinetic effect, are identified in determining the activity of catalyst. We proof that surface with strong binding capability promotes the dissociation reaction, meanwhile impedes the association reaction when the thermodynamic effect is dominant in determining the barrier height. The Brønsted–Evans–Polanyi relation is observed for C–O bond breaking reactions. In addition, kinetic effect also affects the barrier when special transition state exists. The tilt of CO at the transition state for COH formation and chain growth reactions introduces the interaction of atom O with surface. The stronger binding of atom O on Co is crucial to branch the selectivity of Co to chain growth rather than methane. Present study provides a comprehensive picture on the activity and selectivity of catalysts, which is the essential to develop novel catalyst for syngas conversion.
Co-reporter:Cunzhi Zhang
The Journal of Physical Chemistry Letters 2016 Volume 7(Issue 14) pp:2664-2670
Publication Date(Web):June 24, 2016
DOI:10.1021/acs.jpclett.6b01291
Using global particle-swarm optimization method, we report, for the first time, a BeN2 sheet (h-BeN2) with a graphene-like honeycomb lattice but displaying a direct band gap. Symmetry group analysis indicates that the dipole transition is allowed between the conduction band minimum and the valence band maximum. Although the direct band gap of 2.23 eV is close to that (2.14 eV) of MoS2 sheet, the h-BeN2 sheet has additional advantages: the direct band gap feature of the h-BeN2 sheet is quite insensitive to the layer stacking pattern and layer number, in contrast to the well-known direct-to-indirect band gap transition observed in TMDs and h-BN sheets. When rolled up, all the resulting h-BeN2 nanotubes have direct band gaps independent of chirality and diameter. Furthermore, the intrinsic acoustic-phonon-limited carrier mobility of the h-BeN2 sheet can reach ∼105 cm2 V–1 s–1 for electron and ∼104 cm2 V–1 s–1 for hole, which are higher than that of MoS2 and black phosphorus.
Co-reporter:Jiabing YuQiang Sun, Puru Jena
The Journal of Physical Chemistry C 2016 Volume 120(Issue 49) pp:27829-27833
Publication Date(Web):November 22, 2016
DOI:10.1021/acs.jpcc.6b08976
The development of efficient, lightweight, cost-effective, and environmentally friendly thermoelectric materials is critical for energy conversion devices. However, none of the existing thermoelectric materials satisfy these requirements. Herein, we predict a novel carbon-based metal-free thermoelectric material denoted as bct-C80S16 that is composed of a π-conjugated saddle-shaped molecular unit with a negative Gaussian curvature, leading to a low lattice thermal conductivity while maintaining a high charge mobility. The resulting peak figure of merit (ZT) of 2.41 at 1000 K is much larger than those of conventional Bi- and Pb-based thermoelectric materials. Additionally, bct-C80S16 is highly porous and light, with a mass density of 1.11 g/cm3. Such a high thermoelectric performance and low mass density would make this metal-free semiconducting material promising for practical applications in space-based technologies.
Co-reporter:Yu Chen; Min Kan; Qiang Sun;Puru Jena
The Journal of Physical Chemistry Letters 2016 Volume 7(Issue 3) pp:399-405
Publication Date(Web):January 14, 2016
DOI:10.1021/acs.jpclett.5b02770
Motivated by the recent experimental advances in exfoliating Egyptian blue monolayers, we have carried out extensive calculations using density functional theory to understand their geometry, stability, mechanical properties, electronic structures, and magnetism. Upon exfoliation from the bulk, XCuSi4O10 (X = Ca, Sr, and Ba) monolayers are found to change symmetry from tetragonal to orthorhombic. They all satisfy Born criteria and are mechanically stable. Each Cu site carries a magnetic moment of 1.0 μB but with degenerate ferromagnetic and antiferromagnetic coupling states. From Ca to Sr and Ba, as the atomic number increases, the thickness, elastic constants, Young’s moduli, and Poisson’s ratios of the monolayers increase, while the band gaps decrease. Applying strain can tune the magnitude of energy band gaps, but the direct gap feature remains. Complementing the widely studied graphene, MXenes, black phosphorus, and dichalcogenide sheets, the Egyptian blue monolayers add additional features to the family of two-dimensional materials.
Co-reporter:Yawei Li, Haibin Su, Siew Hwa Chan, and Qiang Sun
ACS Catalysis 2015 Volume 5(Issue 11) pp:6658
Publication Date(Web):October 5, 2015
DOI:10.1021/acscatal.5b01165
Graphene-based materials are being hotly pursued for energy and environment applications. Inspired by the recent experimental synthesis of Fe2 dimer supported on graphene (He, Z.; He, K.; Robertson, A. W.; Kirkland, A. I.; Kim, D.; Ihm, J.; Yoon, E.; Lee, G.-D.; Warner, J. H. Nano Lett. 2014, 14, 3766–3772), here using large-scale screening-based density functional theory and microkinetics modeling, we have identified that some transition metal dimers (Cu2, CuMn, and CuNi), when supported on graphene with adjacent single vacancies (labeled as XY@2SV), perform better in CO2 electroreduction with reduced overpotental and enhanced current density. Specifically, Cu2@2SV is catalytically active toward CO production, similar to Au electrodes but distinct from bulk Cu; MnCu@2SV is selective toward CH4 generation, while NiCu@2SV promotes CH3OH production because of the difference in oxophilicity between incorporated Mn and Ni. The advantages of the outstanding selectivity of products, the high dispersity of spatial distribution, and the reduced overpotentials allow these new systems to be promising catalysts, which will motivate more experimental research in this direction to further explore graphene-based materials for CO2 conversion.Keywords: CO2 reduction; density functional theory; electrocatalysis; graphene-supported metal dimers; microkinetics modeling
Co-reporter:Yawei Li, Siew Hwa Chan and Qiang Sun
Nanoscale 2015 vol. 7(Issue 19) pp:8663-8683
Publication Date(Web):16 Apr 2015
DOI:10.1039/C5NR00092K
The conversion of CO2 into fuels and useful chemicals has been intensively pursued for renewable, sustainable and green energy. However, due to the negative adiabatic electron affinity (EA) and large ionization potential (IP), the CO2 molecule is chemically inert, thus making the conversion difficult under normal conditions. Novel catalysts, which have high stability, superior efficiency and low cost, are urgently needed to facilitate the conversion. As the first step to design such catalysts, understanding the mechanisms involved in CO2 conversion is absolutely indispensable. In this review, we have summarized the recent theoretical progress in mechanistic studies based on density functional theory, kinetic Monte Carlo simulation, and microkinetics modeling. We focus on reaction channels, intermediate products, the key factors determining the conversion of CO2 in solid–gas interface thermocatalytic reduction and solid–liquid interface electrocatalytic reduction. Furthermore, we have proposed some possible strategies for improving CO2 electrocatalysis and also discussed the challenges in theory, model construction, and future research directions.
Co-reporter:Min Kan, Hong Gi Nam, Young Hee Lee and Qiang Sun
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 22) pp:14866-14871
Publication Date(Web):08 May 2015
DOI:10.1039/C5CP01649E
The phase stability and phase transition of transition metal dichalcogenide (TMD) monolayer materials have attracted tremendous attention due to their attractive diverse potential applications. Here, first-principles calculations based on density-functional theory are carried out to study the newly synthesized MoTe2 monolayer. A phase different from the semiconducting trigonal prismatic structure and octahedral coordinated structure is found to be stable at room temperature in a free standing state, as evidenced by phonon spectrum analysis and molecular dynamic simulation. Raman vibrations of all the possible phases are calculated to provide additional information for the distinction of different phases in the experiment.
Co-reporter:Guizhi Zhu, Qiang Sun, Yoshiyuki Kawazoe, Puru Jena
International Journal of Hydrogen Energy 2015 Volume 40(Issue 9) pp:3689-3696
Publication Date(Web):9 March 2015
DOI:10.1016/j.ijhydene.2015.01.069
The exploration of two-dimensional (2D) sheets beyond graphene has been gaining increasing interest. In this work, using first-principles calculations combined with grand canonical Monte Carlo (GCMC) simulations we systematically study the stability, electronic structure, optical absorbance and hydrogen adsorption of porphyrin (Por)-based nanosheets. We find these sheets to be thermally and mechanically stable. In addition, their electronic structure can be tuned from semiconducting to metallic by doping different metal atoms, and the sheets can absorb near infrared (NIR) light. We also calculate the hydrogen storage capacities of the MPor (M = Mg, Ca, Sc) at 298 K and 100 bar pressure and find that the hydrogen gravimetric density of ScPor nanosheet can reach 6.71 wt% which represents an enhancement of 45% as compared to the Sc-phthalocyanine sheet. The present study provides new insight into 2D organic nanostructures with potential applications.
Co-reporter:Junyi Liu; Qiang Sun
ChemPhysChem 2015 Volume 16( Issue 3) pp:614-620
Publication Date(Web):
DOI:10.1002/cphc.201402713
Abstract
Based on a recent experimental study on the Ni3C12S12 sheet [J. Am. Chem. Soc. 2013, 135, 2462] and a theoretical study on the Mn3C12S12 sheet [Nanoscale 2013, 5, 10404], by using density functional theory combined with a thermodynamic model, it is shown that when sulfur atoms are replaced by NH groups the resulting Mn3C12N12H12 sheet can exhibit strong ferromagnetism with a Curie temperature of 450 K. The enhanced ferromagnetism is due to two main factors: the reduced lattice constant and nitrogen is more effective in mediating magnetic couplings through p–d exchange interactions. Furthermore, it is also confirmed that the Mn3C12N12H12 sheet is kinetically and thermally stable, and displays half metallicity.
Co-reporter:Yawei Li;Shunhong Zhang;Jiabing Yu;Qian Wang;Puru Jena
Nano Research 2015 Volume 8( Issue 9) pp:2901-2912
Publication Date(Web):2015 September
DOI:10.1007/s12274-015-0795-x
C2 is a well-known pseudo-oxygen unit with an electron affinity of 3.4 eV. We show that it can exhibit metal-ion like behavior when embedded in a porphyrin sheet and form a metal-free two-dimensional material with superior oxygen reduction performance. Here, the positively charged C=C units are highly active for oxygen reduction reaction (ORR) via dissociation pathways with a small energy barrier of 0.09 eV, much smaller than that of other non-platinum group metal (non-PGM) ORR catalysts. Using a microkinetics-based model, we calculated the partial current density to be 3.0 mA/cm2 at 0.65 V vs. a standard hydrogen electrode (SHE), which is comparable to that of the state-of-the-art Pt/C catalyst. We further confirm that the C=C embedded porphyrin sheet is dynamically and thermally stable with a quasi-direct band gap of 1.14 eV. The superior catalytic performance and geometric stability make the metal-free C=C porphyrin sheet ideal for fuel cell applications.
Co-reporter:Jian Zhou and Qiang Sun
Nanoscale 2014 vol. 6(Issue 1) pp:328-333
Publication Date(Web):07 Oct 2013
DOI:10.1039/C3NR04041K
A two-dimensional sheet with long range ferromagnetic (FM) order has been hotly pursued currently. The recent success in synthesizing polymerized Fe-phthalocyanine (poly-FePc) porous sheets paves a possible way to achieve this goal. However, the poly-FePc and its analog poly-CrPc structure are intrinsically antiferromagnetic (AFM). Using first principles combined with Monte-Carlo simulations, we study systematically the carrier-induced magnetic coupling transitions in poly-CrPc and poly-FePc sheets. We show that electron doping can induce stable FM states with Curie temperatures of 130–140 K, while hole doping will enhance the stability of the AFM states. Such changes in magnetic couplings depend on the balance of AFM superexchange and FM p–d exchange.
Co-reporter:Min Kan, Subash Adhikari and Qiang Sun
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 10) pp:4990-4994
Publication Date(Web):22 Jan 2014
DOI:10.1039/C3CP55146F
Using density functional theory combined with Monte Carlo (MC) simulations, we show that the two dimensional (2D) MnS2 and MnSe2 sheets are ideal magnetic semiconductors with long-range magnetic ordering and high magnetic moments (3 μB per unit cell), where all the Mn atoms are ferromagnetically coupled, and the Curie temperatures (TC) estimated for MnS2 and MnSe2 by the MC simulations are 225 and 250 K, respectively, which can be further increased to 330 K and 375 K by applying 5% biaxial tensile strains.
Co-reporter:Guizhi Zhu, Kun Lü, Qiang Sun, Yoshiyuki Kawazoe, Puru Jena
Computational Materials Science 2014 Volume 81() pp:275-279
Publication Date(Web):January 2014
DOI:10.1016/j.commatsci.2013.08.015
•The first study of using multi-scale simulation for H2 storage on Li doped g-C3N4.•The excess gravimetric density of H2 is about 4.50 wt% at T = 298 K and P = 100 bar.•The g-C3N4 porous sheet shows advantages over MOFs and COFs.Due to its porous structure and light mass the recently synthesized triazine-based graphitic C3N4 (g-C3N4) sheet is a promising material for gas storage. First-principles calculations based on density functional theory were used to study the hydrogen storage capacity of Li doped g-C3N4 under ambient thermodynamic conditions. The most stable binding site of Li atom on it is the open-hollow site with a binding energy of 3.26 eV. Based on the force field parameters derived from quantum chemistry calculations, we have further performed grand canonical Monte Carlo (GCMC) simulations to investigate H2 adsorption isotherms on g-C3N4 sheet. We find that the adsorption energy of H2 is 3.48 kcal/mol, and the excess uptake of hydrogen is about 4.50 wt% at 298 K and 100 bar, showing potential as a hydrogen storage material.
Co-reporter:Dr. Guizhi Zhu;Dr. Yawei Li;Dr. Kun Lü; Qiang Sun
ChemPhysChem 2014 Volume 15( Issue 1) pp:126-131
Publication Date(Web):
DOI:10.1002/cphc.201300830
Abstract
Phthalocyanine (Pc) molecules are well-known flexible structural units for 1D nanotubes and 2D nanosheets. First-principles calculations combined with grand canonical Monte Carlo simulations are used to obtain the geometries, electronic structures, optical properties, and hydrogen-storage capacities of nanocages consisting of six Pc molecules with six Mg or Ca atoms. The primitive Pc cage has Th symmetry with twofold degeneracy in the highest occupied molecular orbital (HOMO), and threefold degeneracy in the lowest unoccupied molecular orbital (LUMO); the corresponding HOMO–LUMO gap is found to be 0.97 eV. The MgPc and CaPc cages have Oh symmetry with a HOMO–LUMO gap of 1.24 and 1.13 eV, respectively. Optical absorption spectra suggest that the Pc-based cages can absorb infrared light, which is different from the visible-light absorption in Pc molecules. We further show that the excess uptake of hydrogen on MgPc and CaPc cages at 298 K and 100 bar (1 bar=0.1 MPa) is about 3.49 and 4.74 wt %, respectively. The present study provides new insight into Pc-based nanostructures with potential applications.
Co-reporter:M. Kan, J. Y. Wang, X. W. Li, S. H. Zhang, Y. W. Li, Y. Kawazoe, Q. Sun, and P. Jena
The Journal of Physical Chemistry C 2014 Volume 118(Issue 3) pp:1515-1522
Publication Date(Web):January 3, 2014
DOI:10.1021/jp4076355
As an inorganic cousin of graphene, MoS2 monolayer has attracted considerable attention. However, a full understanding of its structure and stability is still lacking due to its dependence on experimental synthesis conditions. Using first-principle calculations combined with Boltzmann transport equation, we have extensively studied the geometry, energetics, electronic structure, optical absorption, and carrier mobility of various phases of MoS2. We have not only identified the stable phases of a MoS2 monolayer, but also clarified the specific conditions under which different phases are formed. The possible pathways for transitions among different phases are also discussed.
Co-reporter:Guizhi Zhu, Min Kan, Qiang Sun, and Puru Jena
The Journal of Physical Chemistry A 2014 Volume 118(Issue 1) pp:304-307
Publication Date(Web):December 16, 2013
DOI:10.1021/jp4109255
Metal–organic porous sheets, due to their unique atomic configurations and properties, represent a class of materials beyond graphene and BN monolayers. The Mo2–phthalocyanine-based sheet (Mo2Pc) is a new member of this porous organometallic family. Using density functional theory with hybrid functional for exchange–correlation potential, we show that this dimer-based material, unlike conventional organic monolayers that contain isolated metal atoms, possesses unique mechanical, magnetic, electronic, and optical properties due to inherent anisotropy in the structure. Furthermore, it is a semiconductor with a direct band gap of 0.93 eV and is antiferromagnetic with each Mo site carrying a magnetic moment of 0.88 μB. The strong anisotropy in elasticity and infrared light absorption is likely to open new doors for potential applications.
Co-reporter:Xiong Gu and Qiang Sun
Physical Chemistry Chemical Physics 2013 vol. 15(Issue 37) pp:15434-15440
Publication Date(Web):11 Jul 2013
DOI:10.1039/C3CP51200B
Using density functional theory (DFT) and time-dependent DFT in different levels, we have systematically studied new porphyrin-based dyes with A–π–D structures focusing on optical absorption, light harvest efficiency, ground state oxidation potential, excited state oxidation potential, and natural transition orbitals. Compared with existing dyes, we find that these new dyes have wide absorption regions (400–1000 nm) with high molar extinction coefficients, and display good energy level alignment for efficient injection of electrons and fast regeneration of the oxidized dyes.
Co-reporter:M. Kan, J. Zhou, Q. Sun, Y. Kawazoe, and P. Jena
The Journal of Physical Chemistry Letters 2013 Volume 4(Issue 20) pp:3382-3386
Publication Date(Web):September 23, 2013
DOI:10.1021/jz4017848
The Mn atom, because of its special electronic configuration of 3d54s2, has been widely used as a dopant in various two-dimensional (2D) monolayers such as graphene, BN, silicene and transition metal dichalcogenides (TMDs). The distributions of doped Mn atoms in these systems are highly sensitive to the synthesis process and conditions, thus suffering from problems of low solubility and surface clustering. Here we show for the first time that the MnO2 monolayer, synthetized 10 years ago, where Mn ions are individually held at specific sites, exhibits intrinsic ferromagnetism with a Curie temperature of 140 K, comparable to the highest TC value achieved experimentally for Mn-doped GaAs. The well-defined atomic configuration and the intrinsic ferromagnetism of the MnO2 monolayer suggest that it is superior to other magnetic monolayer materials.Keywords: coupling; Curie temperature; magnetism; nanosheet; spintronics;
Co-reporter:Jian Zhou, Qian Wang, Qiang Sun, Yoshiyuki Kawazoe, and Puru Jena
The Journal of Physical Chemistry Letters 2012 Volume 3(Issue 21) pp:3109-3114
Publication Date(Web):October 10, 2012
DOI:10.1021/jz301303t
Motivated by the recent success in synthesizing two-dimensional Fe-phthalocyanine (poly-FePc) porous sheets, we studied strain-induced spin crossover in poly-TMPc (TM = Mn, Fe, Co, and Ni) systems by using first-principle calculations based on density functional theory. A small amount of biaxial tensile strain is found to not only significantly enhance the magnetic moment of the central TM atoms by 2 μB when the strain reaches a critical value, but also the systems undergo low-spin (LS) to high-spin (HS) transition. These systems, however, show different response to strain, namely, poly-FePc sheet becomes ferromagnetic (FM) while poly-MnPc and poly-NiPc sheets become antiferromagnetic (AFM). Poly-CoPc, on the other hand, remains AFM. These predicted results can be observed in suspended poly-TMPc sheets by using scanning tunneling microscope (STM) tips to manipulate strain.Keywords: biaxial tensile strain; crystal field effect; density functional theory; magnetic memory device; single atomic porous sheet;
Co-reporter:Jian Zhou
Journal of the American Chemical Society 2011 Volume 133(Issue 38) pp:15113-15119
Publication Date(Web):August 13, 2011
DOI:10.1021/ja204990j
A two-dimensional (2D) periodic Fe phthalocyanine (FePc) single-layer sheet has very recently been synthesized experimentally (Abel, M.; et al. J. Am. Chem. Soc.2011, 133, 1203), providing a novel pathway for achieving 2D atomic sheets with regularly and separately distributed transition-metal atoms for unprecedented applications. Here we present first-principles calculations based on density functional theory to investigate systematically the electronic and magnetic properties of such novel organometallics (labeled as TMPc, TM = Cr–Zn) as free-standing sheets. Among them, we found that only the 2D MnPc framework is ferromagnetic, while 2D CrPc, FePc, CoPc, and CuPc are antiferromagnetic and 2D NiPc and ZnPc are nonmagnetic. The difference in magnetic couplings for the studied systems is related to the different orbital interactions. Only MnPc displays metallic dxz and dyz orbitals that can hybridize with p electrons of Pc, which mediates the long-range ferromagnetic coupling. Monte Carlo simulations based on the Ising model suggest that the Curie temperature (TC) of the 2D MnPc framework is ∼150 K, which is comparable to the highest TC achieved experimentally, that of Mn-doped GaAs. The present study provides theoretical insight leading to a better understanding of novel phthalocyanine-based 2D structures beyond graphene and BN sheets.
Co-reporter:Jian Zhou ; Qian Wang ; Qiang Sun ;Puru Jena
The Journal of Physical Chemistry C 2011 Volume 115(Issue 13) pp:6136-6140
Publication Date(Web):March 9, 2011
DOI:10.1021/jp1110778
First principles calculations based on density functional theory are carried out to study the effect of lithium functionalization of experimentally synthesized BC3 nanotube on charge transfer, electrostatic potential, and hydrogen storage. Electron-deficient BC3 nanotube is found to promote charge transfer from Li to the substrate when lithiated. The resulting Li ions on the tube surface can effectively polarize hydrogen molecules and improve their binding energy and storage capacity. While each Li site on BC3 nanotube is found to adsorb up to two H2 molecules, zigzag nanotube shows better performance as a hydrogen storage material with good adsorption energy (0.11 eV/H2) and high gravimetric density (6.9%). These data are consistent with the system target set by the DOE for 2010.
Co-reporter:M. M. Wu ; X. Zhong ; Q. Wang ; Q. Sun ; R. Pandey ;P. Jena
The Journal of Physical Chemistry C 2011 Volume 115(Issue 48) pp:23978-23983
Publication Date(Web):November 15, 2011
DOI:10.1021/jp207973b
By use of density functional theory and nonequilibrium Green’s function technique we have studied the electronic structure and transport properties of tubular Janus structures composed of hybridized carbon (C) and boron-nitride (BN) nanotubes (NTs) with carbon caps at both ends. The effect of chirality was probed by focusing on (5, 5) armchair and (9, 0) zigzag structures, both of which have similar radii and are metallic in the infinite length limit. The study has revealed a number of interesting properties: (1) The highest-occupied molecular orbital (HOMO)–lowest-unoccupied molecular orbtial (LUMO) gap is decreased when BN patch is inserted to zigzag structure but increased when it is inserted in the arm chair structure and graphene sheet. (2) The zigzag edges of the heterojunctions lead to anisotropy of frontier orbital distribution where the HOMO and LUMO are respectively predominated by B–C and N–C bonding states. Consequently, one side or one end becomes more reactive than the other. (3) The current in tubular Janus structures is reduced from that in pristine carbon nanotubes. At low bias, the current in the zigzag structure increases with increasing tube length while reverse is the case with the arm chair structure.
Co-reporter:Z. P. Jiang ; X. Zhou ; Qiang Sun ; Qian Wang ;Puru Jena
The Journal of Physical Chemistry C 2010 Volume 114(Issue 45) pp:19202-19205
Publication Date(Web):October 22, 2010
DOI:10.1021/jp1058403
Recent studies of hydrogen storage have focused on lithium metal atoms as dopants in a variety of substrates as Li is the lightest metallic element in the periodic table. In this work, we have explored the role of Li3N nanostructures in hydrogen storage as they possess Li atoms with varying degrees of coordination. We have performed detailed calculations of geometries, electronic structures, and hydrogen adsorption properties of free (Li3N)n (n = 1−7) clusters and those supported on BN nanoribbons by using density functional theory and generalized gradient approximation for the exchange and correlation potential. We found general motifs of (Li3N)n clusters where N sites form polygons for n ≤ 4 and polyhedrons n ≥ 5. The binding energies per formula unit increase with size, whereas the HOMO−LUMO gaps decrease. The HOMO is mainly contributed by Li and the LUMO by N. The bonding between Li and N has both ionic and covalent character. Lithium sites with low coordination are found to have a stronger adsorption energy for hydrogen molecules, which varies in the range of 0.08−0.11 eV/H2. When deposited on a BN nanoribbon, the Li3N molecules show stronger adsorption of hydrogen due to the changes in charge distribution. This suggests that Li3N molecules or small clusters can be introduced in porous substrates for enhancing hydrogen storage.
Co-reporter:J. Zhou;Q. Wang;Q. Sun;P. Jena;X. S. Chen
PNAS 2010 Volume 107 (Issue 7 ) pp:2801-2806
Publication Date(Web):2010-02-16
DOI:10.1073/pnas.0905571107
Using density functional theory, we show that an applied electric field can substantially improve the hydrogen storage properties
of polarizable substrates. This new concept is demonstrated by adsorbing a layer of hydrogen molecules on a number of nanomaterials.
When one layer of H2 molecules is adsorbed on a BN sheet, the binding energy per H2 molecule increases from 0.03 eV/H2 in the field-free case to 0.14 eV/H2 in the presence of an electric field of 0.045 a.u. The corresponding gravimetric density of 7.5 wt% is consistent with the
6 wt% system target set by Department of Energy for 2010. The strength of the electric field can be reduced if the substrate
is more polarizable. For example, a hydrogen adsorption energy of 0.14 eV/H2 can be achieved by applying an electric field of 0.03 a.u. on an AlN substrate, 0.006 a.u. on a silsesquioxane molecule,
and 0.007 a.u. on a silsesquioxane sheet. Thus, application of an electric field to a polarizable substrate provides a novel
way to store hydrogen; once the applied electric field is removed, the stored H2 molecules can be easily released, thus making storage reversible with fast kinetics. In addition, we show that materials
with rich low-coordinated nonmetal anions are highly polarizable and can serve as a guide in the design of new hydrogen storage
materials.
Co-reporter:Qian Wang, Qiang Sun, Puru Jena and Yoshiyuki Kawazoe
ACS Nano 2009 Volume 3(Issue 3) pp:621
Publication Date(Web):March 3, 2009
DOI:10.1021/nn800815e
The capability of AlN nanostructures (nanocages, nanocones, nanotubes, and nanowires) to store hydrogen has been studied using gradient-corrected density functional theory. In contrast to bulk AlN, which has the wurtzite structure and four-fold coordination, the Al sites in AlN nanostructures are unsaturated and have two- and three-fold coordination. Each Al atom is capable of binding one H2 molecule in quasi-molecular form, leading to 4.7 wt % hydrogen, irrespective of the topology of the nanostructures. With the exception of AlN nanotubes, energetics does not support the adsorption of additional hydrogen. The binding energies of hydrogen to these unsaturated metal sites lie in the range of 0.1−0.2 eV/H2 and are ideal for applications under ambient thermodynamic conditions. Furthermore, these materials do not suffer from the clustering problem that often plagues metal-coated carbon nanostructures.Keywords: cage; cone; hydrogen storage; nanostructure with exposed metal sites; tube; wire
Co-reporter:Yu Chen, Qiang Sun and Puru Jena
Journal of Materials Chemistry A 2016 - vol. 4(Issue 26) pp:NaN6361-6361
Publication Date(Web):2016/06/07
DOI:10.1039/C6TC01138A
Complementing the group of two-dimensional (2D) binary phosphorene analogues, we carried out first-principles calculations for α-SiTe and β-SiTe monolayers which are, respectively, black-phosphorene-like and blue-phosphorene-like. We show that both the SiTe monolayers are dynamically, thermally and mechanically stable, although α-SiTe with significant elastic anisotropy is energetically more favorable than β-SiTe. Both monolayers exhibit superior mechanical flexibility and are indirect-gap semiconductors with band gaps of 0.57 and 2.36 eV, respectively. What is even more important is that the α-SiTe monolayer can be tuned from an indirect band gap semiconductor to a direct band gap semiconductor and eventually to a metal when biaxial strains are applied, showing a high degree of flexibility in band engineering which is absent in non-silicon based analogues.
Co-reporter:Min Kan, Hong Gi Nam, Young Hee Lee and Qiang Sun
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 22) pp:NaN14871-14871
Publication Date(Web):2015/05/08
DOI:10.1039/C5CP01649E
The phase stability and phase transition of transition metal dichalcogenide (TMD) monolayer materials have attracted tremendous attention due to their attractive diverse potential applications. Here, first-principles calculations based on density-functional theory are carried out to study the newly synthesized MoTe2 monolayer. A phase different from the semiconducting trigonal prismatic structure and octahedral coordinated structure is found to be stable at room temperature in a free standing state, as evidenced by phonon spectrum analysis and molecular dynamic simulation. Raman vibrations of all the possible phases are calculated to provide additional information for the distinction of different phases in the experiment.
Co-reporter:Xiong Gu and Qiang Sun
Physical Chemistry Chemical Physics 2013 - vol. 15(Issue 37) pp:NaN15440-15440
Publication Date(Web):2013/07/11
DOI:10.1039/C3CP51200B
Using density functional theory (DFT) and time-dependent DFT in different levels, we have systematically studied new porphyrin-based dyes with A–π–D structures focusing on optical absorption, light harvest efficiency, ground state oxidation potential, excited state oxidation potential, and natural transition orbitals. Compared with existing dyes, we find that these new dyes have wide absorption regions (400–1000 nm) with high molar extinction coefficients, and display good energy level alignment for efficient injection of electrons and fast regeneration of the oxidized dyes.
Co-reporter:Min Kan, Subash Adhikari and Qiang Sun
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 10) pp:NaN4994-4994
Publication Date(Web):2014/01/22
DOI:10.1039/C3CP55146F
Using density functional theory combined with Monte Carlo (MC) simulations, we show that the two dimensional (2D) MnS2 and MnSe2 sheets are ideal magnetic semiconductors with long-range magnetic ordering and high magnetic moments (3 μB per unit cell), where all the Mn atoms are ferromagnetically coupled, and the Curie temperatures (TC) estimated for MnS2 and MnSe2 by the MC simulations are 225 and 250 K, respectively, which can be further increased to 330 K and 375 K by applying 5% biaxial tensile strains.
Co-reporter:Hong Fang, Shuo Wang, Junyi Liu, Qiang Sun and Puru Jena
Journal of Materials Chemistry A 2017 - vol. 5(Issue 26) pp:NaN13381-13381
Publication Date(Web):2017/04/06
DOI:10.1039/C7TA01648D
The development of cheap, durable, and safe inorganic solid electrolytes with superionic conductivity is the key for the next generation of all-solid metal-ion batteries. The recent discovery of antiperovskites with composition Li3OA (A = halogen) shows promise in this regard. Here, we demonstrate the potential of a new class of antiperovskites where halogens are replaced by BH4 superhalogens. In addition to maintaining the high ionic conductivity of Li3OA, Li3O(BH4) is lightweight, mechanically flexible, thermodynamically more stable, and electronically more insulating than Li3OA. By mixing BH4 with Cl to make Li3O(BH4)0.5Cl0.5, we further show that the conductivity will be doubled. The Li+-ion conductivity of the new materials is of the order of 10−4 to over 10−3 S cm−1 at room temperature and will be well above 10−2 S cm−1 at higher temperatures below the melting point. The conduction mechanism of the material is revealed by identifying the relationship between the orientational symmetry of the BH4− rotors and the potential surface felt by the lithium ions.
Co-reporter:Junyi Liu, Qiang Sun, Yoshiyuki Kawazoe and Puru Jena
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 13) pp:NaN8784-8784
Publication Date(Web):2015/09/30
DOI:10.1039/C5CP04835D
In addition to spintronics another motivation for exploring ferromagnetic two-dimensional materials is for biomedical applications such as magnetic labeling and hyperthermia treatment of tumors. Unfortunately, the widely studied Mn-containing monolayer is not biocompatible, although it is ferromagnetic. Here using first principles calculations combined with Monte Carlo simulations based on the Ising model, we systematically study a class of 2D ferromagnetic monolayers CrX3 (X = Cl, Br, I). The feasibility of exfoliation from their layered bulk phase is confirmed by the small cleavage energy and high in-plane stiffness. Spin-polarized calculations, combined with self-consistently determined Hubbard U that accounts for strong correlation energy, demonstrate that CrX3 (X = Cl, Br, I) monolayers are ferromagnetic and that Cr is trivalent and carries a magnetic moment of 3 μB; the resulting Cr3+ ions are biocompatible. The corresponding Curie temperatures for CrCl3, CrBr3 and CrI3 are found to be 66, 86, and 107 K, respectively, which can be increased to 323, 314, and 293 K by hole doping. The biocompatibility and ferromagnetism render these Cr-containing trihalide monolayers unique for applications.
Co-reporter:Guizhi Zhu, Junyi Liu, Qiang Sun and Puru Jena
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 27) pp:NaN17874-17874
Publication Date(Web):2016/06/07
DOI:10.1039/C6CP01908K
Motivated by the recent synthesis of bi-coordinated transition metal–organic complexes [Samuel, et al., Chem. Sci., 2015, 6, 3148], we have studied the structure and magnetic properties of a series of bi-coordinated transition metal based nanorings by folding quasi-1D chains. Among the cyclic alkyl(amino)carbine (CAAC) based quasi-1D chains (TM-CAAC, TM = Cr, Mn, Fe, Co, Ni), only Cr-CAAC is found to be ferromagnetic. First-principles calculations combined with Heisenberg–Dirac–van Vleck models were performed to understand the onset of robust ferromagnetism in Cr-based systems. With increasing size, the infrared intensity increases and the exchange energy oscillates. In particular, when the number, n, of TM-CAAC units is even and larger than 3, the magnetic coupling in nanorings is stronger than that in quasi-1D chains. The band gap changes very slowly with size. More importantly, compared with the highly coordinated Cr single molecular magnets, the low coordination of Cr ions enhances magnetic moment and stabilizes ferromagnetic coupling.