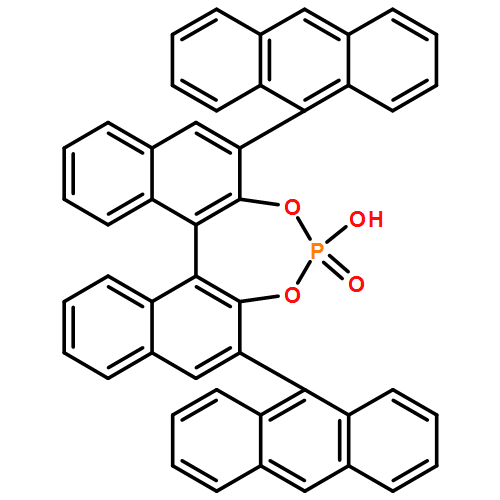
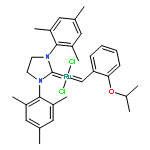
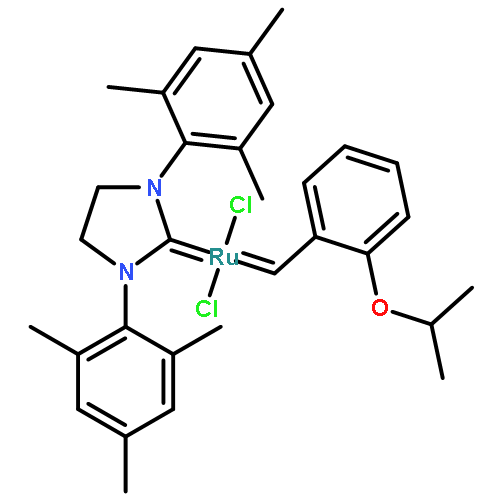
Co-reporter:Ryukichi Takagi and Takehiko Nishi
Organic & Biomolecular Chemistry 2015 vol. 13(Issue 45) pp:11039-11045
Publication Date(Web):11 Sep 2015
DOI:10.1039/C5OB01760B
Asymmetric desymmetrization of 4,4-disubstituted cyclohexadienones via a chiral phosphoric acid-catalyzed enantioselective intermolecular Diels–Alder reaction is developed.
Co-reporter:Ryukichi Takagi, Koumei Yamamoto, Yoshikazu Hiraga, Satoshi Kojima, Manabu Abe
Journal of Organometallic Chemistry 2013 723() pp: 171-175
Publication Date(Web):
DOI:10.1016/j.jorganchem.2012.09.004
Co-reporter:Ryukichi Takagi, Nao Igata, Kazuhiro Yamamoto, Satoshi Kojima
Journal of Organometallic Chemistry 2011 696(8) pp: 1556-1564
Publication Date(Web):
DOI:10.1016/j.jorganchem.2010.12.031
Co-reporter:Ryukichi Takagi, Nao Igata, Kazuhiro Yamamoto, Satoshi Kojima
Journal of Molecular Catalysis A: Chemical 2010 321(1–2) pp: 71-76
Publication Date(Web):
DOI:10.1016/j.molcata.2010.02.004
Co-reporter:Ryukichi Takagi, Yuta Inoue and Katsuo Ohkata
The Journal of Organic Chemistry 2008 Volume 73(Issue 23) pp:9320-9325
Publication Date(Web):November 3, 2008
DOI:10.1021/jo801595y
The construction of a highly functionalized adamantane core of plukenetione-type polycyclic polyprenylated acylphloroglucinols (PPAPs) is described. The method features the construction of the bicyclo[3.3.1]nonane core (3) by successive Michael reactions and the construction of the adamantane core of plukenetione-type PPAPs by acid-catalyzed cyclization of a bicyclo[3.3.1]nonane precursor (2).
Co-reporter:Ryukichi Takagi, Yukiko Miwa, Takashi Nerio, Yuta Inoue, Shuji Matsumura and Katsuo Ohkata
Organic & Biomolecular Chemistry 2007 vol. 5(Issue 2) pp:286-300
Publication Date(Web):08 Dec 2006
DOI:10.1039/B615702E
A method for the practical construction of poly-functionalized bicyclo[3.3.1]nonenones by successive Michael reactions of cyclohexenones 1 with acrylates 2 using K2CO3 and TBAB (n-Bu4N+Br−) was developed. The construction could be carried out in both stepwise and one-pot reactions with similar tendencies in regioselectivity. The α-regioselectivity in the intramolecular Michael reaction agreed with that stereoelectronically expected in intermolecular reactions based upon consideration of the HOMO orbital profile of the enolate I, the precursor to ring-closure, although the reaction site was trisubstituted and prone to steric hindrance in most of the examples presented. For the acetoxymethylacrylates substituted at either the α or γ position, steric hindrance of the substituents (R2 and R3) served as a controlling factor to induce high regiocontrol. Facial selection in the protonation of enolate II, formed upon ring-closure, was also affected by these substituents. In both the intramolecular Michael reaction and the protonation of enolate II, the ammonium counter cation played an important role.
Co-reporter:Ryukichi Takagi and Takehiko Nishi
Organic & Biomolecular Chemistry 2015 - vol. 13(Issue 45) pp:NaN11045-11045
Publication Date(Web):2015/09/11
DOI:10.1039/C5OB01760B
Asymmetric desymmetrization of 4,4-disubstituted cyclohexadienones via a chiral phosphoric acid-catalyzed enantioselective intermolecular Diels–Alder reaction is developed.
Co-reporter:Ryukichi Takagi, Yukiko Miwa, Takashi Nerio, Yuta Inoue, Shuji Matsumura and Katsuo Ohkata
Organic & Biomolecular Chemistry 2007 - vol. 5(Issue 2) pp:NaN300-300
Publication Date(Web):2006/12/08
DOI:10.1039/B615702E
A method for the practical construction of poly-functionalized bicyclo[3.3.1]nonenones by successive Michael reactions of cyclohexenones 1 with acrylates 2 using K2CO3 and TBAB (n-Bu4N+Br−) was developed. The construction could be carried out in both stepwise and one-pot reactions with similar tendencies in regioselectivity. The α-regioselectivity in the intramolecular Michael reaction agreed with that stereoelectronically expected in intermolecular reactions based upon consideration of the HOMO orbital profile of the enolate I, the precursor to ring-closure, although the reaction site was trisubstituted and prone to steric hindrance in most of the examples presented. For the acetoxymethylacrylates substituted at either the α or γ position, steric hindrance of the substituents (R2 and R3) served as a controlling factor to induce high regiocontrol. Facial selection in the protonation of enolate II, formed upon ring-closure, was also affected by these substituents. In both the intramolecular Michael reaction and the protonation of enolate II, the ammonium counter cation played an important role.

![2-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-1H-Pyrrole-1-carboxylic acid 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/141300/141293-28-9.png)
![2-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-1H-Pyrrole-1-carboxylic acid 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/141300/141293-28-9_b.png)




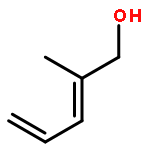




























![Dinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin,4-hydroxy-2,6-bis[2,4,6-tris(1-methylethyl)phenyl]-, 4-oxide, (11bR)-](/data/chemimg/115800/791616-63-2.png)
![Dinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin,4-hydroxy-2,6-bis[2,4,6-tris(1-methylethyl)phenyl]-, 4-oxide, (11bR)-](/data/chemimg/115800/791616-63-2_b.png)