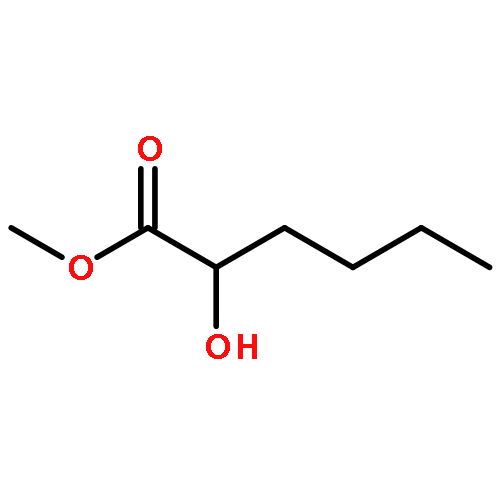
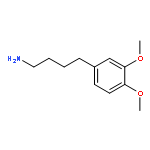
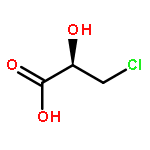
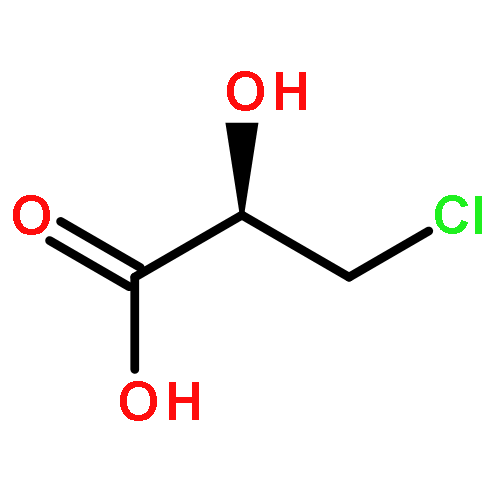
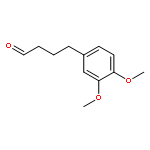
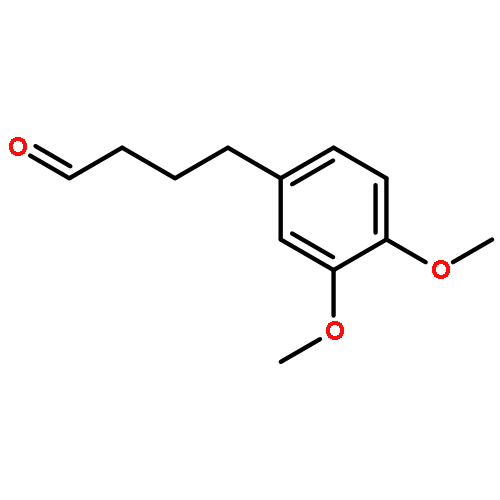
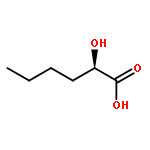
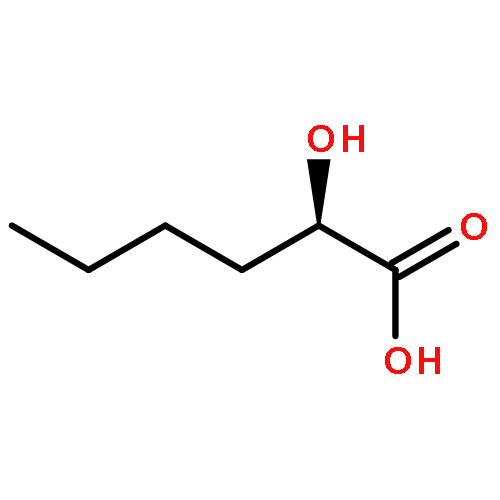
Co-reporter:Akane Kamiyama, Mado Nakajima, Liyou Han, Kei Wada, Masaharu Mizutani, Yukiko Tabuchi, Akiko Kojima-Yuasa, Isao Matsui-Yuasa, Hideyuki Suzuki, Keiichi Fukuyama, Bunta Watanabe, Jun Hiratake
Bioorganic & Medicinal Chemistry 2016 Volume 24(Issue 21) pp:5340-5352
Publication Date(Web):1 November 2016
DOI:10.1016/j.bmc.2016.08.050
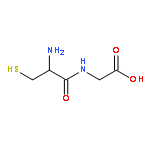
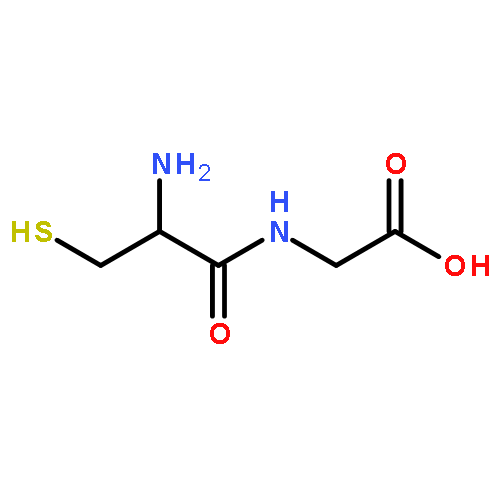
γ-Glutamyl transpeptidase (GGT, EC 2.3.2.2) that catalyzes the hydrolysis and transpeptidation of glutathione and its S-conjugates is involved in a number of physiological and pathological processes through glutathione metabolism and is an attractive pharmaceutical target. We report here the evaluation of a phosphonate-based irreversible inhibitor, 2-amino-4-{[3-(carboxymethyl)phenoxy](methoyl)phosphoryl}butanoic acid (GGsTop) and its analogues as a mechanism-based inhibitor of human GGT. GGsTop is a stable compound, but inactivated the human enzyme significantly faster than the other phosphonates, and importantly did not inhibit a glutamine amidotransferase. The structure–activity relationships, X-ray crystallography with Escherichia coli GGT, sequence alignment and site-directed mutagenesis of human GGT revealed a critical electrostatic interaction between the terminal carboxylate of GGsTop and the active-site residue Lys562 of human GGT for potent inhibition. GGsTop showed no cytotoxicity toward human fibroblasts and hepatic stellate cells up to 1 mM. GGsTop serves as a non-toxic, selective and highly potent irreversible GGT inhibitor that could be used for various in vivo as well as in vitro biochemical studies.
Co-reporter:Mado Nakajima, Bunta Watanabe, Liyou Han, Bun-ichi Shimizu, Kei Wada, Keiichi Fukuyama, Hideyuki Suzuki, Jun Hiratake
Bioorganic & Medicinal Chemistry 2014 Volume 22(Issue 3) pp:1176-1194
Publication Date(Web):1 February 2014
DOI:10.1016/j.bmc.2013.12.034
γ-Glutamyl transpeptidase (GGT) catalyzing the cleavage of γ-glutamyl bond of glutathione and its S-conjugates is involved in a number of physiological and pathological processes through glutathione homeostasis. Defining its Cys-Gly binding site is extremely important not only in defining the physiological function of GGT, but also in designing specific and effective inhibitors for pharmaceutical purposes. Here we report the synthesis and evaluation of a series of glutathione-analogous peptidyl phosphorus esters as mechanism-based inhibitors of human and Escherichia coli GGTs to probe the structural and stereochemical preferences in the Cys-Gly binding site. Both enzymes were inhibited strongly and irreversibly by the peptidyl phosphorus esters with a good leaving group (phenoxide). Human GGT was highly selective for l-aliphatic amino acid such as l-2-aminobutyrate (l-Cys mimic) at the Cys binding site, whereas E. coli GGT significantly preferred l-Phe mimic at this site. The C-terminal Gly and a l-amino acid analogue at the Cys binding site were necessary for inhibition, suggesting that human GGT was highly selective for glutathione (γ-Glu-l-Cys-Gly), whereas E. coli GGT are not selective for glutathione, but still retained the dipeptide (l-AA-Gly) binding site. The diastereoisomers with respect to the chiral phosphorus were separated. Both GGTs were inactivated by only one of the stereoisomers with the same stereochemistry at phosphorus. The strict recognition of phosphorus stereochemistry gave insights into the stereochemical course of the catalyzed reaction. Ion-spray mass analysis of the inhibited E. coli GGT confirmed the formation of a 1:1 covalent adduct with the catalytic subunit (small subunit) with concomitant loss of phenoxide, leaving the peptidyl moiety that presumably occupies the Cys-Gly binding site. The peptidyl phosphonate inhibitors are highly useful as a ligand for X-ray structural analysis of GGT for defining hitherto unidentified Cys-Gly binding site to design specific inhibitors.
Co-reporter:Hideyuki Ikeuchi, Yong-Mo Ahn, Takuya Otokawa, Bunta Watanabe, Lamees Hegazy, Jun Hiratake, Nigel G.J. Richards
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 19) pp:5915-5927
Publication Date(Web):1 October 2012
DOI:10.1016/j.bmc.2012.07.047
An adenylated sulfoximine transition-state analogue 1, which inhibits human asparagine synthetase (hASNS) with nanomolar potency, has been reported to suppress the proliferation of an l-asparagine amidohydrolase (ASNase)-resistant MOLT-4 leukemia cell line (MOLT-4R) when l-asparagine is depleted in the medium. We now report the synthesis and biological activity of two new sulfoximine analogues of 1 that have been studied as part of systematic efforts to identify compounds with improved cell permeability and/or metabolic stability. One of these new analogues, an amino sulfoximine 5 having no net charge at cellular pH, is a better hASNS inhibitor (KI∗ = 8 nM) than 1 and suppresses proliferation of MOLT-4R cells at 10-fold lower concentration (IC50 = 0.1 mM). More importantly, and in contrast to the lead compound 1, the presence of sulfoximine 5 at concentrations above 0.25 mM causes the death of MOLT-4R cells even when ASNase is absent in the culture medium. The amino sulfoximine 5 exhibits different dose-response behavior when incubated with an ASNase-sensitive MOLT-4 cell line (MOLT-4S), supporting the hypothesis that sulfoximine 5 exerts its effect by inhibiting hASNS in the cell. Our work provides further evidence for the idea that hASNS represents a chemotherapeutic target for the treatment of leukemia, and perhaps other cancers, including those of the prostate.
Co-reporter:Hideyuki Ikeuchi, Megan E. Meyer, Yun Ding, Jun Hiratake, Nigel G.J. Richards
Bioorganic & Medicinal Chemistry 2009 Volume 17(Issue 18) pp:6641-6650
Publication Date(Web):15 September 2009
DOI:10.1016/j.bmc.2009.07.071
The first sulfoximine-based inhibitor of human asparagine synthetase (ASNS) with nanomolar potency has been shown to suppress proliferation of asparaginase-resistant MOLT-4 cells in the presence of l-asparaginase. This validates literature hypotheses concerning the viability of human ASNS as a target for new drugs against acute lymphoblastic leukemia and ovarian cancer. Developing structure–function relationships for this class of human ASNS inhibitors has proven difficult, however, primarily because of the absence of rapid synthetic procedures for constructing highly functionalized sulfoximines. We now report conditions for the efficient preparation of these compounds by coupling sulfoxides and sulfamides in the presence of a rhodium catalyst. Access to this methodology has permitted the construction of two new adenylated sulfoximines, which were expected to exhibit similar binding affinity and better bioavailability than the original human ASNS inhibitor. Steady-state kinetic characterization of these compounds, however, has revealed the importance of a localized negative charge on the inhibitor that mimics that of the phosphate group in a key acyl-adenylate reaction intermediate. These experiments place an important constraint on the design of sulfoximine libraries for screening experiments to obtain ASNS inhibitors with increased potency and bioavailability.
Co-reporter:Jun Hiratake
The Chemical Record 2005 Volume 5(Issue 4) pp:
Publication Date(Web):22 JUL 2005
DOI:10.1002/tcr.20045
Carefully designed molecules that are intimately related to the reaction mechanism of enzymes are often highly selective and potent inhibitors that serve as extremely useful chemical probes for understanding the reaction mechanism and structure of enzymes. This article describes the design, synthesis, and applications of specific inhibitors of two mechanistically distinct groups of enzymes, ATP-dependent amide ligases and Ser- and Thr-hydrolases. Our strategy is based on the premise that stable analogues of the transition state (transition-state analogues) are highly potent inhibitors that serve as good mechanistic probes, and that a key structure of a good inhibitor of one enzyme is also utilized for the inhibitors of other enzymes that share the same chemistry in their catalyzed reactions, irrespective of the degree of structural similarity and evolutionary link between the enzymes. According to these principles, we designed and synthesized a series of phosphinate- and sulfoximine-based transition-state analogue inhibitors of glutathione synthetase, γ-glutamylcysteine synthetase and asparagine synthetase. For the second group of enzymes, we synthesized a γ-monofluorophosphono glutamate analogue for mechanism-based affinity labeling of γ-glutamyltranspeptidase and fluorescent phosphonic acid esters for the active-site titration of lipase. These inhibitors were used successfully as ligands for detailed kinetic analyses, X-ray crystallography, and mass analysis of the enzymes to identify the key amino acid residues responsible for catalysis and substrate recognition in the transition state. © 2005 The Japan Chemical Journal Forum and Wiley Periodicals, Inc. Chem Rec 5: 209–228; 2005: Published online in Wiley InterScience (www.interscience.wiley.com) DOI 10.1002/tcr.20045
Co-reporter:Kazuko Inoue, Jun Hiratake, Masaharu Mizutani, Masayasu Takada, Mikio Yamamoto, Kanzo Sakata
Carbohydrate Research 2003 Volume 338(Issue 14) pp:1477-1490
Publication Date(Web):4 July 2003
DOI:10.1016/S0008-6215(03)00201-5
An affinity adsorbent for β-glycosidases has been prepared by using β-glycosylamidine as a ligand. β-Glucosylamidine and β-galactosylamidine, highly potent and selective inhibitors of β-glucosidases and β-galactosidases, respectively, were immobilized by a novel one-pot procedure involving the addition of a β-glycosylamine and 2-iminothiolane·HCl simultaneously to a matrix modified with maleimido groups via an appropriate spacer to give an affinity adsorbent for β-glucosidases and β-galactosidases, respectively. This one-pot procedure enables various β-glycosylamidine ligands to be formed and immobilized conveniently according to the glycon substrate specificities of the enzymes. A crude enzyme extract from tea leaves (Camellia sinensis) and a β-galactosidase from Penicillium multicolor were chromatographed directly on each affinity adsorbent to give a β-glucosidase and a β-galactosidase to apparent homogeneity in one step by eluting the column with glucose or by a gradient NaCl elution, respectively. The β-glucosidase and β-galactosidase were inhibited competitively by a soluble form of the corresponding β-glycosylamidine ligand with an inhibition constant (Ki) of 2.1 and 0.80 μM, respectively. Neither enzyme was bound to the adsorbent with a mismatched ligand, indicating that the binding of the glycosidases was of specific nature that corresponds to the glycon substrate specificity of the enzymes. The ease of preparation and the selective nature of the affinity adsorbent should promise a large-scale preparation of the affinity adsorbent for the purification and removal of specific glycosidases according to their glycon substrate specificities.β-Glycosylamidine was used as a ligand for affinity chromatography of β-glycosidases to effect one-step purification of β-glycosidases according to the glycon substrate specificity.
Co-reporter:Wenfei Guo, Jun Hiratake, Koichi Ogawa, Mikio Yamamoto, Seung-Jin Ma, Kanzo Sakata
Bioorganic & Medicinal Chemistry Letters 2001 Volume 11(Issue 4) pp:467-470
Publication Date(Web):26 February 2001
DOI:10.1016/S0960-894X(00)00706-X
β-d-Glycosylamidines, in which a glycon is connected via an N-glycoside linkage with a substituted amidine (aglycon), were synthesized in two steps from the corresponding sugars and served as stable and potent β-glycosidase inhibitors with high selectivity according to the glycon- and α, β-specificities of the enzymes.The synthesis and evaluation of β-d-glycosylamidines 1a–c as potent and selective β-glycosidase inhibitors (Ki=0.1 μM) are reported.