Co-reporter:M. Sc. Hassan Abul-Futouh;Dr. Laith R. Almazahreh;Dr. Takahiro Sakamoto; Dr. Nhu Y. T. Stessman; Dr. Dennis L. Lichtenberger; Dr. Richard S. Glass;Dr. Helmar Görls; Dr. Mohammad El-Khateeb; Dr. Philippe Schollhammer; Dr. Grzegorz Mloston; Dr. Wolfgang Weig
Chemistry - A European Journal 2017 Volume 23(Issue 2) pp:346-359
Publication Date(Web):2017/01/05
DOI:10.1002/chem.201603843
AbstractAnalogues of the [2Fe-2S] subcluster of hydrogenase enzymes in which the central group of the three-atom chain linker between the sulfur atoms is replaced by GeR2 and SnR2 groups are studied. The six-membered FeSCECS rings in these complexes (E=Ge or Sn) adopt an unusual conformation with nearly co-planar SCECS atoms perpendicular to the Fe-Fe core. Computational modelling traces this result to the steric interaction of the Me groups with the axial carbonyls of the Fe2(CO)6 cluster and low torsional strain for GeMe2 and SnMe2 moieties owing to the long C−Ge and C−Sn bonds. Gas-phase photoelectron spectroscopy of these complexes shows a shift of ionization potentials to lower energies with substantial sulfur orbital character and, as supported by the computations, an increase in sulfur character in the predominantly metal–metal bonding HOMO. Cyclic voltammetry reveals that the complexes follow an ECE-type reduction mechanism (E=electron transfer and C=chemical process) in the absence of acid and catalysis of proton reduction in the presence of acid. Two cyclic tetranuclear complexes featuring the sulfur atoms of two Fe2S2(CO)6 cores bridged by CH2SnR2CH2, R=Me, Ph, linkers were also obtained and characterized.
Co-reporter:Takuhei Yamamoto, Jixun Dai, Neil E. Jacobsen, Malika Ammam, Gabriel B. Hall, Olivier Mozziconacci, Christian Schöneich, George S. Wilson, and Richard S. Glass
Organic Letters 2016 Volume 18(Issue 15) pp:3522-3525
Publication Date(Web):July 12, 2016
DOI:10.1021/acs.orglett.6b01309
The electrochemical oxidation of thioethers is shown to be facilitated by neighboring amide participation. 1H NMR spectroscopic analysis in acetonitrile solution of two conformationally constrained compounds with such facilitation shows that two-electron participation by the amide π2 orbital can occur to stabilize the developing sulfur radical cation.
Co-reporter:Olivier Mozziconacci, Ganga Viswanathan Bhagavathy, Takuhei Yamamoto, George S. Wilson, Richard S. Glass, Christian Schöneich
Tetrahedron 2016 Volume 72(Issue 48) pp:7770-7789
Publication Date(Web):1 December 2016
DOI:10.1016/j.tet.2016.08.075
Oxidation of Met affects the stability of proteins, and was identified as a step in the beta amyloid-dependent pathogenesis of Alzheimer's disease. One-electron oxidation of Met is facilitated through stabilization of sulfide radical cations with electron-rich heteroatoms. The formation of such 2-center-3-electron bonds, formed between sulfide radical cations and amides, leads to pronounced product selectivity during biologically relevant oxidation conditions. Conformationally constrained methionine analogs embedded within a norbornane framework, i.e., 2,6-endo, endo- and 2,6-exo, endo-pyrrolidine amide thiomethyl bicyclo[2.2.1]heptanes were synthesized. Oxidation of both methionine analogs in the Fenton oxidation yielded some sulfoxide. In addition, the oxidation of the endo, endo-derivative generated a vinyl sulfide while the exo, endo-derivative was converted into a ketone, indicating a selective influence of a sulfur-oxygen 2-center-3-electron bond on product formation. Mechanistic details of product formation were investigated through the incorporation of stable isotopes.
Co-reporter:Takuhei Yamamoto, Malika Ammam, Sue A. Roberts, George S. Wilson, Richard S. Glass
Tetrahedron 2016 Volume 72(Issue 20) pp:2527-2534
Publication Date(Web):19 May 2016
DOI:10.1016/j.tet.2016.03.040
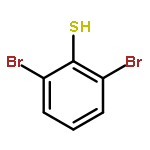
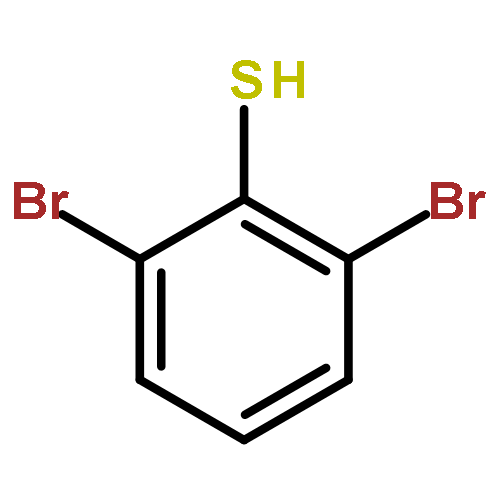
S-tert-Butyl m-terphenyl thioethers have been efficiently synthesized by Suzuki–Miyaura coupling reactions with 2,6-dibromo-S-tert-butylthio benzene. Selective monocoupling could be achieved with o-substituted boronic acids. This facilitated the synthesis of unsymmetrical S-tert-butyl m-terphenyl thioethers and bis(S-tert-butyl m-terphenyl thioether)s. The study of their electrochemistry showed facilitated oxidations resulting from through-space π⋯S⋯π interactions.
Co-reporter:G. Joel Meyer, Gabriel B. Hall, Elliott R. Smith, Takahiro Sakamoto, Dennis L. Lichtenberger, Richard S. Glass
Polyhedron 2015 Volume 86() pp:125-132
Publication Date(Web):28 January 2015
DOI:10.1016/j.poly.2014.06.050
A series of conformationally constrained 2,6-bisferrocenylphenyl thioethers were synthesized via Suzuki–Miyaura cross coupling reactions. Structural information was obtained using X-ray crystallography and dynamic 1H NMR spectroscopic studies, showing highly constrained m-terphenyl systems. Interaction of the ferrocene moieties through space mediated by the sulfur were studied by ultra-violet photoelectron spectroscopy (UPS), cyclic voltammetry, differential pulse voltammetry, UV–Vis–NIR spectroscopy and DFT computations. Electrochemical results show two, fully reversible 1e− redox processes for the ferrocenes where the separation of peaks is affected by both solvent and supporting electrolyte, suggesting significant electrostatic interaction which is further confirmed in the gas phase by UPS studies.Graphical abstractGeometrically constrained 2,6-diferrocenylphenylthioethers were synthesized and characterized. Through space interaction between the ferrocene moieties mediated by sulfur were evaluated by electrochemistry, photoelectron spectroscopy and UV–Vis–NIR spectroscopy of the mixed valence species. Enhanced interaction mediated by sulfur was ascribed to electrostatic interaction enhanced by the polarizability of sulfur.
Co-reporter:Nicolas P.-A. Monney, Thomas Bally, Takuhei Yamamoto, and Richard S. Glass
The Journal of Physical Chemistry A 2015 Volume 119(Issue 52) pp:12990-12998
Publication Date(Web):December 4, 2015
DOI:10.1021/acs.jpca.5b09665
Electronic absorption spectra and quantum chemical calculations of the radical cations of m-terphenyl tert-butyl thioethers, where the S–t-Bu bond is forced to be perpendicular to the central phenyl ring, show the occurrence of through-space [π···S···π]+ bonding interactions which lead to a stabilization of the thioether radical cations. In the corresponding methyl derivatives there is a competition between delocalization of the hole that is centered on a p-AO of the S atom into the π-system of the central phenyl ring or through space into the flanking phenyl groups, which leads to a mixture of planar and perpendicular conformations in the radical cation. Adding a second m-terphenyl tert-butyl thioether moiety does not lead to further delocalization; the spin and charge remain in one of the two halves of the radical cation. These findings have interesting implications with regard to the role of methionines as hopping stations in electron transfer through proteins.
Co-reporter:Gabriel B. Hall ; Rudresha Kottani ; Greg A. N. Felton ; Takuhei Yamamoto ; Dennis H. Evans ; Richard S. Glass ;Dennis L. Lichtenberger
Journal of the American Chemical Society 2014 Volume 136(Issue 10) pp:4012-4018
Publication Date(Web):February 14, 2014
DOI:10.1021/ja500087m
Reductive cleavage of disulfide bonds is an important step in many biological and chemical processes. Whether cleavage occurs stepwise or concertedly with electron transfer is of interest. Also of interest is whether the disulfide bond is reduced directly by intermolecular electron transfer from an external reducing agent or mediated intramolecularly by internal electron transfer from another redox-active moiety elsewhere within the molecule. The electrochemical reductions of 4,4′-bipyridyl-3,3′-disulfide (1) and the di-N-methylated derivative (22+) have been studied in acetonitrile. Simulations of the cyclic voltammograms in combination with DFT (density functional theory) computations provide a consistent model of the reductive processes. Compound 1 undergoes reduction directly at the disulfide moiety with a substantially more negative potential for the first electron than for the second electron, resulting in an overall two-electron reduction and rapid cleavage of the S–S bond to form the dithiolate. In contrast, compound 22+ is reduced at less negative potential than 1 and at the dimethyl bipyridinium moiety rather than at the disulfide moiety. Most interesting, the second reduction of the bipyridinium moiety results in a fast and reversible intramolecular two-electron transfer to reduce the disulfide moiety and form the dithiolate. Thus, the redox-active bipyridinium moiety provides a low energy pathway for reductive cleavage of the S–S bond that avoids the highly negative potential for the first direct electron reduction. Following the intramolecular two-electron transfer and cleavage of the S–S bond the bipyridinium undergoes two additional reversible reductions at more negative potentials.
Co-reporter:Mohammad K. Harb;Ahmad Daraosheh;Helmar Görls;Elliott R. Smith;G. Joel Meyer;Matthew T. Swenson;Takahiro Sakamoto;Dennis L. Lichtenberger;Dennis H. Evans;Mohammad El-khateeb;Wolfgang Weig
Heteroatom Chemistry 2014 Volume 25( Issue 6) pp:592-606
Publication Date(Web):
DOI:10.1002/hc.21216
ABSTRACT
Models of [FeFe]-hydrogenases containing diselenolato ligands with different bridge linker length have been prepared: Fe2(μ-Se(CH2)4Se-μ)(CO)6 (4DS), and Fe2(μ-Se(CH2)5Se-μ)(CO)6 (5DS) as well as dithiolato Fe2(μ-S(CH2)4S-μ)(CO)6 (4DT) and compared with Fe2(μ-S(CH2)3S-μ)(CO)6 (PDT) and Fe2(μ-Se(CH2)3Se-μ)(CO)6 (PDS). Compounds 4DT, PDS, 4DS, and 5DS were characterized by spectroscopic techniques including NMR, IR, mass spectrometry, ultraviolet photoelectron spectroscopy (UPS), elemental analysis, and X-ray crystal structure analysis. Combinations of electrochemical measurements, UPS, and density functional theory calculations indicate that oxidations of these five compounds are not significantly affected by chalcogen character but instead are governed by linker length. Cations for all compounds are calculated to adopt a bridged CO “rotated” structure with a vacant site on one of the Fe centers. In 4DT, 4DS, and 5DS, the alkane linker forms an agostic interaction with the vacant site on the rotated Fe. The reduction potentials for these compounds shift positively on average 0.16 V for each carbon added to the alkane linker with shifts being as large as 0.23 V between PDT and 4DT, and as small as 0.09 V between 4DS and 5DS. Catalytic reduction of protons from acetic acid in CH2Cl2 occurs at −1.79 and −1.86 V for PDT and 4DT and −2.02, −2.09, and −2.04 V for PDS, 4DS, and 5DS, indicating that chalcogen character is the primary factor that affects catalytic potential. On average the S-containing compounds catalyze proton reduction at potentials, which are 0.23 V less negative than the Se-containing compounds in this study.
Co-reporter:Nicolas P.-A. Monney, Thomas Bally, Ganga S. Bhagavathy, and Richard S. Glass
Organic Letters 2013 Volume 15(Issue 19) pp:4932-4935
Publication Date(Web):September 23, 2013
DOI:10.1021/ol402126f
The oxidation potential of thioethers constrained to be near aromatic rings is lowered, due to an antibonding interaction between the p-type sulfur lone pair with the neighboring phenyl π-system which on removal of an electron becomes a new kind of 3-electron S∴π bonding that reveals itself in the photoelectron spectrum and by an electronic transition involving the orbitals participating in the S∴π bond.
Co-reporter:Gabriel B. Hall, Jinzhu Chen, Charles A. Mebi, Noriko Okumura, Matthew T. Swenson, Stephanie E. Ossowski, Uzma I. Zakai, Gary S. Nichol, Dennis L. Lichtenberger, Dennis H. Evans, and Richard S. Glass
Organometallics 2013 Volume 32(Issue 21) pp:6605-6612
Publication Date(Web):October 7, 2013
DOI:10.1021/om400913p
Noninnocent ligands that are electronically coupled to active catalytic sites can influence the redox behavior of the catalysts. A series of (μ-dithiolato)Fe2(CO)6 complexes, in which the sulfur atoms of the μ-dithiolato ligand are bridged by 5-substituted (Me, OMe, Cl, t-Bu)-1,4-benzoquinones, 1,4-naphthoquinone, or 1,4-anthraquinone, have been synthesized and characterized. In addition, the bis-phosphine complex derived from the 1,4-naphthoquinone-bridged complex has also been prepared and characterized. Cyclic voltammetry of these complexes shows two reversible one-electron reductions at potentials 0.2 to 0.5 V less negative than their corresponding parent quinones. In the presence of acetic acid two-electron reductions of the complexes result in conversion of the quinones to hydroquinone moieties. EPR spectroscopic and computational studies of the one-electron-reduced complexes show electron delocalization from the semiquinones to the 2Fe2S moieties, thereby revealing the “noninnocent” behavior of these ligands with these catalysts.
Co-reporter:Takuhei Yamamoto;Pi-Yu Chen;Guangxin Lin;Anna B&x142;och-Mechkour;Neil E. Jacobsen;Thomas Bally
Journal of Physical Organic Chemistry 2012 Volume 25( Issue 10) pp:878-882
Publication Date(Web):
DOI:10.1002/poc.2939
Variable temperature 1H NMR spectroscopic studies of 2, 6-di(o-anisyl) anisole show syn and anti atropisomers at low temperature. The barrier for interconverting these isomers by rotation about the aryl-aryl bond, found by fitting the experimental data, is 41.2 kJ/mol. Copyright © 2012 John Wiley & Sons, Ltd.

![2H-Thieno[3,4-b][1,4]dioxepin-3-methanol, 3,4-dihydro-3-methyl-](/data/chemimg/123200/426263-16-3.png)
![2H-Thieno[3,4-b][1,4]dioxepin-3-methanol, 3,4-dihydro-3-methyl-](/data/chemimg/123200/426263-16-3_b.png)









![Benzene, 1,3-dibromo-2-[(1,1-dimethylethyl)thio]-](http://img.cochemist.com/ccimg/62300/62261-15-8.png)
![Benzene, 1,3-dibromo-2-[(1,1-dimethylethyl)thio]-](http://img.cochemist.com/ccimg/62300/62261-15-8_b.png)