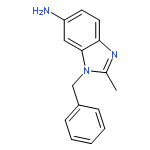
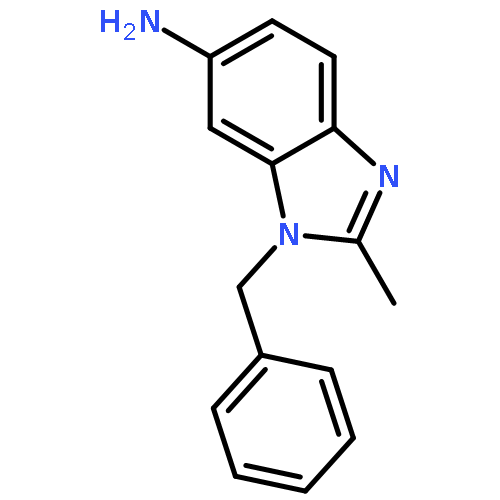
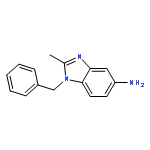
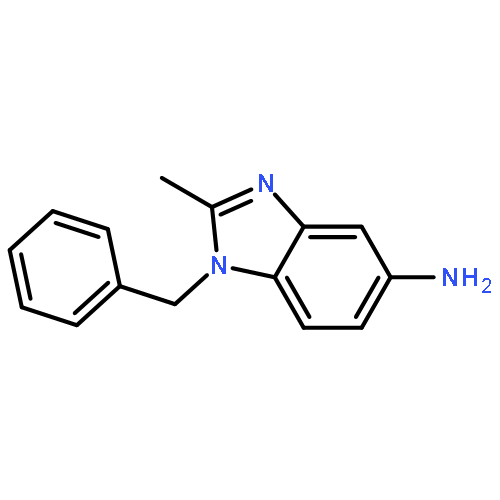
Co-reporter:Prinka Singla, Vijay Luxami, Raja Singh, Vibha Tandon, Kamaldeep Paul
European Journal of Medicinal Chemistry 2017 Volume 126(Volume 126) pp:
Publication Date(Web):27 January 2017
DOI:10.1016/j.ejmech.2016.09.093
•A Series of pyrazolo[3,4-d]pyrimidine-benzimidazole conjugate has been synthesized.•Compounds were evaluated in vitro for 60 human cancer cell lines.•Four compounds showed excellent antiproliferative activities.•Calf thymus-DNA interactions with compounds have been studied.•Compounds have also been investigated for bovine serum albumin-binding activity.A series of new pyrazolo[3,4-d]pyrimidine possessing 4-(1H-benzimidazol-2-yl)-phenylamine moiety at C4 position and primary as well as secondary amines at C6 position has been designed and synthesized. Their antitumor activities were evaluated against a panel of 60 human cancer cell lines at National Cancer Institute (NCI). Six compounds displayed potent and broad spectrum anticancer activities at 10 μM. Compounds 8, 12, 14 and 17 proved to be the most active and efficacious candidate in this series, with mean GI50 values of 1.30 μM, 1.43 μM, 2.38 μM and 2.18 μM, respectively against several cancer cell lines. Further biological evaluation of these compounds suggested that these compounds induce apoptosis and inhibit human topoisomerase (Topo) IIα as a possible intracellular target. UV-visible and fluorescence studies of these compounds revealed strong interaction with ct-DNA and bovine serum albumin (BSA).Download high-res image (166KB)Download full-size image
Co-reporter:Richa Goel, Vijay Luxami, Kamaldeep Paul
Journal of Photochemistry and Photobiology A: Chemistry 2017 Volume 348(Volume 348) pp:
Publication Date(Web):1 November 2017
DOI:10.1016/j.jphotochem.2017.08.009
•A series of triazole based imidazo[1,2-a]pyrazine-benzimidazole analogues have been synthesized.•Compounds were synthesized through click and Suzuki reactions.•Quantum yields of the synthesized compounds were determined.•FRET efficiency of the molecule has been used for the ratiometric detection of pyrophosphate.Triazole tethered imidazo[1,2-a]pyrazine-benzimidazole conjugates 13-38 has been synthesized by click and Suzuki-Miyaura cross coupling reactions at C-8 and C-6 positions, respectively. The research findings clearly predicted that by modification of electronic structure of the receptor, the sensitivity of the recognition process could be modified. Compound 24 with hydroxyphenyl substituent, showed stronger binding to the pyrophosphate than other compounds. Compound 24 has been used as selective probe for ratiometric detection of pyrophosphate amongst the other anions. The binding event of compound 24 toward PPi has been successfully evaluated by absorption and emission spectroscopy as well as NMR titration method. The compound 24 showed H-bonding assisted facilitation of FRET phenomenon in the presence of PPi.Download high-res image (196KB)Download full-size image
Co-reporter:Prinka Singla, Vijay Luxami, Kamaldeep Paul
European Journal of Medicinal Chemistry 2016 Volume 117() pp:59-69
Publication Date(Web):19 July 2016
DOI:10.1016/j.ejmech.2016.03.088
•A series of triazine-benzimidazole analogs has been synthesized.•Compounds were evaluated in vitro for their antitumor activity.•Four compounds showed excellent activity towards 60 human cancer cell lines.•Interaction with bovine serum albumin has been determined.A novel series of triazine-benzimidazole analogs has been designed and synthesized for their in vitro anticancer activities. Four compounds (6, 16, 17 and 20) were identified as highly potent anticancer agents against 60 human cancer cell lines with GI50 in the nanomolar range. To improve the drug applications toward cancer cells, there is a need to couple these compounds to some carrier macromolecules. Following this approach, the interaction between triazine-benzimidazole analogues and bovine serum albumin (BSA) has been investigated with UV–Visible and fluorescence spectroscopic methods under physiological conditions. The observed fluorescence quenching indicates that these compounds could efficiently bind with BSA and be transported to the target site.
Co-reporter:Richa Goel, Vijay Luxami and Kamaldeep Paul
RSC Advances 2016 vol. 6(Issue 44) pp:37664-37671
Publication Date(Web):05 Apr 2016
DOI:10.1039/C6RA07861C
Novel cassettes capable of energy transfer involving simple synthetic methods viz., copper catalyzed azide–alkyne cycloaddition (click reaction) at the C-8 position and palladium catalyzed Suzuki–Miyaura cross coupling at the C-6 position have been represented. The resulting imidazo[1,2-a]pyrazine-triazole bridged coumarin cassettes are capable of energy transfer from a donor core to an acceptor moiety.
Co-reporter:Prinka Singla, Vijay Luxami and Kamaldeep Paul
RSC Advances 2016 vol. 6(Issue 18) pp:14741-14750
Publication Date(Web):28 Jan 2016
DOI:10.1039/C5RA24001H
Triazine–benzimidazole analogues with different substitutions of primary and secondary amines as well as aryl groups were synthesized and characterized by 1H, 13C NMR and mass spectrometry. Interactions of these compounds with ct-DNA were explored by spectroscopic and viscometric techniques. These results and molecular modelling studies showed that compounds 7, 9–11, 16 and 21 interacted with ct-DNA through groove binding and not intercalation.
Co-reporter:Alka Sharma;Vijay Luxami;Sanjai Saxena
Archiv der Pharmazie 2016 Volume 349( Issue 3) pp:193-201
Publication Date(Web):
DOI:10.1002/ardp.201500281
A series of benzimidazole-based quinazoline derivatives with different substitutions of primary and secondary amines at the C2 position (1–12) were evaluated for their Aurora kinase inhibitory activities. All compounds except for 3 and 6 showed good activity against Aurora kinase inhibitors, with IC50 values in the range of 0.035–0.532 μM. The ligand efficiency (LE) of the compounds with Aurora A kinase was also determined. The structure–activity relationship and the quantitative structure–activity relationship revealed that the Aurora inhibitory activities of these derivatives primarily depend on the different substitutions of the amine present at the C2 position of the quinazoline core. Molecular docking studies in the active binding site also provided theoretical support for the experimental biological data acquired. The current study identifies a novel class of Aurora kinase inhibitors, which can further be used for the treatment of cancer.
Co-reporter:Prinka Singla, Vijay Luxami, Kamaldeep Paul
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 2) pp:518-523
Publication Date(Web):15 January 2016
DOI:10.1016/j.bmcl.2015.11.083
A series of triazine–benzimidazoles with 4-fluoroaniline substitution has been designed and synthesized. These compounds were further substituted with different primary and secondary amines. The structures of newly synthesized compounds were confirmed by 1H, 13C NMR, mass spectrometry and, in case of compound 18, by single crystal X-ray diffraction analysis. The newly synthesized compounds were evaluated against 60 human tumor cell lines at one dose and five dose concentration levels. Compounds 7, 8 and 22 have been found to be the most active antitumor agents with GI50 values of 1.77, 1.94 and 2.87 μM, respectively. The synthesized compounds were then evaluated for their inhibitory activity to mammalian dihydrofolate reductase. Compound 22 was depicted as the most active compound for the inhibition of dihydrofolate reductase with IC50 value of 2.0 nM. DNA binding studies were also revealed strong interacting properties of triazine derivatives towards calf thymus-DNA.
Co-reporter:Alka Sharma;Vijay Luxami
Journal of Heterocyclic Chemistry 2016 Volume 53( Issue 1) pp:241-248
Publication Date(Web):
DOI:10.1002/jhet.2330
An efficient palladium-catalyzed C–O and subsequent C–C bond formation of 2,4-dichloroquinazoline have been described. The designed strategy results in the synthesis of novel 2-arylated quinazolin-4-ones framework with various aryl/heteroaryl boronic acids in moderate to good yields along with 2,4-diarylated quinazolines. This methodology offers a direct transformation of aryl halides to aryl alcohols/ketone as well as the straight forward application to generate a wide variety of monoaryl and diaryl quinazoline.
Co-reporter:Richa Goel, Vijay Luxami and Kamaldeep Paul
Organic & Biomolecular Chemistry 2015 vol. 13(Issue 12) pp:3525-3555
Publication Date(Web):10 Dec 2014
DOI:10.1039/C4OB01380H
Imidazo[1,2-a]pyrazine acts as a versatile scaffold in organic synthesis and drug development. This review article is an effort to compile the progress made in synthetic methods and to illustrate its reactivity and multifarious biological activity. This review is mainly based on the pattern and position of the substitution, and should help the scientific community to bring about future developments.
Co-reporter:Alka Sharma, Vijay Luxami, Kamaldeep Paul
European Journal of Medicinal Chemistry 2015 Volume 93() pp:414-422
Publication Date(Web):26 March 2015
DOI:10.1016/j.ejmech.2015.02.036
•A new series of regioisomeric purine-benzimidazole hybrids.•Single X-ray determination of compound 19.•In vitro evaluation of synthesized compounds for their antitumor activity.•In vitro evaluation of Aurora kinase inhibitors.•Molecular modeling in active sites of Aurora A kinase.In an effort to identify novel compounds for the treatment of cancer, a diverse array of potential bioactive hybrid, purine-benzimidazole was synthesized in good yields through nucleophilic substitution at C6 position of purine ring with versatile cyclic amines at C2 position. The structures of newly prepared compounds were confirmed by IR, 1H, 13C NMR, mass spectroscopy and, in case of 19, by single crystal X-ray diffraction analysis. The newly synthesized compounds were evaluated against 60 human tumour cell lines at one dose concentration level. Compound 6 exhibited significant growth inhibition and was evaluated as 60 cell panel at five dose concentration levels. Compound 6 proved to be 1.25 fold more active than the positive control 5-FU, with GI50 value of 18.12 μM (MG-MID). Interaction of the compounds with Aurora-A enzyme involved in the process of propagation of cancer, has also been investigated. Compound 6 showed selectivity towards Aurora-A kinase inhibition with IC50 value of 0.0l μM. Molecular docking studies in the active binding site provided theoretical support for the experimental biological data acquired.
Co-reporter:Alka Sharma, Vijay Luxami, Kamaldeep Paul
European Journal of Medicinal Chemistry 2015 Volume 96() pp:381
Publication Date(Web):26 May 2015
DOI:10.1016/j.ejmech.2015.04.029
Co-reporter:Richa Goel, Vijay Luxami and Kamaldeep Paul
RSC Advances 2015 vol. 5(Issue 47) pp:37887-37895
Publication Date(Web):07 Apr 2015
DOI:10.1039/C5RA00584A
A new series of imidazo[1,2-a]pyrazine–coumarin hybrids have been synthesized by the combination of two biologically active moieties, imidazo[1,2-a]pyrazine and coumarin, followed by the Suzuki–Miyaura cross coupling reaction for monoarylation at the C6 position and symmetrical/unsymmetrical diarylation at the C3 and C6 positions. These compounds were further screened for their in vitro antitumor activities.
Co-reporter:Meenakshi Verma, Vijay Luxami and Kamaldeep Paul
RSC Advances 2015 vol. 5(Issue 52) pp:41803-41813
Publication Date(Web):01 May 2015
DOI:10.1039/C5RA00925A
A novel series of 2-allyl-6-substituted-benzo[de]isoquinoline-1,3-diones has been synthesized and evaluated against 60 human tumor cell lines for their in vitro antitumor activities. Compound 6b proved to be the most active member at a single dose concentration of 10 μM and broad spectrum of antitumor activity with GI50, TGI and LC50 values of 84.2 nM, 27.6 μM and 89.3 μM respectively at five dose concentration levels. The DNA binding properties of this compound has been investigated by a UV-Vis and fluorescence spectrophotometer as well as thermal denaturation experiments. Molecular docking studies of compound 6b have also supported the corresponding biological data.
Co-reporter:Richa Goel, Vijay Luxami and Kamaldeep Paul
RSC Advances 2015 vol. 5(Issue 99) pp:81608-81637
Publication Date(Web):15 Sep 2015
DOI:10.1039/C5RA14795F
Imidazo[1,2-a]pyrimidine has been receiving significant attention in the synthetic chemistry community through different chemosynthetic methodologies viz., multicomponent reactions, condensation reactions, intramolecular cyclizations, tandem reaction, carbon–hydrogen, carbon–carbon, carbon–nitrogen bond formation, aza-Michael–Mannich reaction, chiral compounds synthesis etc. The mechanisms for the selected reactions are also discussed to observe the formation of this heterocyclic moiety. This review comprehensively summarizes the recently reported various synthetic approaches along with its functionalization at 3-positions to construct this privileged scaffold for further use in the development of new chemosynthetic strategies and drug development due to its wide range of applications in medicinal chemistry.
Co-reporter:Prinka Singla, Vijay Luxami, Kamaldeep Paul
Bioorganic & Medicinal Chemistry 2015 23(8) pp: 1691-1700
Publication Date(Web):
DOI:10.1016/j.bmc.2015.03.012
Co-reporter:Richa Goel, Vijay Luxami and Kamaldeep Paul
RSC Advances 2014 vol. 4(Issue 19) pp:9885-9892
Publication Date(Web):19 Dec 2013
DOI:10.1039/C3RA47192F
Palladium catalyzed Suzuki–Miyaura cross-coupling reactions are reported for the synthesis of monoarylated (at the C8 position) and symmetrical diarylated (at the C6 and C8 positions) imidazo[1,2-a]pyrazines. Monoarylated products have also been used to synthesize unsymmetrical C6/C8 diarylated products. These compounds were screened for in vitro antitumor activities against a preliminary tumor cell line panel assay.
Co-reporter:Prinka Singla, Vijay Luxami and Kamaldeep Paul
RSC Advances 2014 vol. 4(Issue 24) pp:12422-12440
Publication Date(Web):18 Dec 2013
DOI:10.1039/C3RA46304D
Great advances in elucidating molecular structures allow the precise determination of the interactions between a protein and a therapeutic agent. Enzyme inhibitors are used as a therapeutic agent with organic molecules, that interact with their targets through the weak linkages of hydrogen bonding and van der Waals interactions. These reduce the undesirable side effects and allow more non-specific interactions with non-target molecules. Benzimidazole acts as an enzyme inhibitor that may interact with different proteins and enzymes and has inspired chemists to carry out various structural variations of it. This review discusses the development of distinct benzimidazoles with an array of enzyme inhibitors viz., aurora kinase inhibitors, cyclin-dependent kinase inhibitors, mitogen activated protein kinase inhibitors, polo like kinase inhibitors, Tie kinase inhibitors, lymphocyte specific kinase inhibitors etc., also highlighting the molecular interaction with enzyme inhibitors. Various derivatives of benzimidazole, with different inhibitory activities, have been described on the basis of substitution around the central moiety, with an aim to help medicinal chemists to develop structure–activity relationships. The reviews in the literature till now are focused only on the biological activities of benzimidazole viz., antiviral, anticancer and antifungal, but the present review focuses on the latest work, describing the inhibitor aspects and the potential of the benzimidazole ring. This discussion will further help in the development of novel benzimidazole compounds.
Co-reporter:Kamaldeep Paul, Alka Sharma, Vijay Luxami
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 2) pp:624-629
Publication Date(Web):15 January 2014
DOI:10.1016/j.bmcl.2013.12.005
By combining the structural features of quinazoline and benzimidazole, new hybrid regioisomeric molecules with substituted primary amines have been synthesized. Evaluation of these molecules over 60 cancer cell line panel has identified three molecules as most potent anticancer agents. Compound 10 showed ten and eleven folds more activity than respective quinazoline and benzimidazole class of compounds with GI50 value of 1.64 μM. Compound 11 (GI50 value of 0.81 μM) showed almost twenty and twenty-two fold more activity than quinazoline and benzimidazole analogue, respectively while compound 12 (GI50 value of 4.52 μM) has four fold more activity than quinazoline and benzimidazole analogue. In vitro evaluation of compound 11 exhibited remarkable anticancer activity towards colon cancer cell lines and prostate cancer cell lines at five dose concentrations with GI50 values of 0.34 and 0.31 μM, respectively.
Co-reporter:Kamaldeep Paul, Shweta Bindal, Vijay Luxami
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 12) pp:3667-3672
Publication Date(Web):15 June 2013
DOI:10.1016/j.bmcl.2012.12.071
A series of novel coumarin–benzimidazole hybrids, 3-(1H-benzo[d]imidazol-2-yl)-7-(substituted amino)-2H-chromen-2-one derivatives of biological interest were synthesized. Six out of the newly synthesized compounds were screened for in vitro antitumor activity against preliminary 60 tumor cell lines panel assay. A significant inhibition for cancer cells was observed with compound 8 (more than 50% inhibition) compared with other compounds and active known drug 5-fluorouracil (in some cell lines) as positive control. Compound 8 displayed appreciable anticancer activities against leukemia, colon cancer and breast cancer cell lines.
Co-reporter:Alka Sharma, Vijay Luxami, Kamaldeep Paul
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 11) pp:3288-3294
Publication Date(Web):1 June 2013
DOI:10.1016/j.bmcl.2013.03.107
A series of novel regioisomeric hybrids of quinazoline/benzimidazole viz. (3-allyl-2-methyl-3H-benzimidazol-5-yl)-(2-substituted-quinazolin-4-yl)-amine and (1-allyl-2-methyl-1H-benzimidazol-5-yl)-(2-substituted-quinazolin-4-yl)-amine of biological interest were synthesized. All the synthesized compounds were well characterized by 1H and 13C NMR as well as mass spectroscopy. The newly synthesized compounds were screened for in vitro antitumor activities against 60 tumor cell lines panel assay. A significant inhibition for cancer cells were observed with compound 9 and also more active against known drug 5-fluorouracil (5-FU) in some tumor cell lines. Compound 9 displayed appreciable anticancer activity against leukemia, colon, melanoma, renal and breast cancer cell lines.
Co-reporter:Prinka Singla, Vijay Luxami, Kamaldeep Paul
Journal of Photochemistry and Photobiology B: Biology (March 2017) Volume 168() pp:156-164
Publication Date(Web):March 2017
DOI:10.1016/j.jphotobiol.2017.02.009
Co-reporter:Richa Goel, Sheryl Sharma, Kamaldeep Paul, Vijay Luxami
Sensors and Actuators B: Chemical (July 2017) Volume 246() pp:776-782
Publication Date(Web):July 2017
DOI:10.1016/j.snb.2017.02.090
Co-reporter:Richa Goel, Vijay Luxami and Kamaldeep Paul
Organic & Biomolecular Chemistry 2015 - vol. 13(Issue 12) pp:NaN3555-3555
Publication Date(Web):2014/12/10
DOI:10.1039/C4OB01380H
Imidazo[1,2-a]pyrazine acts as a versatile scaffold in organic synthesis and drug development. This review article is an effort to compile the progress made in synthetic methods and to illustrate its reactivity and multifarious biological activity. This review is mainly based on the pattern and position of the substitution, and should help the scientific community to bring about future developments.

![IMIDAZO[1,2-A]PYRAZINE, 6,8-BIS(4-CHLOROPHENYL)-](http://img.cochemist.com/ccimg/852200/852101-93-0.png)
![IMIDAZO[1,2-A]PYRAZINE, 6,8-BIS(4-CHLOROPHENYL)-](http://img.cochemist.com/ccimg/852200/852101-93-0_b.png)
![Imidazo[1,2-a]pyrazine, 8-(4-chlorophenyl)-6-(4-methoxyphenyl)-](http://img.cochemist.com/ccimg/852200/852101-92-9.png)
![Imidazo[1,2-a]pyrazine, 8-(4-chlorophenyl)-6-(4-methoxyphenyl)-](http://img.cochemist.com/ccimg/852200/852101-92-9_b.png)
![Imidazo[1,2-a]pyrazine, 6-(4-chlorophenyl)-8-(4-methoxyphenyl)-](http://img.cochemist.com/ccimg/852200/852101-89-4.png)
![Imidazo[1,2-a]pyrazine, 6-(4-chlorophenyl)-8-(4-methoxyphenyl)-](http://img.cochemist.com/ccimg/852200/852101-89-4_b.png)
![IMIDAZO[1,2-A]PYRAZINE, 6,8-BIS(4-METHOXYPHENYL)-](http://img.cochemist.com/ccimg/852200/852101-88-3.png)
![IMIDAZO[1,2-A]PYRAZINE, 6,8-BIS(4-METHOXYPHENYL)-](http://img.cochemist.com/ccimg/852200/852101-88-3_b.png)
![IMIDAZO[1,2-A]PYRAZINE, 6,8-DIPHENYL-](http://img.cochemist.com/ccimg/852200/852101-63-4.png)
![IMIDAZO[1,2-A]PYRAZINE, 6,8-DIPHENYL-](http://img.cochemist.com/ccimg/852200/852101-63-4_b.png)
![1H-BENZ[DE]ISOQUINOLINE-1,3(2H)-DIONE, 6-(HEXYLAMINO)-2-(2-PROPENYL)-](http://img.cochemist.com/ccimg/391300/391232-25-0.png)
![1H-BENZ[DE]ISOQUINOLINE-1,3(2H)-DIONE, 6-(HEXYLAMINO)-2-(2-PROPENYL)-](http://img.cochemist.com/ccimg/391300/391232-25-0_b.png)
![1H-Benz[de]isoquinoline-1,3(2H)-dione, 6-(butylamino)-2-(2-propenyl)-](http://img.cochemist.com/ccimg/391300/391232-24-9.png)
![1H-Benz[de]isoquinoline-1,3(2H)-dione, 6-(butylamino)-2-(2-propenyl)-](http://img.cochemist.com/ccimg/391300/391232-24-9_b.png)








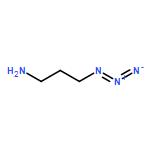
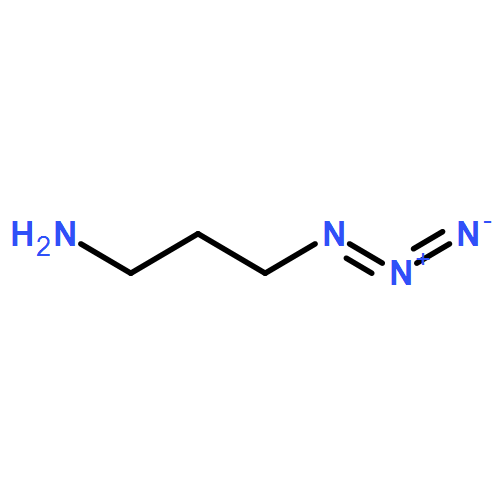
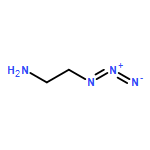
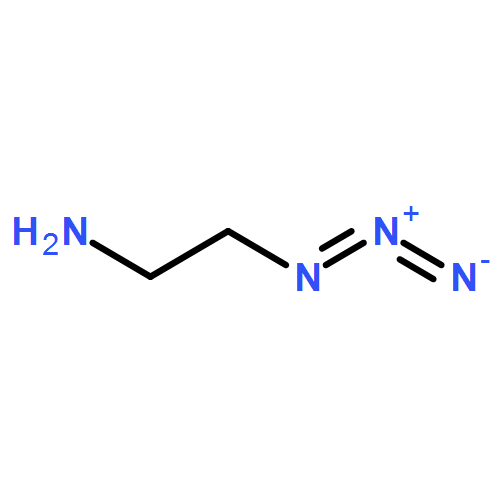


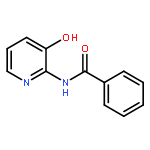
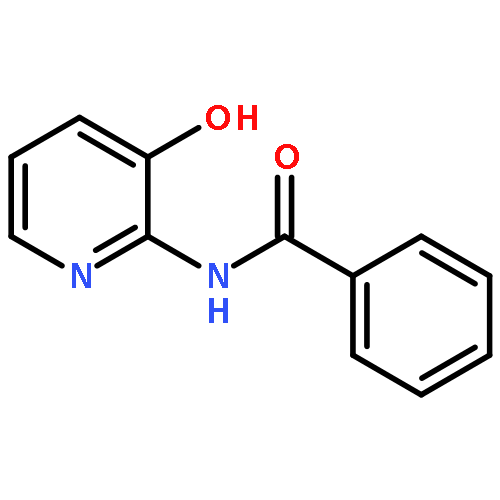
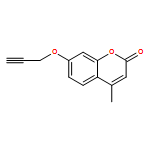
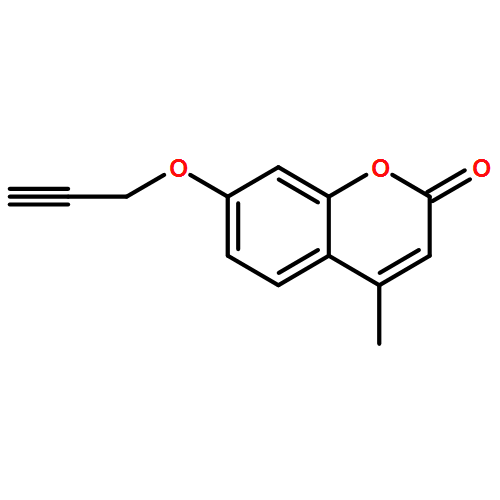
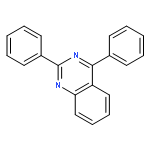
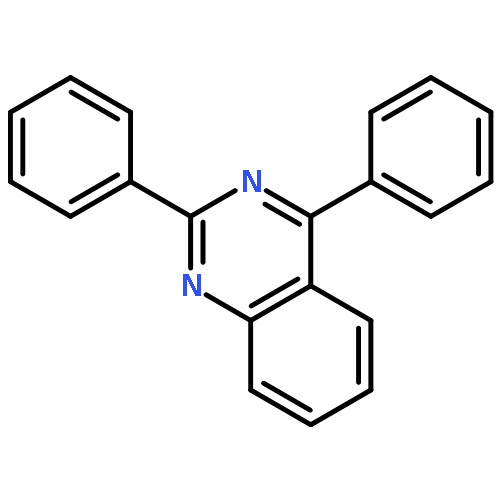








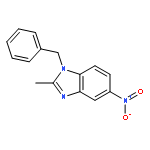
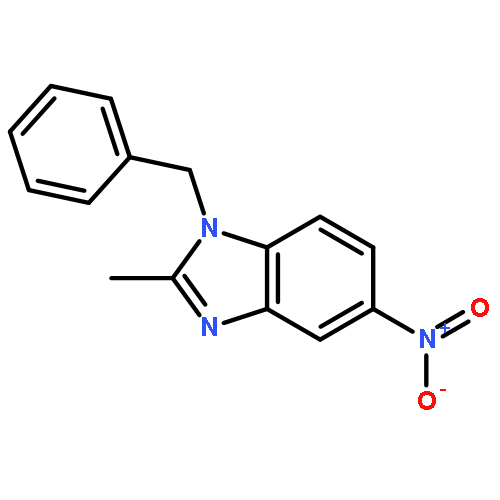


![1-(2,4-DIMETHYLPHENYL)-1H-PYRAZOLO[3,4-D]PYRIMIDIN-4-OL](http://img.cochemist.com/ccimg/7500/7455-77-8.png)
![1-(2,4-DIMETHYLPHENYL)-1H-PYRAZOLO[3,4-D]PYRIMIDIN-4-OL](http://img.cochemist.com/ccimg/7500/7455-77-8_b.png)




![4-(1H-Benzo[d]imidazol-2-yl)aniline](http://img.cochemist.com/ccimg/3000/2963-77-1.png)
![4-(1H-Benzo[d]imidazol-2-yl)aniline](http://img.cochemist.com/ccimg/3000/2963-77-1_b.png)