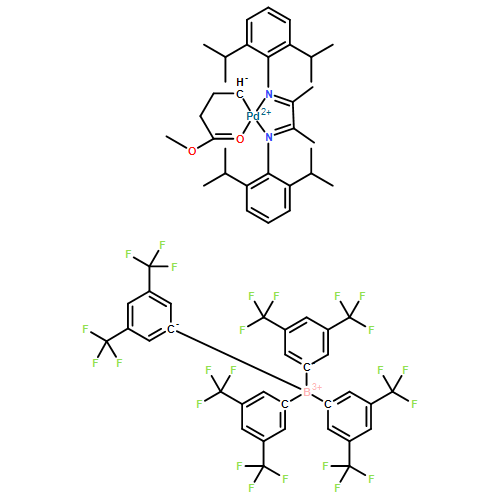
Co-reporter:Yoshio Nishimura, Jaeyoon Chung, Hurik Muradyan, and Zhibin Guan
Journal of the American Chemical Society October 25, 2017 Volume 139(Issue 42) pp:14881-14881
Publication Date(Web):October 9, 2017
DOI:10.1021/jacs.7b08826
Here we introduce silyl ether linkage as a novel dynamic covalent motif for dynamic material design. Through introduction of a neighboring amino moiety, we show that the silyl ether exchange rate can be accelerated by almost three orders of magnitude. By incorporating such silyl ether linkages into covalently cross-linked polymer networks, we demonstrate dynamic covalent network polymers displaying both malleability and reprocessability. The malleability of the networks is studied by monitoring stress relaxation at varying temperature, and their topology freezing temperatures are determined. The tunable dynamic properties coupled with the high thermal stability and reprocessability of silyl ether-based networks open doors to many potential applications for this family of materials.
Co-reporter:James A. Neal;Dr. Nathan J. Oldenhuis;Andrea L. Novitsky;Emil M. Samson;William J. Thrift; Regina Ragan; Zhibin Guan
Angewandte Chemie International Edition 2017 Volume 56(Issue 49) pp:15575-15579
Publication Date(Web):2017/12/04
DOI:10.1002/anie.201707587
AbstractMechanical gradients are often employed in nature to prevent biological materials from damage by creating a smooth transition from strong to weak that dissipates large forces. Synthetic mimics of these natural structures are highly desired to improve distribution of stresses at interfaces and reduce contact deformation in manmade materials. Current synthetic gradient materials commonly suffer from non-continuous transitions, relatively small gradients in mechanical properties, and difficult syntheses. Inspired by the polychaete worm jaw, we report a novel approach to generate stiffness gradients in polymeric materials via incorporation of dynamic monodentate metal–ligand crosslinks. Through spatial control of metal ion content, we created a continuous mechanical gradient that spans over a 200-fold difference in stiffness, approaching the mechanical contrast observed in biological gradient materials.
Co-reporter:James A. Neal;Dr. Nathan J. Oldenhuis;Andrea L. Novitsky;Emil M. Samson;William J. Thrift; Regina Ragan; Zhibin Guan
Angewandte Chemie 2017 Volume 129(Issue 49) pp:15781-15785
Publication Date(Web):2017/12/04
DOI:10.1002/ange.201707587
AbstractMechanical gradients are often employed in nature to prevent biological materials from damage by creating a smooth transition from strong to weak that dissipates large forces. Synthetic mimics of these natural structures are highly desired to improve distribution of stresses at interfaces and reduce contact deformation in manmade materials. Current synthetic gradient materials commonly suffer from non-continuous transitions, relatively small gradients in mechanical properties, and difficult syntheses. Inspired by the polychaete worm jaw, we report a novel approach to generate stiffness gradients in polymeric materials via incorporation of dynamic monodentate metal–ligand crosslinks. Through spatial control of metal ion content, we created a continuous mechanical gradient that spans over a 200-fold difference in stiffness, approaching the mechanical contrast observed in biological gradient materials.
Co-reporter:Mark E. Johnson, Judy Shon, Brian M. Guan, Joseph P. Patterson, Nathan J. Oldenhuis, Alexander C. Eldredge, Nathan C. Gianneschi, and Zhibin Guan
Bioconjugate Chemistry 2016 Volume 27(Issue 8) pp:1784
Publication Date(Web):July 25, 2016
DOI:10.1021/acs.bioconjchem.6b00216
We report the synthesis and study of fluorocarbon (FC) modified polyethylenimine (PEI) for the purpose of siRNA delivery. Low-molecular-weight PEI (Mn = 600) was functionalized with fluorocarbon epoxides of varying length. All FC-modified samples with greater than 2.0 equiv of FC epoxide per PEI induced potent gene silencing in vitro. Compared to hydrocarbon (HC) analogues, the FC vectors showed greater general silencing efficacy, higher cell uptake, and reduced association with serum components. Collectively, the data suggest that modification of polyamines with FCs is a promising approach for the discovery of novel vectors for siRNA delivery.
Co-reporter:Davoud Mozhdehi, James A. Neal, Scott C. Grindy, Yves Cordeau, Sergio Ayala, Niels Holten-Andersen, and Zhibin Guan
Macromolecules 2016 Volume 49(Issue 17) pp:6310-6321
Publication Date(Web):August 29, 2016
DOI:10.1021/acs.macromol.6b01626
Tunable mechanical response under dynamic and static loading is desirable for many technological applications. Traditionally, mechanical performance of polymeric materials is controlled by modulating structural (i.e., molecular weight, chain packing, or cross-link density) or temporal parameters (such as kinetics of the exchange of dynamic cross-linkers). Metal–ligand interactions are uniquely suited to control both structural and temporal parameters as the thermodynamics and kinetics of mechanically active cross-linkers can be varied by careful selection of metal without significant synthetic modification of the polymer backbone. Here, we have demonstrated that it is possible to engineer desired mechanical properties in a metallopolymer with a high degree of tunability by simply changing the type and amount of added metal. Specifically, we cross-linked an imidazole-containing brush copolymer system with the divalent cations of zinc, copper, and cobalt. Using rheology and tensile experiments, we have correlated the emergent mechanical properties to the stoichiometric ratio of ligand to metal as well as the coordination number and ligand exchange mechanism of the imidazole–metal cross-links. In contrary to the general view that unbound free ligands are normally regarded as mechanically inactive dangling chains in metallopolymer networks, this study clearly shows that they can play a critical role in stress distribution and chain relaxation. Importantly, this work shows for the first time that it is possible to simultaneously control both the structure of networks and the temporal response of bulk materials using dynamic association of weak and monodentate ligands with transition metals.
Co-reporter:Xiangqing Jia;Tobias Friedberger;Chuan Qin;Zheng Huang
Science Advances 2016 Volume 2(Issue 6) pp:e1501591
Publication Date(Web):17 Jun 2016
DOI:10.1126/sciadv.1501591
A catalytic alkane metathesis provides a mild and selective degradation of polyethylene wastes into valuable fuels and waxes.
Co-reporter:Alexander C. Eldredge, Mark E. Johnson, Nathan J. Oldenhuis, and Zhibin Guan
Biomacromolecules 2016 Volume 17(Issue 10) pp:3138
Publication Date(Web):August 26, 2016
DOI:10.1021/acs.biomac.6b00635
In this study, we report a new dipeptide functionalization strategy for developing new dendritic bolaamphiphile vectors for efficient siRNA transfection. A focused library of dipeptides was constructed using four amino acids: l-arginine, l-histidine, l-lysine, and l-tryptophan. The dipeptides were coupled to two dendritic bolaamphiphile scaffolds that we developed previously, allowing us to quickly access a focused library of discrete vectors with multivalent dendritic dipeptide functionalities. The resulting discrete bolaamphiphiles were screened for siRNA delivery in vitro in HEK-293 and HeLa cells. Bolaamphiphiles functionalized with dipeptides containing Lys or Arg and either His or Trp were the most effective for in vitro siRNA delivery. Necessary cationic charge to ensure efficient siRNA binding are provided by Arg and Lys residues, whereas endosomal escape is provided through pH responsive buffering of His or membrane interactions of Trp. The most effective vectors (F10 HR/RH) exhibited greater than 75% gene silencing in multiple cell lines and exhibited serum stability.
Co-reporter:Gregory A. Williams;Ryohei Ishige;Olivia R. Cromwell;Jaeyoon Chung;Atsushi Takahara
Advanced Materials 2015 Volume 27( Issue 26) pp:3934-3941
Publication Date(Web):
DOI:10.1002/adma.201500927
Co-reporter:Amir Hashemi, Nicolas Jouault, Gregory A. Williams, Dan Zhao, Kevin J. Cheng, Jeffrey W. Kysar, Zhibin Guan, and Sanat K. Kumar
Nano Letters 2015 Volume 15(Issue 8) pp:5465-5471
Publication Date(Web):July 20, 2015
DOI:10.1021/acs.nanolett.5b01859
It is now well accepted that the addition of nanoparticles (NPs) can strongly affect the thermomechanical properties of the polymers into which they are incorporated. In the solid (glassy) state, previous work has implied that optimal mechanical properties are achieved when the NPs are well dispersed in the matrix and when there is strong interfacial binding between the grafted NPs and the polymer matrix. Here we provide strong evidence supporting the importance of intermolecular interactions through the use of NPs grafted with polymers that can hydrogen bond with the matrix, yielding to significant improvements in the measured mechanical properties. Our finding thus supports the previously implied central role of strong interfacial binding in optimizing the mechanical properties of polymer nanocomposites.
Co-reporter:Nobuhiko Hosono; Aaron M. Kushner; Jaeyoon Chung; Anja R. A. Palmans; Zhibin Guan;E. W. Meijer
Journal of the American Chemical Society 2015 Volume 137(Issue 21) pp:6880-6888
Publication Date(Web):May 6, 2015
DOI:10.1021/jacs.5b02967
Atomic force microscopy (AFM)-based single-molecule force spectroscopy (SMFS) is applied to single-chain polymeric nanoparticles (SCPNs) to acquire information about the internal folding structure of SCPNs and inherent kinetic parameters of supramolecular self-assembling motifs embedded into the SCPNs. The SCPNs used here are polyacrylate-based polymers carrying 2-ureido-4-[1H]-pyrimidinone (UPy) or benzene-1,3,5-tricarboxamide (BTA) pendants that induce an intramolecular chain collapse into nanoparticles consisting of one polymer chain only via internal supramolecular cross-linking. The SCPN is stretched by an AFM cantilever to unfold mechanically, which allows measuring of force–extension profiles of the SCPNs. Consecutive peaks observed in the force profiles are attributed to rupture events of self-assembled UPy/BTA units in the SCPNs. The force profiles have been analyzed statistically for a series of polymers with different UPy/BTA incorporation densities. The results provide insights into the internal conformation of SCPNs, where the folding structure can be changed with the incorporation density of UPy/BTA. In addition, dynamic loading rate analysis allows the determination of kinetic parameters of BTA self-assembly, which has not been accessible by any other method. This study offers a rational tool for understanding the folding structure, kinetics, and pathway of two series of SCPNs.
Co-reporter:James A. Neal; Davoud Mozhdehi
Journal of the American Chemical Society 2015 Volume 137(Issue 14) pp:4846-4850
Publication Date(Web):March 19, 2015
DOI:10.1021/jacs.5b01601
Polymers that repair themselves after mechanical damage can significantly improve their durability and safety. A major goal in the field of self-healing materials is to combine robust mechanical and efficient healing properties. Here, we show that incorporation of sacrificial bonds into a self-repairable network dramatically improves the overall mechanical properties. Specifically, we use simple secondary amide side chains to create dynamic energy dissipative hydrogen bonds in a covalently cross-linked polymer network, which can self-heal via olefin cross-metathesis. We envision that this straightforward sacrificial bonding strategy can be employed to improve mechanical properties in a variety of self-healing systems.
Co-reporter:Olivia R. Cromwell; Jaeyoon Chung
Journal of the American Chemical Society 2015 Volume 137(Issue 20) pp:6492-6495
Publication Date(Web):May 6, 2015
DOI:10.1021/jacs.5b03551
Despite numerous strategies involving dynamic covalent bond exchange for dynamic and self-healing materials, it remains a challenge to be able to tune the malleability and self-healing properties of bulk materials through simple small molecule perturbations. Here we describe the use of tunable rates of boronic ester transesterification to tune the malleability and self-healing efficiencies of bulk materials. Specifically, we used two telechelic diboronic ester small molecules with variable transesterification kinetics to dynamically cross-link 1,2-diol-containing polymer backbones. The sample cross-linked with fast-exchanging diboronic ester showed enhanced malleability and accelerated healing compared to the slow-exchanging variant under the same conditions. Our report demonstrates the possibility of transferring small molecule kinetics to dynamic properties of bulk solid material and may serve as a guide for the rational design of tunable dynamic materials.
Co-reporter:Hanxiang Zeng, Cathleen Schlesener, Olivia Cromwell, Markus Hellmund, Rainer Haag, and Zhibin Guan
Biomacromolecules 2015 Volume 16(Issue 12) pp:
Publication Date(Web):November 16, 2015
DOI:10.1021/acs.biomac.5b01196
The development of safe and effective delivery vectors is a great challenge for the medicinal application of RNA interference (RNAi). In this study, we aimed to develop new synthetic transfection agents based on dendritic polyglycercol (dPG), which has shown great biocompatibility in several biomaterial applications. Histidine and aromatic amino acids were conjugated to the amine-terminated dPGs through amide bonds. We systematically tuned the amino acid combination, functionalization ratio, ligand density, and dPG core size to find optimal vectors. It was found that histidine–tryptophan-functionalized dPGs exhibited improved delivery efficiency and greatly reduced toxicity over simple amine-terminated dPGs. Furthermore, the optimized vectors exhibited strong siRNA binding and high transfection efficiency in serum containing media. The results indicate that the current amino acid-functionalized dPG system is a promising candidate for in vivo siRNA delivery applications.
Co-reporter:Zhongkai Wang, Feng Jiang, Yaqiong Zhang, Yezi You, Zhigang Wang, and Zhibin Guan
ACS Nano 2015 Volume 9(Issue 1) pp:271
Publication Date(Web):December 31, 2014
DOI:10.1021/nn506960f
Human skin exhibits highly nonlinear elastic properties that are essential to its physiological functions. It is soft at low strain but stiff at high strain, thereby protecting internal organs and tissues from mechanical trauma. However, to date, the development of materials to mimic the unique mechanical properties of human skin is still a great challenge. Here we report a bioinspired design of nanostructured elastomers combining two abundant plant-based biopolymers, stiff cellulose and elastic polyisoprene (natural rubber), to mimic the mechanical properties of human skin. The nanostructured elastomers show highly nonlinear mechanical properties closely mimicking that of human skin. Importantly, the mechanical properties of these nanostructured elastomers can be tuned by adjusting cellulose content, providing the opportunity to synthesize materials that mimic the mechanical properties of different types of skins. Given the simplicity, efficiency, and tunability, this design may provide a promising strategy for creating artificial skin for both general mechanical and biomedical applications.Keywords: human skin; mechanical processing; mechanical property mimicking; microphase separation; multiphase polymer; nonlinear elasticity;
Co-reporter:Hanxiang Zeng, Mark E. Johnson, Nathan J. Oldenhuis, Timothy N. Tiambeng, and Zhibin Guan
ACS Central Science 2015 Volume 1(Issue 6) pp:303
Publication Date(Web):August 14, 2015
DOI:10.1021/acscentsci.5b00233
Development of safe and effective delivery vectors is a critical challenge for the application of RNA interference (RNAi)-based biotechnologies. In this study we show the rational design of a series of novel dendritic peptide bolaamphiphile vectors that demonstrate high efficiency for the delivery of small interfering RNA (siRNA) while exhibiting low cytotoxicity and hemolytic activity. Systematic investigation into structure–property relationships revealed an important correlation between molecular design, self-assembled nanostructure, and biological activity. The unique bolaamphiphile architecture proved a key factor for improved complex stability and transfection efficiency. The optimal vector contains a fluorocarbon core and exhibited enhanced delivery efficiency to a variety of cell lines and improved serum resistance when compared to hydrocarbon analogues and lipofectamine RNAiMAX. In addition to introducing a promising new vector system for siRNA delivery, the structure–property relationships and “fluorocarbon effect” revealed herein offer critical insight for further development of novel materials for nucleic acid delivery and other biomaterial applications.
Co-reporter:Yulin Chen, Zhibin Guan
Polymer 2015 Volume 69() pp:249-254
Publication Date(Web):9 July 2015
DOI:10.1016/j.polymer.2015.03.023
•Brush copolymers with PMMA backbone and H-bonding PA-amide brushes were prepared.•The brush copolymers were able to self-assemble into spherical nanoparticles.•The materials showed thermoplastic elastomers with tunable mechanical properties.•The materials show a combination of strong mechanical and self-healing properties.We synthesized a series of brush copolymers having glassy polymethylmethacrylate (PMMA) backbone and flexible polyacrylate-amide (PA-amide) brushes that exhibit thermoplastic elastomer properties. Importantly, the dynamic hydrogen bonds in the soft PA-amide matrix enables the material to self-heal after mechanical damage at room temperature without the need of any external stimulus.Figure optionsDownload full-size imageDownload as PowerPoint slide
Co-reporter:Davoud Mozhdehi ; Sergio Ayala ; Olivia R. Cromwell
Journal of the American Chemical Society 2014 Volume 136(Issue 46) pp:16128-16131
Publication Date(Web):October 28, 2014
DOI:10.1021/ja5097094
A new self-healing multiphase polymer is developed in which a pervasive network of dynamic metal–ligand (zinc–imidazole) interactions are programmed in the soft matrix of a hard/soft two-phase brush copolymer system. The mechanical and dynamic properties of the materials can be tuned by varying a number of molecular parameters (e.g., backbone/brush degree of polymerization and brush density) as well as the ligand/metal ratio. Following mechanical damage, these thermoplastic elastomers show excellent self-healing ability under ambient conditions without any intervention.
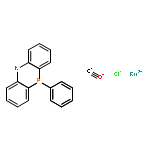
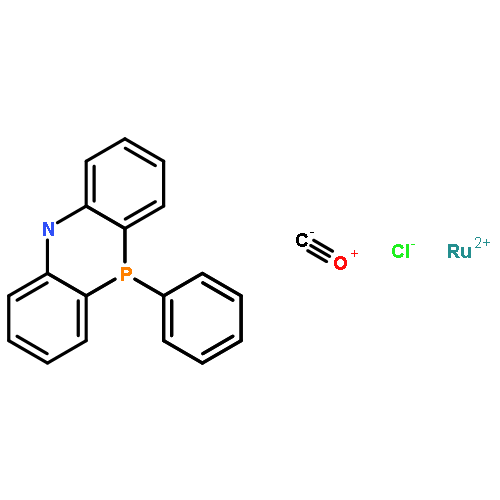
Co-reporter:Nathan J. Oldenhuis ; Vy M. Dong
Journal of the American Chemical Society 2014 Volume 136(Issue 36) pp:12548-12551
Publication Date(Web):August 29, 2014
DOI:10.1021/ja5058482
A commercially available ruthenium(II) PNP-type pincer catalyst (Ru-Macho) promotes the formation of α-chiral tert-butanesulfinylamines from racemic secondary alcohols and Ellman’s chiral tert-butanesulfinamide via a hydrogen borrowing strategy. The formation of α-chiral tert-butanesulfinylamines occurs in yields ranging from 31% to 89% with most examples giving >95:5 dr.
Co-reporter:Yulin Chen and Zhibin Guan
Chemical Communications 2014 vol. 50(Issue 74) pp:10868-10870
Publication Date(Web):28 Jul 2014
DOI:10.1039/C4CC03168G
Triblock copolymers having glassy PMMA blocks and dynamic hydrogen bonding blocks were synthesized by sequential atom transfer radical polymerization (ATRP). The dynamic triblock copolymers self-assemble into nanocomposite materials exhibiting a combination of mechanical strength, toughness, and self-healing capability.
Co-reporter:Nathan J. Oldenhuis, Vy M. Dong, Zhibin Guan
Tetrahedron 2014 70(27–28) pp: 4213-4218
Publication Date(Web):
DOI:10.1016/j.tet.2014.03.085
Co-reporter:Tobias Friedberger, Joseph W. Ziller, and Zhibin Guan
Organometallics 2014 Volume 33(Issue 8) pp:1913-1916
Publication Date(Web):April 16, 2014
DOI:10.1021/om5001343
The synthesis, characterization, and ethylene polymerization behavior of novel RuIV(η3:η3-C10H16)(OPO) (OPO = bis(arenesulfonato)phosphine) complexes is reported here. Upon activation with AlMe3-depleted methylaluminoxane (dMAO), the Ru(IV) precursors were able to produce polyethylene with activities up to 1182 h–1 turnover frequency (TOF). The polymers were highly linear with a low degree of branching (<12 methyl branches per 1000 C) and had high molecular weights (up to Mp = 289 kg mol–1) with a bimodal molecular weight distribution. The polymerization activity increased with decreasing donor strength of the OPO ligand.
Co-reporter:Hanxiang Zeng ; Hannah C. Little ; Timothy N. Tiambeng ; Gregory A. Williams
Journal of the American Chemical Society 2013 Volume 135(Issue 13) pp:4962-4965
Publication Date(Web):March 15, 2013
DOI:10.1021/ja400986u
In this study, we designed and synthesized a biodegradable dendronized polypeptide (denpol) platform for delivery of small interfering RNA (siRNA). The novel denpol architecture combines the multivalency of dendrimers and conformational flexibility of linear polymers for optimal siRNA binding. Multifunctional amino acids were incorporated onto the dendrons and the structure was tuned both systematically and combinatorially to select optimal vectors. By screening a focused library, we identified several denpols that can effectively deliver siRNA to NIH 3T3 cells in vitro and exhibit minimal toxicity. For comparison, the best-performing denpol showed significantly improved transfection efficiency over Lipofectamine in serum-containing media. Fluorescence intracellular trafficking studies indicated that amphiphilicity is important for cell uptake and that the buffering capacity of histidine facilitates endosomal membrane rupture and therefore enhances the transfection efficiency. The combination of high delivery efficiency in serum and low cytotoxicity suggests the denpol system as a promising new carrier for siRNA delivery.
Co-reporter:Miguel A. Camacho-Fernandez, Max Yen, Joseph W. Ziller and Zhibin Guan
Chemical Science 2013 vol. 4(Issue 7) pp:2902-2906
Publication Date(Web):07 May 2013
DOI:10.1039/C3SC50676B
We report here the first direct observation of a cationic ruthenium complex catalyzing ethylene insertion polymerization. An arene-tethered ruthenium complex (η6-C6H5(CH2)3SCH3RuCl2) (3) was synthesized and shown to be able to catalyze ethylene polymerization upon activation with AlMe2Cl. For mechanistic studies, we synthesized the homologous dimethylated η6-C6H5(CH2)3SCH3Ru(CH3)2 (4) complex, which upon activation with the Brookhart acid ([H(Et2O)2]+ [BAr′4]−) was also active for ethylene insertion polymerization. 1H-NMR and mass spectrometry (MS) studies provide direct evidence for a ruthenium cationic [η6-C6H5(CH2)3SCH3Ru(oligomer)]+ complex as the active species during polymerization. This has unambiguously shown for the first time a ruthenium complex as the active species for catalyzing olefin insertion polymerization.
Co-reporter:Davoud Mozhdehi and Zhibin Guan
Chemical Communications 2013 vol. 49(Issue 85) pp:9950-9952
Publication Date(Web):28 Aug 2013
DOI:10.1039/C3CC45419C
We describe in this manuscript a new design of supramolecular amino acids (SAAs) for directing peptide folding in organic environments. The incorporated supramolecular motif has a strong driving force to dimerize in a sequence- and orientation-specific manner. By introducing such SAAs into the primary sequence of peptides, the specific and directional dimerization of the supramolecular units should facilitate the folding of the peptides. Our approach may provide a general strategy to program secondary structures in organic media.
Co-reporter:Yulin Chen and Zhibin Guan
Polymer Chemistry 2013 vol. 4(Issue 18) pp:4885-4889
Publication Date(Web):13 Mar 2013
DOI:10.1039/C3PY00078H
New core–shell nanoparticles were synthesized by grafting a poly(acrylate amide) shell onto a cross-linked polystyrene nanoparticle (NP) via atom transfer radical polymerization (ATRP). The core–shell NPs self assemble into self-healing materials in which the dynamic hydrogen-bonding shell confers self-healing properties while the hard cross-linked core provides stiffness.
Co-reporter:Sophia W. Liao, Jeffrey Rawson, Keiko Omori, Kohei Ishiyama, Davoud Mozhdehi, Alina R. Oancea, Taihei Ito, Zhibin Guan, Yoko Mullen
Biomaterials 2013 34(16) pp: 3984-3991
Publication Date(Web):
DOI:10.1016/j.biomaterials.2013.02.007
Co-reporter:Hiromitsu Urakami, Jens Hentschel, Kellie Seetho, Hanxiang Zeng, Kanika Chawla, and Zhibin Guan
Biomacromolecules 2013 Volume 14(Issue 10) pp:
Publication Date(Web):September 18, 2013
DOI:10.1021/bm401039r
Nanogels have attracted much attention lately because of their many potential applications, including as nanocarriers for drug and gene delivery. Most nanogels reported previously, however, are not biodegradable, and their synthesis often requires the use of surfactants. Herein we report a surfactant-free method for the preparation of biodegradable, biocompatible, and stimuli-responsive cationic nanogels. The nanogels were synthesized by simply coaservating linear polymer precursors in mixed solvents followed by in situ cross-linking with homobifunctional cross-linkers. The versatility of this approach has been demonstrated by employing two different polymers and various cross-linkers to prepare nanogel particles with diameters ranging from 170 to 220 nm. Specifically, disulfide-containing tetralysine (TetK)- and oligoethylenimine (OEI)-based prepolymers were prepared and the subsequent nanogels were formed by covalently cross-linking the polymer coacervate phase. Nanogel particles are responsive to pH changes, increasing in size and zeta-potential with concomitant lowering of solution pH. Furthermore, as revealed by AFM imaging, nanogel particles were degradable in the presence of glutathione at concentrations similar to those in intracellular environment (10 mM). Both the nanogel and the polymer precursors were determined to exhibit minimal cytotoxicity against fibroblast 3T3 cells by flow cytometric analyses and fluorescent imaging. This study demonstrates a new surfactant-free method for preparing biodegradable, biocompatible, and stimuli-responsive nanogels as potential nanocarriers for the delivery of drugs and genes.
Co-reporter:Yi-Xuan Lu ; François Tournilhac ; Ludwik Leibler
Journal of the American Chemical Society 2012 Volume 134(Issue 20) pp:8424-8427
Publication Date(Web):May 8, 2012
DOI:10.1021/ja303356z
Covalently cross-linked polymers have many technological applications for their excellent properties, but they suffer from the lack of processability and adaptive properties. We report a simple, efficient method of generating adaptive cross-linked polymers via olefin metathesis. By introducing a very low level of the Grubbs’ second-generation Ru metathesis catalyst, a chemically cross-linked polybutadiene network becomes malleable at room temperature while retaining its insolubility. The stress relaxation capability increases with increasing level of catalyst loading. In sharp contrast, catalyst-free control samples with identical network topology and cross-linking density do not show any adaptive properties. This chemistry should offer a possibility to combine the dimensional stability and solvent resistance of cross-linked polymers and the processability/adaptibility of thermoplastics.
Co-reporter:Yi-Xuan Lu
Journal of the American Chemical Society 2012 Volume 134(Issue 34) pp:14226-14231
Publication Date(Web):August 6, 2012
DOI:10.1021/ja306287s
In this article, we demonstrate transition-metal-catalyzed olefin metathesis as a simple, effective method for healing polymers via dynamic exchange of strong carbon–carbon double bonds. Upon introducing a very low level of the Grubbs’ second-generation Ru metathesis catalyst into cross-linked polybutadiene (PBD) network, the material self-heals effectively at various conditions under moderate pressures. In sharp contrast, catalyst-free control samples with identical network topology and cross-linking density show minimal healing. The healing efficiency of the materials was carefully investigated under different concentrations of the Ru catalyst, compression pressures, and temperatures. It is demonstrated for the first time that a bulk polymer could effectively heal via dynamic covalent bond formation at sub-ambient temperature. The Ru-loaded PBD samples not only heal well with themselves but also with control samples without any catalyst. Furthermore, a completely Ru-free PBD network can heal effectively upon simply applying a very small amount of Ru catalyst only at the fracture surface. The simplicity and effectiveness of this self-healing approach make it potentially applicable to a wide range of olefin-containing polymers.
Co-reporter:Ying-Wei Yang, Jens Hentschel, Yi-Chun Chen, Mark Lazari, Hanxiang Zeng, R. Michael van Dam and Zhibin Guan
Journal of Materials Chemistry A 2012 vol. 22(Issue 3) pp:1100-1106
Publication Date(Web):17 Nov 2011
DOI:10.1039/C1JM14131G
We report the design and synthesis of a new perfluoropolyether-based material, which has liquid-like viscosity and can be cured into a tough, highly durable elastomer when “clicked” with selected tri-pod organic small molecules. This highly fluorinated elastomer exhibits remarkable resistance to a variety of organic solvents, water, heat and even harsh acidic and basic conditions. Whereas PDMS-based microfluidic devices are commonly used for aqueous based applications, their limited chemical resistance and high swellability in many common organic solvents make it unfeasible for microfluidic applications involving organic solvents and/or harsh conditions. With excellent chemical resistance and low swellability, our newly synthesized fluoro-elastomers will hopefully provide an alternative material for organic based microfluidic devices. Furthermore, the alkyne–azide “click” chemistry employed in curing not only provides high efficiency of synthesis and ease of device fabrication, but, more importantly, produces 1,2,3-triazole linkages that are very stable against harsh acidic or basic conditions. This work has great potential to expand microfluidics to a series of novel applications especially in organic and medicinal chemistry.
Co-reporter:Guobin Sun, Jens Hentschel, and Zhibin Guan
ACS Macro Letters 2012 Volume 1(Issue 5) pp:585
Publication Date(Web):April 18, 2012
DOI:10.1021/mz300069h
We report an efficient catalytic synthesis of “necklace” polymers, polyethylene (PE) denpols, utilizing the chain-walking polymerization (CWP). The approach is based on the design of a linear multivalent chain-walking catalyst that can initiate hyperbranched polymerization of ethylene for in situ formation of multiple dendritic PEs covalently tethered to the linear polymer backbone. The simplicity and efficiency of this approach makes it promising for facile preparation of large soluble nanostructures for various potential applications.
Co-reporter:Chris S. Popeney, Maike C. Lukowiak, Christoph Böttcher, Boris Schade, Pia Welker, Dorothea Mangoldt, Gesine Gunkel, Zhibin Guan, and Rainer Haag
ACS Macro Letters 2012 Volume 1(Issue 5) pp:564
Publication Date(Web):April 13, 2012
DOI:10.1021/mz300083y
A water-soluble molecular transporter with a dendritic core–shell nanostructure has been prepared by a tandem coordination, ring-opening, hyperbranched polymerization process. Consisting of hydrophilic hyperbranched polyglycerol shell grafted from hydrophobic dendritic polyethylene core, the transporter has a molecular weight of 951 kg/mol and a hydrodynamic diameter of 17.5 ± 0.9 nm, as determined by static and dynamic light scattering, respectively. Based on evidence from fluorescence spectroscopy, light scattering, and electron microscopy, the core–shell copolymer transports the hydrophobic guests pyrene and Nile red by a unimolecular transport mechanism. Furthermore, it was shown that the core–shell copolymer effectively transports the hydrophobic dye Nile red into living cells under extremely high and biologically relevant dilution conditions, which is in sharp contrast to a small molecule amphiphile. These results suggest potential applicability of such core–shell molecular transporters in the administration of poorly water-soluble drugs.
Co-reporter:Dr. Jens Hentschel;Dr. Aaron M. Kushner;Dr. Joseph Ziller ;Dr. Zhibin Guan
Angewandte Chemie 2012 Volume 124( Issue 42) pp:10713-10717
Publication Date(Web):
DOI:10.1002/ange.201204840
Co-reporter:Dr. Jens Hentschel;Dr. Aaron M. Kushner;Dr. Joseph Ziller ;Dr. Zhibin Guan
Angewandte Chemie International Edition 2012 Volume 51( Issue 42) pp:10561-10565
Publication Date(Web):
DOI:10.1002/anie.201204840
Co-reporter:Kanika Chawla, Ting-bin Yu, Lisa Stutts, Max Yen, Zhibin Guan
Biomaterials 2012 33(26) pp: 6052-6060
Publication Date(Web):
DOI:10.1016/j.biomaterials.2012.04.058
Co-reporter:Hanxiang Zeng
Journal of the American Chemical Society 2011 Volume 133(Issue 5) pp:1159-1161
Publication Date(Web):January 4, 2011
DOI:10.1021/ja106958s
We report a direct synthesis of polyamides via catalytic dehydrogenation of diols and diamines. A PNN pincer ruthenium complex, the Milstein catalyst, was used for this reaction and polyamides with number average molecular weight from ∼10 to 30 kDa could be obtained from a wide variety of diols and diamines bearing aliphatic or aromatic, linear or cyclic spacers. Because of the high catalytic selectivity of primary amine over secondary amine, polyamines could be conveniently incorporated into linear polyamides without tedious protection/deprotection steps. Compared with conventional condensation method, this catalytic system avoids the requirement of stoichiometric preactivation or in situ activation reagents and provides a much cleaner process with high atomic economy.
Co-reporter:Kanika Chawla, Ting−Bin Yu, Sophia W. Liao, and Zhibin Guan
Biomacromolecules 2011 Volume 12(Issue 3) pp:
Publication Date(Web):February 8, 2011
DOI:10.1021/bm100980w
Saccharide−peptide hydrogels have been developed in our laboratory as new synthetic extracellular matrices for regenerative medicine applications. In this work, we have expanded on our previously reported system and applied copolymerization of cysteine (Cys) and vinyl sulfone (VS)-functionalized saccharide−peptide polymers via Michael-type addition for encapsulation and 3D culture of cells. Specifically, our aims were to (1) develop a novel hydrogel platform, which could be applied for encapsulating and culturing mesenchymal stem cells (MSCs) in a 3D environment, (2) characterize the tunable properties of the hydrogel, specifically, degradation, mechanical, and gel network properties, and (3) determine the biocompatibility of the saccharide−peptide hydrogel material with MSCs. Hydrogel mechanical properties were tunable by varying the VS:Cys ratio (= 0.5, 1, or 2) as well as the pH (6, 7, or 8) of the cross-linking components. Stiffer gels were formed at VS:Cys = 1 and pH 6 or 7. Gels formed at pH 8 or with excess Cys (VS:Cys = 0.5) or VS (VS:Cys = 2) were significantly softer. Cross-linking pH and VS:Cys ratio also had an effect on the degradation behavior of the VS:Cys gels, with higher cross-linking pH resulting in an accelerated loss of mass. On the basis of environmental scanning electron microscopy (ESEM) analysis and fluorescence microscopy, all hydrogels appeared to exhibit porous gel networks. MSCs cultured in monolayer and exposed to soluble Cys or VS copolymers (0.1−5 mg/mL) did not exhibit measurable cytotoxicity. In addition, MSCs were cultured in 3D for up to 14 days in vitro without deleterious effects on cell viability. In summary, we have established and characterized a tunable 3D saccharide−peptide hybrid copolymer hydrogel platform for culturing MSCs. Future studies will focus on utilizing the hydrogel system for controlling the differentiation of MSCs.
Co-reporter:Dr. Aaron M. Kushner ;Dr. Zhibin Guan
Angewandte Chemie 2011 Volume 123( Issue 39) pp:9190-9223
Publication Date(Web):
DOI:10.1002/ange.201006496
Abstract
Unter Verwendung der zwanzig natürlichen Aminosäuren als Grundbausteine haben biologische Systeme während der Jahrmillionen unter evolutionärem und umweltbedingtem Druck stabile und leichte Peptid-basierte Materialien entwickelt. Diese Materialien übertreffen ihre synthetischen Gegenstücke in mehrfacher Hinsicht: 1) Multifunktionalität/Abstimmbarkeit, 2) Anpassungsfähigkeit/Reizempfindlichkeit, 3) Synthese und Weiterverarbeitung bei Umgebungsbedingungen in wässrigem Medium und 4) Wiederverwertung und biologische Abbaubarkeit. Diese besonderen Eigenschaften können durch eine “Bottom-up”-Synthese und die hierarchische Organisation auf verschiedenen Längenskalen erreicht werden. Das Arbeitsgebiet der “Biomimikry” beschäftigt sich damit, die Gestaltungsprinzipien natürlicher Materialien und deren molekulare Bausteine aufzuklären und zu kopieren. Hier beschreiben wir, was bereits über Struktur und molekulare Mechanismen natürlicher polymerer Materialien erforscht wurde, sowie den Fortschritt in Richtung synthetischer “Nachbildungen” dieser bemerkenswerten Systeme.
Co-reporter:Dr. Aaron M. Kushner ;Dr. Zhibin Guan
Angewandte Chemie International Edition 2011 Volume 50( Issue 39) pp:9026-9057
Publication Date(Web):
DOI:10.1002/anie.201006496
Abstract
Under eons of evolutionary and environmental pressure, biological systems have developed strong and lightweight peptide-based polymeric materials by using the 20 naturally occurring amino acids as principal monomeric units. These materials outperform their man-made counterparts in the following ways: 1) multifunctionality/tunability, 2) adaptability/stimuli-responsiveness, 3) synthesis and processing under ambient and aqueous conditions, and 4) recyclability and biodegradability. The universal design strategy that affords these advanced properties involves “bottom-up” synthesis and modular, hierarchical organization both within and across multiple length-scales. The field of “biomimicry”—elucidating and co-opting nature’s basic material design principles and molecular building blocks—is rapidly evolving. This Review describes what has been discovered about the structure and molecular mechanisms of natural polymeric materials, as well as the progress towards synthetic “mimics” of these remarkable systems.
Co-reporter:Chris S. Popeney, Chris M. Levins, and Zhibin Guan
Organometallics 2011 Volume 30(Issue 8) pp:2432-2452
Publication Date(Web):March 30, 2011
DOI:10.1021/om200193r
The synthesis of Ni(II) and Pd(II) cyclophane-based α-diimine olefin polymerization catalysts bearing a range of electron-donating or -withdrawing groups is described. Substituent effects were confirmed by measurement of CO infrared stretching frequencies in Pd(II) carbonyl complexes. Polymerizations with ethylene were investigated in detail involving determination of catalyst productivity and thermal stability, especially at elevated temperature, as well as analysis of polymer molecular weight and microstructure. The Ni(II) catalysts, formed by in situ treatment of tetraarylborate salts of [Ni(diimine)(acac)]+ with triisobutylaluminum, exhibited little variation in productivity or thermal stability across the substitution series, but the resulting polymers showed an increase in both molecular weight and branching density for catalysts with increasingly electron-withdrawing character. The chloride-substituted Pd(II) analogue, however, was notable in its markedly higher productivity and thermal stability at elevated temperature compared to the other substituted Pd(II) cyclophane catalysts, which otherwise showed little variation, like the Ni(II) catalysts. Unlike the previously studied acyclic Pd(II) α-diimine catalysts, a strong tendency toward higher molecular weight polymer with more electron-deficient catalysts was noted. An alternative dissociative chain transfer mechanism is proposed to account for this difference, as well as the generally lower than expected molecular weight of the cyclophane catalysts. As a further explanation for the unusual behavior of these catalysts, NMR evidence of a stabilizing ligand−metal H-agostic interaction in the case of the Pd(II) system is also reported.
Co-reporter:Yulin Chen
Journal of the American Chemical Society 2010 Volume 132(Issue 13) pp:4577-4579
Publication Date(Web):March 17, 2010
DOI:10.1021/ja9104446
Bioinspired modular synthesis of elastin-mimic polymers (EMPs) is achieved via Cu-catalyzed alkyne-azide cyclization (CuAAC). By changing the module, EMPs with different secondary structures determined by circular dichroism (CD) spectra in trifluoroethanol (TFE) solution are obtained. The EMPs are characterized by measuring the lower critical solution temperatures (LSCTs) and the bulk mechanic properties under the conditions of both dry and hydrated forms. The unique molecular design enables us to probe mechanistic questions and assess the structure−property relationship of the EMPs. Our results indicate that, instead of a highly organized secondary structure, hydrophobic hydration is critical for the elasticity of EMPs.
Co-reporter:Drexel H. Camacho and Zhibin Guan
Chemical Communications 2010 vol. 46(Issue 42) pp:7879-7893
Publication Date(Web):20 Sep 2010
DOI:10.1039/C0CC01535K
The innovation of polyolefin with unique architecture, composition and topology continues to inspire polymer chemists. An exciting recent direction in the polyolefin field is the design of new catalysts based on late-transition metals. In this review, we highlight recent developments in rationally designing late-transition metal catalysts for olefin polymerization. The examples described in this review showcase the power of the design of well-defined late-metal catalysts for tailored polyolefin synthesis, which may usher in a new era in the polymer industry.
Co-reporter:Yi-Xuan Lu, Zhu-Ming Shi, Zhan-Ting Li and Zhibin Guan
Chemical Communications 2010 vol. 46(Issue 47) pp:9019-9021
Publication Date(Web):05 Nov 2010
DOI:10.1039/C0CC03689G
Inspired by arylamide-based oligomeric foldermers that are stabilized by intramolecular hydrogen bonding, a series of polyamides with intramolecular hydrogen-bonding motifs were synthesized via polycondensation reactions. These polymers can fold into helical conformation different from their linear control. The chirality of helical conformation can further be tuned via acid–base complexation using chiral residues.
Co-reporter:Guobin Sun and Zhibin Guan
Macromolecules 2010 Volume 43(Issue 23) pp:9668-9673
Publication Date(Web):November 10, 2010
DOI:10.1021/ma1017617
A tandem polymerization strategy has been applied to the synthesis of novel core−shell soft nanoparticles having tunable thermosensitivity. These nanoparticles have amphiphilic dendritic core−shell structures, containing dendritic polyethylene (PE) as the hydrophobic core and a layer of poly(oligo((ethylene glycol) methacrylate)s (poly(OEGMA)s) as the hydrophilic shell. The dendritic PE core was first synthesized by chain walking polymerization, which was subsequently grafted with multiple poly(OEGMA) arms by atom transfer radical polymerization (ATRP) of OEGMAs. By varying the composition of the poly(OEGMA) grafts, the thermosensitivity of the core−shell nanoparticles can be tuned with the lower critical solution temperature (LCST) temperature ranging from 20 to 60 °C. Taking advantage of the thermosensitivity and amphiphilicity, the synthesized core−shell nanoparticles show interesting thermosensitive encapsulation of hydrophobic small molecules, which can be utilized for efficient separation of hydrophobic compounds from aqueous solution. This unique type of core−shell nanoparticles may find potential applications in drug formulation and delivery, water treatment, and other nanotechnology applications.
Co-reporter:Zhu-Ming Shi, Jin Huang, Zhi Ma, Xin Zhao, Zhibin Guan and Zhan-Ting Li
Macromolecules 2010 Volume 43(Issue 14) pp:6185-6192
Publication Date(Web):June 28, 2010
DOI:10.1021/ma100952h
This paper reports a foldamer-based approach to modulating the thermal and mechanical properties of cross-linked n-butyl methacrylate copolymers. Intramolecularly hydrogen-bonding-induced folded aromatic amide segments were designed and prepared, which reacted with a salicylaldehyde-bearing prepolymer to form cross-linked copolymers. As a control, analogous aromatic cross-links incapable of forming discrete folded structures due to lack of specific intramolecular H-bonding were also prepared and incorporated into copolymers. From the two series of cross-linked copolymers, 18 films were prepared and characterized by dynamic mechanical analysis (DMA) as well as creep/recovery experiments. We show that, compared to the control, the discrete folded cross-links substantially improve the mechanical properties of the copolymers. We attribute this enhancement to the ability of the folded cross-links to reversibly reveal the hidden length on extension via dissipative cleavage of the intramolecular H-bonds.
Co-reporter:Zhibin Guan
Chemistry – An Asian Journal 2010 Volume 5( Issue 5) pp:1058-1070
Publication Date(Web):
DOI:10.1002/asia.200900749
Abstract
Catalysis continues to spark new imagination and excitement of synthetic chemists. Discovery of new catalytic processes often makes seemingly impossible transformations proceed magically at high efficiency. In modern polymer synthesis, an exciting direction in recent years is the development of transition-metal-catalyzed polymerizations for controlling polymer topology. In this Focus Review, we highlight a few recent examples for catalytic syntheses of polyolefins having cyclic, block, and dendritic topologies, respectively. These examples showcase the power and efficiency of transition-metal catalysis for accessing various polymer topologies ranging from simple cyclic to complex globular dendritic polymers.
Co-reporter:Chris S. Popeney and Zhibin Guan
Macromolecules 2010 Volume 43(Issue 9) pp:4091-4097
Publication Date(Web):April 14, 2010
DOI:10.1021/ma100220n
The effect of electron-donating and -withdrawing groups on the ligands of Pd(II) α-diimine olefin polymerization catalysts on catalyst stability, activity, and polymer molecular weight is investigated. The polyethylene molecular weight and the productivity of catalysts bearing substituted bis(aryl)dimethyldiazabutadiene (Me2DAB) and bis(aryl)acenaphthenequinonediimine (BIAN) ligands were analyzed over time at room temperature and 40 °C to monitor catalyst stability and chain transfer processes. The introduction of electron-donating groups led to a dramatic increase in polymer molecular weight, with polymer chains still growing after 24 h of polymerization. The amino-substituted Me2DAB analogue afforded polymer of more than twice the molecular weight compared to the polymer made with the unsubstituted analogue after 24 h of polymerization. The unsubstituted catalysts and those bearing electron-withdrawing groups, however, reached a maximal molecular weight, generally lower, after a comparatively short time, which was presumably due to higher chain transfer rates. Electron-donating groups also provided increased stability to the catalysts leading to longer catalyst lifetimes. Both of these effects are likely due to stabilization of the reactive, electron-deficient, and coordinatively unsaturated alkyl agostic intermediate, the reactivity of which is key to both chain transfer and decomposition processes.
Co-reporter:Guobin Sun and Zhibin Guan
Macromolecules 2010 Volume 43(Issue 11) pp:4829-4832
Publication Date(Web):April 23, 2010
DOI:10.1021/ma100367q
Co-reporter:Dora L. Guzmán;Arlo Randall;Pierre Baldi;
Proceedings of the National Academy of Sciences 2010 107(5) pp:1989-1994
Publication Date(Web):January 13, 2010
DOI:10.1073/pnas.0905796107
Resolving molecular determinants of mechanical stability of proteins is crucial in the rational design of advanced biomaterials
for use in biomedical and nanotechnological applications. Here we present an interdisciplinary study combining bioinformatics
screening, steered molecular dynamics simulations, protein engineering, and single-molecule force spectroscopy that explores
the mechanical properties of a macro domain protein with mixed α + β topology. The unique architecture is defined by a single seven-stranded β-sheet in the core of the protein flanked by five α-helices. Unlike mechanically stable proteins studied thus far, the macro domain provides the distinct advantage of having
the key load-bearing hydrogen bonds (H bonds) buried in the hydrophobic core protected from water attacks. This feature allows
direct measurement of the force required to break apart the load-bearing H bonds under locally hydrophobic conditions. Steered
molecular dynamics simulations predicted extremely high mechanical stability of the macro domain by using constant velocity
and constant force methods. Single-molecule force spectroscopy experiments confirm the exceptional mechanical strength of
the macro domain, measuring a rupture force as high as 570 pN. Furthermore, through selective deletion of shielding peptide
segments, we examined the same key H bonds under hydrophilic environments in which the β-strands are exposed to solvent and verify that the high mechanical stability of the macro domain results from excellent shielding
of the load-bearing H bonds from competing water. Our study reveals that shielding water accessibility to the load-bearing
strands is a critical molecular determinant for enhancing the mechanical stability of proteins.
Co-reporter:Aaron M. Kushner ; John D. Vossler ; Gregory A. Williams
Journal of the American Chemical Society 2009 Volume 131(Issue 25) pp:8766-8768
Publication Date(Web):June 9, 2009
DOI:10.1021/ja9009666
Natural materials employ many elegant strategies to achieve mechanical properties required for survival under varying environmental conditions. Thus these remarkable biopolymers and nanocomposites often not only have a combination of mechanical properties such as high modulus, toughness, and elasticity, but also exhibit adaptive and stimuli-responsive properties. Inspired by skeletal muscle protein titin, we have synthesized a biomimetic modular polymer that not only closely mimics the modular multidomain structure of titin, but also manifests an exciting combination of mechanical properties, as well as adaptive properties such as self-healing and temperature-responsive shape-memory properties.
Co-reporter:Chris S. Popeney
Journal of the American Chemical Society 2009 Volume 131(Issue 34) pp:12384-12393
Publication Date(Web):August 10, 2009
DOI:10.1021/ja904471v
A detailed mechanistic investigation of the copolymerization of ethylene and methyl acrylate (MA) by a Pd(II) cyclophane-based α-diimine catalyst is reported. Our previous observations of unusually high incorporations of acrylates in copolymerization using this catalyst (J. Am. Chem. Soc. 2007, 129, 10062) prompted us to conduct a full mechanistic study on ethylene/MA copolymerization, which indicates a dramatic departure from normal Curtin−Hammett kinetic behavior as observed in copolymerization using the normal Brookhart type of Pd(II) α-diimine catalysts. Further investigation reveals that this contrasting behavior originates from the axial blocking effect of the cyclophane ligand hindering olefin substitution and equilibration. In equilibrium studies of ethylene with nitriles, the cyclophane catalyst was found to more strongly favor the linearly binding nitrile ligands as compared to the standard acyclic Pd(II) α-diimine catalysts. Ethylene exchange rates in the complexes [(N∧N)PdMe(C2H4)]+ (N∧N = diimine) were measured by 2D EXSY NMR spectroscopy and found to be over 100 times slower in the cyclophane case. Measurement of the slow equilibration of ethylene, methyl acrylate, and 4-methoxystyrene in cyclophane-based Pd(II) olefin complexes by 1H NMR and fitting of the obtained kinetic plots allowed for the estimation of exchange rates and equilibrium constants of the olefins. After extrapolation to typical polymerization temperature, ΔG⧧ = 20.6 and 16.4 kcal/mol for ethylene-methyl acrylate exchange in the forward (ethylene displacement by methyl acrylate) and reverse directions, respectively. These values are of similar magnitude to the previously determined migratory insertion barriers of ethylene (ΔG⧧ = 18.9 kcal/mol) and methyl acrylate (ΔG⧧ = 16.3 kcal/mol) under equivalent conditions, but contrast strongly to the rapid olefin exchange seen in the Brookhart acyclic catalyst. The large barrier to olefin exchange hinders olefin pre-equilibrium, decreasing the cyclophane catalyst’s ability to preferentially incorporate one monomer (in this case ethylene) over the other, thus giving rise to high comonomer incorporations.
Co-reporter:Ting-Bin Yu;JaneZ. Bai Dr.
Angewandte Chemie International Edition 2009 Volume 48( Issue 6) pp:1097-1101
Publication Date(Web):
DOI:10.1002/anie.200805009
Co-reporter:Ting-Bin Yu;JaneZ. Bai Dr.
Angewandte Chemie 2009 Volume 121( Issue 6) pp:1117-1121
Publication Date(Web):
DOI:10.1002/ange.200805009
Co-reporter:Chris S. Popeney, Arnold L. Rheingold and Zhibin Guan
Organometallics 2009 Volume 28(Issue 15) pp:4452-4463
Publication Date(Web):July 6, 2009
DOI:10.1021/om900302r
The synthesis and characterization of Ni(II) and Pd(II) α-diimine olefin polymerization catalysts bearing a fluorinated cyclophane-based ligand were performed. Fluorine was placed in such a manner as to interact with the metal center from the axial direction. The catalysts were active in the polymerization of ethylene, showing substantial differences in both catalytic behavior and polymer size and structure as compared to their nonfluorinated analogues. Both catalysts afforded polymer of comparatively low branching density and high molecular weight. The Ni(II) catalysts, from precursor [Ni(acetylacetonato)(F-Cyc)]+ salts (F-Cyc = fluorinated cyclophane), exhibited enhanced thermal stability by remaining active after 70 min with little loss in polymerization activity at 105 °C. The Pd(II) catalysts from salts of [Pd(F-Cyc)Me(NCR)]+ (NCR = nitrile) afforded polymer of molecular weights far higher than the nonfluorinated analogue. Additionally, polymerization activity was directly related to ethylene feed pressure for the Pd(II) system, and NMR analysis could not detect the presence of bound olefin, indicating that the polymerization proceeded via different kinetics involving an olefin-free 14 e− complex as the catalyst resting state. Furthermore, NMR 1H−19F coupling data provide clear evidence that the fluorine atoms were indeed interacting with the metal axial site. The unusual properties of these new complexes are thus attributed to stabilization of the highly reactive 14 e− intermediate by donation of the fluorine lone pair to the metal center.
Co-reporter:Hiromitsu Urakami and Zhibin Guan
Biomacromolecules 2008 Volume 9(Issue 2) pp:
Publication Date(Web):January 26, 2008
DOI:10.1021/bm701180r
An efficient living ring-opening polymerization (ROP) of a permethoxylated ϵ-caprolactone [(OMe)CL] catalyzed by yttrium(III) isopropoxide was developed for the synthesis of degradable protein-resistant polymers [P(OMe)CL]. The lactone monomer was efficiently prepared from a reduced sugar, d-dulcitol. Kinetic studies of the ROP revealed a linear dependence of ln[M]0/[M] on polymerization time as well as a linear correlation between the number-averaged molecular weight (Mn) and monomer conversion; both support it is a living polymerization. A series of block copolymers of our permethoxylated lactone with ϵ-caprolactone [P(OMe)CL-b-PCL] were synthesized and fully characterized. In thermal analyses only single Tgs were observed in all the block copolymers, suggesting that P(OMe)CL and PCL blocks are fully miscible. Finally, surface plasmon resonance (SPR) sensograms demonstrated that both P(OMe)CL and the P(OMe)CL-b-PCL block copolymers exhibit excellent resistance to fibrinogen and lysozyme.
Co-reporter:Guanghui Chen, Philip L. Felgner and Zhibin Guan
Biomacromolecules 2008 Volume 9(Issue 7) pp:
Publication Date(Web):June 3, 2008
DOI:10.1021/bm7013476
Here we present an efficient synthesis of functional dendritic polymers carrying internal fluorescence labels for bioconjugation. Specifically, dendritic polymers having pyrene as fluorescence label in the core and N-hydroxysuccinimide (NHS) functional groups at the periphery were synthesized by coupling heterobifunctional PEG to hydroxyl functionalized dendritic polyethylene core. The dendritic polyethylene cores containing one pyrene label per polymer molecule were prepared through a one-step transition-metal-catalyzed polymerization using a pyrene-labeled Pd(II)-α-diimine chain walking catalyst. A series of pyrene-labeled dendritic scaffolds were obtained with different molecular weights and sizes. NHS active end groups were introduced to the periphery of the dendritic scaffolds through end-group functionalization. Those NHS-functionalized dendritic scaffolds were successfully used to conjugate a model protein, ovalbumin, to yield protein−polymer conjugates carrying multiple copies of protein attached to each scaffold.
Co-reporter:Mark Metzke and Zhibin Guan
Biomacromolecules 2008 Volume 9(Issue 1) pp:
Publication Date(Web):December 14, 2007
DOI:10.1021/bm701013y
Here we describe structure–property studies on our carbohydrate-derived side-chain ether polymers as protein-resistant biomaterials. A series of side-chain ether polymers, including two polyesters and two polyamides, were prepared by condensation polymerization of monomers derived from simple carbohydrates. The two side-chain permethoxylated polyesters having different stereochemical repeating units demonstrate excellent resistance toward nonspecific protein adsorption as shown by surface plasmon resonance, indicating that the polymer stereochemistry does not have much effect on its protein-resistant properties. The introduction of amide bonds to polymer backbones leads to more pronounced effects. While the polymer degradation stability is significantly enhanced by replacing ester with amide linkages, the protein resistance for the polymer is greatly reduced by introduction of amide bonds. Finally, our results suggest that free hydroxyl and amide groups, while both are hydrogen-bond donors, seem to have different effects on protein resistant properties for polymers. It appears that free amide groups have more detrimental effect on protein resistance than free hydroxyl groups. These results show that the protein-resistant properties of this family of polymers can be tailored by modifying the backbone and side chain functionalities. In combination with the biodegradability and functionalizability, this family of carbohydrate-derived polymers shows promise as versatile biomaterials for biomedical applications.
Co-reporter:Zhibin Guan
Polymer International 2007 Volume 56(Issue 4) pp:
Publication Date(Web):9 FEB 2007
DOI:10.1002/pi.2245
This mini review uses two examples to illustrate the importance of supramolecular chemistry in natural and biomimetic polymers for advanced mechanical properties. In the first example, dragline silk, one of the strongest and toughest natural fibers, uses intermolecular weak forces to self-assemble into nanocomposites composed of β-sheet nanocrystals imbedded in an amorphous matrix. In the second example, a load-bearing protein, the sarcomere muscle protein titin, uses intramolecular weak forces to fold into repetitive modules for combined strength, toughness, and elasticity. Both examples show vividly that many natural polymers can combine important mechanical properties through programming supramolecular weak interactions into covalently formed biopolymers. This mini review attempts to summarize the current understanding of the molecular mechanisms that contribute to the exceptional mechanical properties for each natural polymer. Following that, the efforts from others and the author's own laboratory on developing synthetic polymers to mimic these natural counterparts are discussed. Copyright © 2007 Society of Chemical Industry
Co-reporter:Keunchan Oh and Zhibin Guan
Chemical Communications 2006 (Issue 29) pp:3069-3071
Publication Date(Web):13 Jun 2006
DOI:10.1039/B606185K
Alkyne–azide cycloaddition (“click” chemistry) between two peptide strands derivatized with terminal azide and alkyne, respectively, provides an efficient convergent synthesis of triazole ring-based new β-turn mimics.
Co-reporter:Mark Metzke, Naphtali O'Connor, Soumen Maiti, Edward Nelson,Zhibin Guan
Angewandte Chemie International Edition 2005 44(40) pp:6529-6533
Publication Date(Web):
DOI:10.1002/anie.200501944
Co-reporter:Mark Metzke;Naphtali O'Connor;Soumen Maiti Dr.;Edward Nelson Dr. Dr.
Angewandte Chemie 2005 Volume 117(Issue 40) pp:
Publication Date(Web):15 SEP 2005
DOI:10.1002/ange.200501944
Ganz natürlich: Eine neue Klasse synthetischer Biomaterialien (die abgebildeten Saccharid-Peptid-Hybridcopolymere) ist biologisch abbaubar, ungiftig und nicht-immunogen. Die kationischen Saccharid-Peptid-Hybridcopolymere erwiesen sich auch als wirksam beim Verdichten von Plasmid-DNA und ihrem Einschleusen in Zellen.
Co-reporter:Drexel H. Camacho Dr.;Eric V. Salo;Joseph W. Ziller Dr. Dr.
Angewandte Chemie 2004 Volume 116(Issue 14) pp:
Publication Date(Web):24 MAR 2004
DOI:10.1002/ange.200353226
Vorteile durch Design: Makrocyclische Cyclophanliganden erhöhen die thermische Stabilität von neuen Katalysatoren für die Polymerisation von Ethen zu Polyethylen mit hohem Molekulargewicht. Entscheidend für die hohe Aktivität und die Stabilität ist die strategische Platzierung des Zentralmetalls im Kern der Cyclophanarchitektur (siehe Bild; Pd rot, Cl grün, N blau, C grau).
Co-reporter:Drexel H. Camacho Dr.;Eric V. Salo;Joseph W. Ziller Dr. Dr.
Angewandte Chemie 2004 Volume 116(Issue 23) pp:
Publication Date(Web):1 JUN 2004
DOI:10.1002/ange.200490071
Co-reporter:Drexel H. Camacho Dr.;Eric V. Salo;Joseph W. Ziller Dr. Dr.
Angewandte Chemie International Edition 2004 Volume 43(Issue 14) pp:
Publication Date(Web):24 MAR 2004
DOI:10.1002/anie.200353226
Better by design: Exploitation of the macrocyclic architecture of a cyclophane-based ligand provides new highly active catalysts with improved thermal stability for ethylene polymerization. The strategic positioning of the metal center at the core of the cyclophane-based ligand is the key to the observed high activity and thermal stability, and to the high molecular weight of the polyethylene (see picture; red: Pd, green: Cl, blue: N, gray: C).
Co-reporter:Drexel H. Camacho Dr.;Eric V. Salo;Joseph W. Ziller Dr. Dr.
Angewandte Chemie International Edition 2004 Volume 43(Issue 23) pp:
Publication Date(Web):1 JUN 2004
DOI:10.1002/anie.200490071
Co-reporter:Zhibin Guan
Journal of Polymer Science Part A: Polymer Chemistry 2003 Volume 41(Issue 22) pp:3680-3692
Publication Date(Web):9 OCT 2003
DOI:10.1002/pola.10969
An Erratum has been published for this article in J Polym Sci Part A: Polym Chem (2004) 42 213
In this article, recent examples are reviewed of late-transition-metal catalysis applied to polymer topology control. By the judicious selection or design of late-transition-metal catalysts, polymers with a broad range of topologies, including linear, short-chain-branched, hyperbranched, dendritic, and cyclic topologies, have been successfully synthesized. A distinctive advantage of the catalyst approach is that polymers with complex topologies can be prepared in one pot from simple commercial monomers. A fundamental difference of the catalyst approach with respect to other approaches is that the polymer topology is controlled by the catalysts instead of the monomer structure. In our own laboratory, we have successfully used two strategies to control the polymer topology with late-transition-metal catalysts. In the first strategy, hyperbranched polymers are prepared by the direct free-radical polymerization of divinyl monomers through control of the competition between propagation and chain transfer with a cobalt chain-transfer catalyst. In the second strategy, polyethylene topology is successfully controlled by the regulation of the competition between propagation and chain walking with the Brookhart PdII-α-bisimine catalyst. © 2003 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem 41: 3680–3692, 2003
Co-reporter:Zhibin Guan
Chemistry - A European Journal 2002 Volume 8(Issue 14) pp:
Publication Date(Web):20 JUN 2002
DOI:10.1002/1521-3765(20020715)8:14<3086::AID-CHEM3086>3.0.CO;2-L
Chain-walking catalysts are demonstrated to control the branching topology of polyethylene by tuning the competition between monomer insertion and chain isomerization (walking). The topology of the polyethylene was controlled from linear with moderate branching to hyperbranched to dendritic. Although overall branching number and distribution of short chain branching are relatively constant, the topology of the polyethylene changes from linear polyethylene with moderate branches at high ethylene pressures to a dendritic polyethylene at low pressures. This provides a straightforward one-pot process for making polymers with a full range of tunable topologies.
Co-reporter:Ying-Wei Yang, Jens Hentschel, Yi-Chun Chen, Mark Lazari, Hanxiang Zeng, R. Michael van Dam and Zhibin Guan
Journal of Materials Chemistry A 2012 - vol. 22(Issue 3) pp:NaN1106-1106
Publication Date(Web):2011/11/17
DOI:10.1039/C1JM14131G
We report the design and synthesis of a new perfluoropolyether-based material, which has liquid-like viscosity and can be cured into a tough, highly durable elastomer when “clicked” with selected tri-pod organic small molecules. This highly fluorinated elastomer exhibits remarkable resistance to a variety of organic solvents, water, heat and even harsh acidic and basic conditions. Whereas PDMS-based microfluidic devices are commonly used for aqueous based applications, their limited chemical resistance and high swellability in many common organic solvents make it unfeasible for microfluidic applications involving organic solvents and/or harsh conditions. With excellent chemical resistance and low swellability, our newly synthesized fluoro-elastomers will hopefully provide an alternative material for organic based microfluidic devices. Furthermore, the alkyne–azide “click” chemistry employed in curing not only provides high efficiency of synthesis and ease of device fabrication, but, more importantly, produces 1,2,3-triazole linkages that are very stable against harsh acidic or basic conditions. This work has great potential to expand microfluidics to a series of novel applications especially in organic and medicinal chemistry.
Co-reporter:Miguel A. Camacho-Fernandez, Max Yen, Joseph W. Ziller and Zhibin Guan
Chemical Science (2010-Present) 2013 - vol. 4(Issue 7) pp:NaN2906-2906
Publication Date(Web):2013/05/07
DOI:10.1039/C3SC50676B
We report here the first direct observation of a cationic ruthenium complex catalyzing ethylene insertion polymerization. An arene-tethered ruthenium complex (η6-C6H5(CH2)3SCH3RuCl2) (3) was synthesized and shown to be able to catalyze ethylene polymerization upon activation with AlMe2Cl. For mechanistic studies, we synthesized the homologous dimethylated η6-C6H5(CH2)3SCH3Ru(CH3)2 (4) complex, which upon activation with the Brookhart acid ([H(Et2O)2]+ [BAr′4]−) was also active for ethylene insertion polymerization. 1H-NMR and mass spectrometry (MS) studies provide direct evidence for a ruthenium cationic [η6-C6H5(CH2)3SCH3Ru(oligomer)]+ complex as the active species during polymerization. This has unambiguously shown for the first time a ruthenium complex as the active species for catalyzing olefin insertion polymerization.
Co-reporter:Davoud Mozhdehi and Zhibin Guan
Chemical Communications 2013 - vol. 49(Issue 85) pp:NaN9952-9952
Publication Date(Web):2013/08/28
DOI:10.1039/C3CC45419C
We describe in this manuscript a new design of supramolecular amino acids (SAAs) for directing peptide folding in organic environments. The incorporated supramolecular motif has a strong driving force to dimerize in a sequence- and orientation-specific manner. By introducing such SAAs into the primary sequence of peptides, the specific and directional dimerization of the supramolecular units should facilitate the folding of the peptides. Our approach may provide a general strategy to program secondary structures in organic media.
Co-reporter:Yulin Chen and Zhibin Guan
Chemical Communications 2014 - vol. 50(Issue 74) pp:NaN10870-10870
Publication Date(Web):2014/07/28
DOI:10.1039/C4CC03168G
Triblock copolymers having glassy PMMA blocks and dynamic hydrogen bonding blocks were synthesized by sequential atom transfer radical polymerization (ATRP). The dynamic triblock copolymers self-assemble into nanocomposite materials exhibiting a combination of mechanical strength, toughness, and self-healing capability.
Co-reporter:Drexel H. Camacho and Zhibin Guan
Chemical Communications 2010 - vol. 46(Issue 42) pp:NaN7893-7893
Publication Date(Web):2010/09/20
DOI:10.1039/C0CC01535K
The innovation of polyolefin with unique architecture, composition and topology continues to inspire polymer chemists. An exciting recent direction in the polyolefin field is the design of new catalysts based on late-transition metals. In this review, we highlight recent developments in rationally designing late-transition metal catalysts for olefin polymerization. The examples described in this review showcase the power of the design of well-defined late-metal catalysts for tailored polyolefin synthesis, which may usher in a new era in the polymer industry.
Co-reporter:Yi-Xuan Lu, Zhu-Ming Shi, Zhan-Ting Li and Zhibin Guan
Chemical Communications 2010 - vol. 46(Issue 47) pp:NaN9021-9021
Publication Date(Web):2010/11/05
DOI:10.1039/C0CC03689G
Inspired by arylamide-based oligomeric foldermers that are stabilized by intramolecular hydrogen bonding, a series of polyamides with intramolecular hydrogen-bonding motifs were synthesized via polycondensation reactions. These polymers can fold into helical conformation different from their linear control. The chirality of helical conformation can further be tuned via acid–base complexation using chiral residues.
.jpg)













































































































































































































































![Propanoic acid, 2-[[(ethylthio)thioxomethyl]thio]-2-methyl-](/data/chemimg/113500/881037-62-3.png)
![Propanoic acid, 2-[[(ethylthio)thioxomethyl]thio]-2-methyl-](/data/chemimg/113500/881037-62-3_b.png)








![Poly[[8,48-bis(octyloxy)-3,6,13,36,43,46,53,76-octaoxo-2,5,12,35,42,4
5,52,75-octaazapentacyclo[75.3.1.17,11.137,41.147,51]tetraoctaconta-1(81
),7,9,11(84),37,39,41(83),47,49,51(82),77,79-dodecaene-38,78-diyl]ox
y-1,11-undecanediyloxycarbonylimino-1,4-phenylene(1-methylethyliden
e)-1,4-phenyleneiminocarbonyloxy-1,11-undecanediyl]](http://img.cochemist.com/ccimg/863500/863418-01-3.png)
![Poly[[8,48-bis(octyloxy)-3,6,13,36,43,46,53,76-octaoxo-2,5,12,35,42,4
5,52,75-octaazapentacyclo[75.3.1.17,11.137,41.147,51]tetraoctaconta-1(81
),7,9,11(84),37,39,41(83),47,49,51(82),77,79-dodecaene-38,78-diyl]ox
y-1,11-undecanediyloxycarbonylimino-1,4-phenylene(1-methylethyliden
e)-1,4-phenyleneiminocarbonyloxy-1,11-undecanediyl]](http://img.cochemist.com/ccimg/863500/863418-01-3_b.png)






![[1,1':3',1''-TERPHENYL]-2'-AMINE, 4,4''-DIETHENYL-](http://img.cochemist.com/ccimg/679900/679835-72-4.png)
![[1,1':3',1''-TERPHENYL]-2'-AMINE, 4,4''-DIETHENYL-](http://img.cochemist.com/ccimg/679900/679835-72-4_b.png)
![[1,1':3',1''-TERPHENYL]-4,4''-DICARBOXALDEHYDE, 2'-AMINO-](http://img.cochemist.com/ccimg/679900/679835-70-2.png)
![[1,1':3',1''-TERPHENYL]-4,4''-DICARBOXALDEHYDE, 2'-AMINO-](http://img.cochemist.com/ccimg/679900/679835-70-2_b.png)












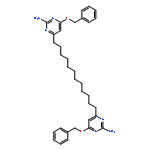
![POLY[[6-(PHENYLMETHOXY)-2,4-PYRIMIDINEDIYL]-1,12-DODECANEDIYL[6-(PHENYLMETHOXY)-4,2-PYRIMIDINEDIYL]IMINOCARBONYLIMINO-1,12-DODECANEDIYLIMINOCARBONYLIMINO]](http://img.cochemist.com/ccimg/532500/532403-62-6.png)
![POLY[[6-(PHENYLMETHOXY)-2,4-PYRIMIDINEDIYL]-1,12-DODECANEDIYL[6-(PHENYLMETHOXY)-4,2-PYRIMIDINEDIYL]IMINOCARBONYLIMINO-1,12-DODECANEDIYLIMINOCARBONYLIMINO]](http://img.cochemist.com/ccimg/532500/532403-62-6_b.png)




























![L-Lysine, N2,N6-bis[N2,N6-bis[(1,1-dimethylethoxy)carbonyl]-L-lysyl]-](http://img.cochemist.com/ccimg/185900/185816-27-7.png)
![L-Lysine, N2,N6-bis[N2,N6-bis[(1,1-dimethylethoxy)carbonyl]-L-lysyl]-](http://img.cochemist.com/ccimg/185900/185816-27-7_b.png)






![2-methoxy-N-[(1R)-1-phenylethyl]acetamide](http://img.cochemist.com/ccimg/163000/162929-44-4.png)
![2-methoxy-N-[(1R)-1-phenylethyl]acetamide](http://img.cochemist.com/ccimg/163000/162929-44-4_b.png)








![L-Lysine, N2-[(1,1-dimethylethoxy)carbonyl]-L-lysyl-, methyl ester](http://img.cochemist.com/ccimg/135200/135112-46-8.png)
![L-Lysine, N2-[(1,1-dimethylethoxy)carbonyl]-L-lysyl-, methyl ester](http://img.cochemist.com/ccimg/135200/135112-46-8_b.png)










![Benzene, 1,3-bis[2-(2-nitrophenyl)ethenyl]-](http://img.cochemist.com/ccimg/103300/103228-78-0.png)
![Benzene, 1,3-bis[2-(2-nitrophenyl)ethenyl]-](http://img.cochemist.com/ccimg/103300/103228-78-0_b.png)




![Benzene, [3-(methylthio)propyl]-](http://img.cochemist.com/ccimg/87300/87231-07-0.png)
![Benzene, [3-(methylthio)propyl]-](http://img.cochemist.com/ccimg/87300/87231-07-0_b.png)






















![L-Valine, N-[N-[(1,1-dimethylethoxy)carbonyl]glycyl]-, methyl ester](http://img.cochemist.com/ccimg/58900/58871-93-5.png)
![L-Valine, N-[N-[(1,1-dimethylethoxy)carbonyl]glycyl]-, methyl ester](http://img.cochemist.com/ccimg/58900/58871-93-5_b.png)








![L-Proline, 1-[N-[(1,1-dimethylethoxy)carbonyl]-L-valyl]-, methyl ester](http://img.cochemist.com/ccimg/38100/38017-75-3.png)
![L-Proline, 1-[N-[(1,1-dimethylethoxy)carbonyl]-L-valyl]-, methyl ester](http://img.cochemist.com/ccimg/38100/38017-75-3_b.png)
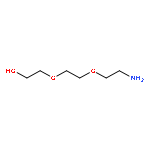
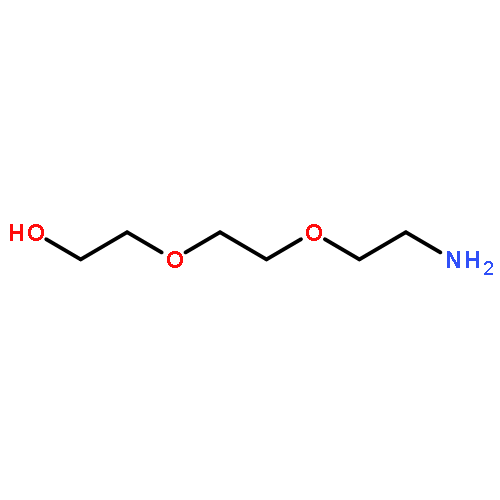




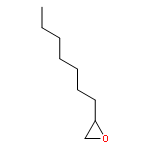
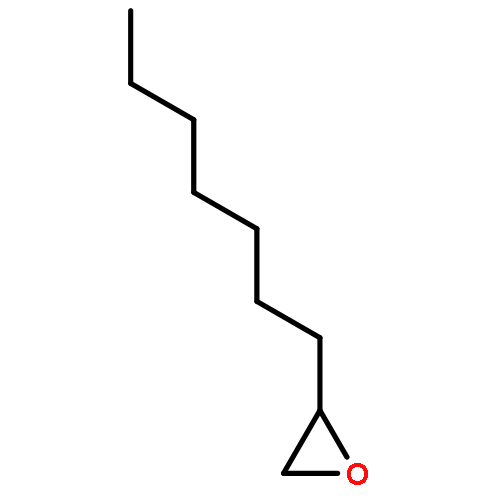
](http://img.cochemist.com/ccimg/27000/26913-06-4.png)
](http://img.cochemist.com/ccimg/27000/26913-06-4_b.png)






![(Z)-cis-9-oxabicyclo[6.1.0]non-4-ene](http://img.cochemist.com/ccimg/19800/19740-90-0.png)
![(Z)-cis-9-oxabicyclo[6.1.0]non-4-ene](http://img.cochemist.com/ccimg/19800/19740-90-0_b.png)





















![N'-[2-[2-(2-AMINOETHYLAMINO)ETHYLAMINO]ETHYL]ETHANE-1,2-DIAMINE;PENTAHYDROCHLORIDE](http://img.cochemist.com/ccimg/5000/4961-41-5.png)
![N'-[2-[2-(2-AMINOETHYLAMINO)ETHYLAMINO]ETHYL]ETHANE-1,2-DIAMINE;PENTAHYDROCHLORIDE](http://img.cochemist.com/ccimg/5000/4961-41-5_b.png)



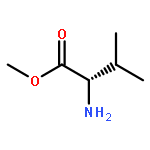
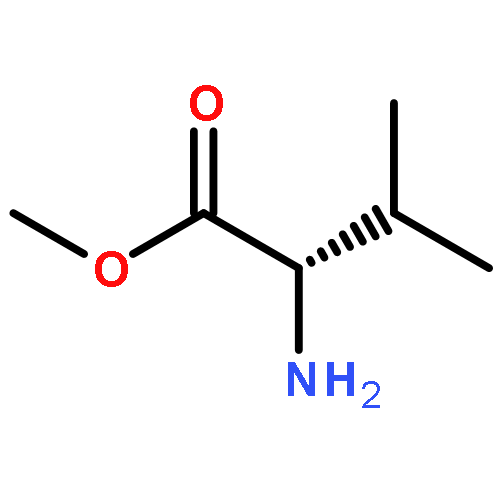






![methyl N6-[(1,1-dimethylethoxy)carbonyl]-N2-[(phenylmethoxy)carbonyl]-L-lysinate](http://img.cochemist.com/ccimg/2400/2389-49-3.png)
![methyl N6-[(1,1-dimethylethoxy)carbonyl]-N2-[(phenylmethoxy)carbonyl]-L-lysinate](http://img.cochemist.com/ccimg/2400/2389-49-3_b.png)
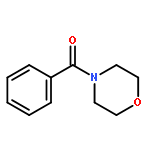
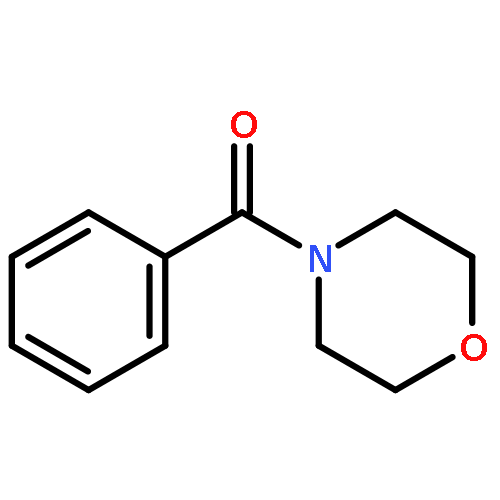


![DICHLORO-[4-(TRIFLUOROMETHYL)PHENYL]PHOSPHANE](http://img.cochemist.com/ccimg/800/774-99-2.png)
![DICHLORO-[4-(TRIFLUOROMETHYL)PHENYL]PHOSPHANE](http://img.cochemist.com/ccimg/800/774-99-2_b.png)













































































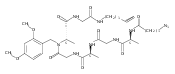












![Ruthenium,[6-[[bis(1,1-dimethylethyl)phosphino-kP]methylene]-N,N-diethyl-1,6-dihydro-2-pyridinemethanaminato-kN1,kN2]carbonylhydro-, (SP-5-52)-](http://img.cochemist.com/ccimg/864000/863971-63-5.png)
![Ruthenium,[6-[[bis(1,1-dimethylethyl)phosphino-kP]methylene]-N,N-diethyl-1,6-dihydro-2-pyridinemethanaminato-kN1,kN2]carbonylhydro-, (SP-5-52)-](http://img.cochemist.com/ccimg/864000/863971-63-5_b.png)












![Glycine, N-[2-(octyloxy)-5-[(1-oxo-10-undecenyl)amino]benzoyl]-](http://img.cochemist.com/ccimg/807700/807631-50-1.png)
![Glycine, N-[2-(octyloxy)-5-[(1-oxo-10-undecenyl)amino]benzoyl]-](http://img.cochemist.com/ccimg/807700/807631-50-1_b.png)
![Benzoic acid, 5-nitro-2-[[11-(phenylmethoxy)undecyl]oxy]-](http://img.cochemist.com/ccimg/807700/807631-39-6.png)
![Benzoic acid, 5-nitro-2-[[11-(phenylmethoxy)undecyl]oxy]-](http://img.cochemist.com/ccimg/807700/807631-39-6_b.png)
![BENZOIC ACID, 5-NITRO-2-[[11-(PHENYLMETHOXY)UNDECYL]OXY]-, METHYL ESTER](http://img.cochemist.com/ccimg/807700/807631-38-5.png)
![BENZOIC ACID, 5-NITRO-2-[[11-(PHENYLMETHOXY)UNDECYL]OXY]-, METHYL ESTER](http://img.cochemist.com/ccimg/807700/807631-38-5_b.png)
![Urea, N-[6-(3-butenyl)-1,4-dihydro-4-oxo-2-pyrimidinyl]-N'-2-propenyl-](http://img.cochemist.com/ccimg/628800/628718-42-3.png)
![Urea, N-[6-(3-butenyl)-1,4-dihydro-4-oxo-2-pyrimidinyl]-N'-2-propenyl-](http://img.cochemist.com/ccimg/628800/628718-42-3_b.png)


![GLYCINE, N-[5-AMINO-2-(OCTYLOXY)BENZOYL]-, ETHYL ESTER](http://img.cochemist.com/ccimg/267900/267897-52-9.png)
![GLYCINE, N-[5-AMINO-2-(OCTYLOXY)BENZOYL]-, ETHYL ESTER](http://img.cochemist.com/ccimg/267900/267897-52-9_b.png)
![Glycine, N-[5-nitro-2-(octyloxy)benzoyl]-, ethyl ester](http://img.cochemist.com/ccimg/267900/267897-50-7.png)
![Glycine, N-[5-nitro-2-(octyloxy)benzoyl]-, ethyl ester](http://img.cochemist.com/ccimg/267900/267897-50-7_b.png)








![Benzene, [[(11-bromoundecyl)oxy]methyl]-](http://img.cochemist.com/ccimg/144900/144878-51-3.png)
![Benzene, [[(11-bromoundecyl)oxy]methyl]-](http://img.cochemist.com/ccimg/144900/144878-51-3_b.png)
























![[1,1':3',1''-Terphenyl]-4,4''-dicarboxaldehyde, 2'-amino-5'-methyl-](http://img.cochemist.com/ccimg/679900/679835-69-9.png)
![[1,1':3',1''-Terphenyl]-4,4''-dicarboxaldehyde, 2'-amino-5'-methyl-](http://img.cochemist.com/ccimg/679900/679835-69-9_b.png)




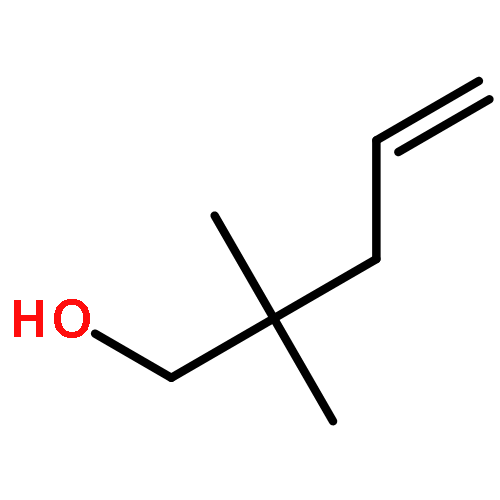











![[1,1':3',1''-Terphenyl]-2'-amine, 4,4''-diethenyl-5'-methyl-](http://img.cochemist.com/ccimg/679900/679835-71-3.png)
![[1,1':3',1''-Terphenyl]-2'-amine, 4,4''-diethenyl-5'-methyl-](http://img.cochemist.com/ccimg/679900/679835-71-3_b.png)




![5H-Benzo[a]phenoxazin-5-one,9-(diethylamino)-](http://img.cochemist.com/ccimg/7400/7385-67-3.png)
![5H-Benzo[a]phenoxazin-5-one,9-(diethylamino)-](http://img.cochemist.com/ccimg/7400/7385-67-3_b.png)