Co-reporter:Thomas Lanyon-Hogg, Markus Ritzefeld, Naoko Masumoto, Anthony I. Magee, Henry S. Rzepa, and Edward W. Tate
The Journal of Organic Chemistry 2015 Volume 80(Issue 9) pp:4370-4377
Publication Date(Web):February 25, 2015
DOI:10.1021/acs.joc.5b00205
2-Substituted N-acyl-piperidine is a widespread and important structural motif, found in approximately 500 currently available structures, and present in nearly 30 pharmaceutically active compounds. Restricted rotation of the acyl substituent in such molecules can give rise to two distinct chemical environments. Here we demonstrate, using NMR studies and density functional theory modeling of the lowest energy structures of 5-acyl-6,7-dihydrothieno[3,2-c]pyridine derivatives, that the amide E:Z equilibrium is affected by non-covalent interactions between the amide oxygen and adjacent aromatic protons. Structural predictions were used to design molecules that promote either the E- or Z-amide conformation, enabling preparation of compounds with a tailored conformational ratio, as proven by NMR studies. Analysis of the available X-ray data of a variety of published N-acyl-piperidine-containing compounds further indicates that these molecules are also clustered in the two observed conformations. This finding emphasizes that directed conformational isomerism has significant implications for the design of both small molecules and larger amide-containing molecular architectures.
Co-reporter:Anukul Jana, Volker Huch, Henry S. Rzepa, and David Scheschkewitz
Organometallics 2015 Volume 34(Issue 11) pp:2130-2133
Publication Date(Web):December 24, 2014
DOI:10.1021/om501286g
We report the synthesis and isolation of a stable complex containing the formally neutral Fe2Ge2 motif, which is stabilized by the coordination of an N-heterocyclic carbene to the germanium and of carbon monoxide to the iron center. [(NHCiPr2Me2)GeFe(CO)4]2 is obtained by reduction of the NHCiPr2Me2-coordinated dichlorogermylene adduct of Fe(CO)4, which in turn is obtained from the reaction of Fe2(CO)9 with GeCl2·NHCiPr2Me2 (NHCiPr2Me2 = 1,3-diisopropyl-4,5-dimethylimidazol-2-ylidene). The solid-state structure of the title compound reveals two distinct coordination modes for the Fe(CO)4 fragments: bridging (π-type) and terminal (σ-type). In solution, the rapid equilibrium between the two modes was resolved by NMR at −35 °C. Reaction with propylene sulfide at room temperature affords the sulfide-bridged digermanium complex with two terminal Fe(CO)4 moieties.
Co-reporter:Matthew J. Harvey, Nicholas J. Mason, and Henry S. Rzepa
Journal of Chemical Information and Modeling 2014 Volume 54(Issue 10) pp:2627-2635
Publication Date(Web):August 29, 2014
DOI:10.1021/ci500302p
We discuss the concept of recasting the data-rich scientific journal article into two components, a narrative and separate data components, each of which is assigned a persistent digital object identifier. Doing so allows each of these components to exist in an environment optimized for purpose. We make use of a poorly-known feature of the handle system for assigning persistent identifiers that allows an individual data file from a larger file set to be retrieved according to its file name or its MIME type. The data objects allow facile visualization and retrieval for reuse of the data and facilitates other operations such as data mining. Examples from five recently published articles illustrate these concepts.
Co-reporter:Dr. David Danovich; Sason Shaik; Henry S. Rzepa; Roald Hoffmann
Angewandte Chemie International Edition 2013 Volume 52( Issue 23) pp:5926-5928
Publication Date(Web):
DOI:10.1002/anie.201302350
Co-reporter:Fanny L. Cherblanc, Ya-Pei Lo, Wouter A. Herrebout, Patrick Bultinck, Henry S. Rzepa, and Matthew J. Fuchter
The Journal of Organic Chemistry 2013 Volume 78(Issue 23) pp:11646-11655
Publication Date(Web):September 27, 2013
DOI:10.1021/jo401316a
The stereochemistry of the desulfurization products of chiral natural and synthetic 3,6-epidithiodiketopiperazines (ETPs) is specified inconsistently in the literature. Qualitative mechanisms have been put forward to explain apparently divergent stereochemical pathways, but the quantitative feasibility of such mechanistic pathways has not been assessed. We report a computational study revealing that desulfurization of ETPs should occur universally with retention of configuration. While the majority of stereochemically assigned and reassigned cases fit this model, until now desulfurization of the synthetic gliotoxin analogue shown has remained assigned as proceeding via inversion of configuration. Through detailed chiroptical studies comparing experimentally obtained optical rotation values, electronic circular dichroism spectra, and vibrational circular dichroism spectra to their computationally simulated counterparts as well as chemical derivatization studies, we have unambiguously demonstrated that contrary to its current assignment in the literature, the desulfurization of this synthetic ETP also proceeds with retention of configuration.
Co-reporter:Dr. Sason Shaik;Dr. Henry S. Rzepa;Dr. Roald Hoffmann
Angewandte Chemie International Edition 2013 Volume 52( Issue 10) pp:3020-3033
Publication Date(Web):
DOI:10.1002/anie.201208206
Co-reporter: Henry S. Rzepa
Chemistry - A European Journal 2013 Volume 19( Issue 15) pp:4932-4937
Publication Date(Web):
DOI:10.1002/chem.201102942
Abstract
Density functional calculations of 1H NMR spectra and reaction barriers at the ωB97XD/6-311G(d,p)/continuum water level do not support the claimed identification of encarcerated 1,3-dimethylcyclobutadiene in either the solid state or aqueous solution, as reported by Barboiu et al (Chem. Eur. 2011, 17, 10021). Instead, previous suggestions that the species identified in the solid state is in fact 2-oxabicyclo[2.2.0]hex-5-en-3-one (the Dewar lactone Me22) are reaffirmed. Analysis of the ground-state electronic structure of this species indicates an unusual π-anomeric effect is promoting a Dunitz-like chemical reaction pathway leading to the eventual elimination of carbon dioxide and formation of 1,3-dimethylcyclobutadiene.
Co-reporter:Henry S. Rzepa and Charlotte S. M. Allan
Journal of Chemical Education 2010 Volume 87(Issue 2) pp:221-228
Publication Date(Web):January 12, 2010
DOI:10.1021/ed800058c
Our understanding of carbonium ions as intermediates in chemical reaction mechanisms derives from the early work of Julius Stieglitz and the more famous Hans Meerwein, the latter studying the racemization of isobornyl chloride when treated with Lewis acids. This review analyzes how key mechanistic concepts for this reaction evolved and gives the pedagogy a modern slant based on results obtained from accurate quantum mechanical calculations. Thus, originally thought of as involving ionization of the C−Cl bond to form a carbocation that then undergoes a transannular hydride shift, an analysis of modern calculations (using AIM- and ELF-based quantum electronic topology) reveals that a more appropriate description of the bonding at the transition state for this transfer is a nonclassical 3-center-2-electron interaction. We see how other concepts important to organic chemistry such as symmetric intermediates and stereoelectronic control emerge along the way.Keywords (Audience): Second-Year Undergraduate; Upper-Division Undergraduate; Keywords (Domain): Organic Chemistry; Keywords (Pedagogy): Textbooks/Reference Books; Keywords (Topic): Carbocations; Enantiomers; Molecular Modeling;
Co-reporter:Chaitanya S. Wannere, Henry S. Rzepa, B. Christopher Rinderspacher, Ankan Paul, Charlotte S. M. Allan and Henry F. Schaefer III, Paul v. R. Schleyer
The Journal of Physical Chemistry A 2009 Volume 113(Issue 43) pp:11619-11629
Publication Date(Web):July 28, 2009
DOI:10.1021/jp902176a
Higher-order aromatic charged Möbius-type annulenes have been Lkrealized computationally. These charged species are based on strips with more than one electronic half-twist, as defined by their linking numbers. The B3LYP/6-311+G(d,p) optimized structures and properties of annulene rings with such multiple half-twists (C12H122+, C12H122−, C14H14, C18H182+, C18H182−, C21H21+, C24H242−, C28H282+, and C28H282−) have the nearly equal C−C bond lengths, small dihedral angles around the circuits, stabilization energies, and nucleus-independent chemical shift values associated with aromaticity. The topology and nature of Möbius annulene systems are analyzed in terms of the torus curves defined by electron density functions (ρ(r)π, ELFπ) constructed using only the occupied π-MOs. The π-torus subdivides into a torus knot for annulenes defined by an odd linking number (Lk = 1, 3π) and a torus link for those with an even linking number (Lk = 2, 4π). The torus topology is shown to map onto single canonical π-MOs only for even values of Lk. Incomplete and misleading descriptions of the topology of π-electronic Möbius systems with an odd number of half twists result when only signed orbital diagrams are considered, as is often done for the iconic single half twist system.
Co-reporter:D. Christopher Braddock and Henry S. Rzepa
Journal of Natural Products 2008 Volume 71(Issue 4) pp:728-730
Publication Date(Web):February 23, 2008
DOI:10.1021/np0705918
Revised structures proposed previously for obtusallenes V−VII (5–7) have been confirmed on the basis of computed GIAO-DFT 13C NMR chemical shifts.
Co-reporter:Gabriel A. Asseily, Robert P. Davies, Henry S. Rzepa and Andrew J. P. White
New Journal of Chemistry 2005 vol. 29(Issue 2) pp:315-319
Publication Date(Web):06 Dec 2004
DOI:10.1039/B411873A
Two new thioether interhalogen adducts, (PhCH2)2S·ICl (1) and (PhCH2)2S·IBr (2) have been prepared and characterised in the solid-state, the prior being the first crystallographically characterised example of a thioether ICl charge transfer complex. Theoretical calculations have been carried out on a related series of thioether IX adducts (X = F, Cl, Br, I) and reveal the computed geometries for these systems, in particular the S–I and I–X distances, to vary significantly based on the choice of basis set and type of density functional hybridisation model employed. In order to model these electron rich adducts accurately we demonstrate the importance of using high quality basis sets and density functional hybridisation models which are able to handle electron correlation accurately, and also the importance of lattice effects.


![Cyclohexanamine, N,N'-[bis(4-fluorophenyl)-1,2-ethanediylidene]bis-](http://img.cochemist.com/ccimg/884100/884050-45-7.png)
![Cyclohexanamine, N,N'-[bis(4-fluorophenyl)-1,2-ethanediylidene]bis-](http://img.cochemist.com/ccimg/884100/884050-45-7_b.png)
![1,3,5,7,9-Decapentaene, 5,6-bis[(S)-fluoroiodomethyl]-, (3Z,5E,7Z)-](http://img.cochemist.com/ccimg/869400/869360-86-1.png)
![1,3,5,7,9-Decapentaene, 5,6-bis[(S)-fluoroiodomethyl]-, (3Z,5E,7Z)-](http://img.cochemist.com/ccimg/869400/869360-86-1_b.png)


![Tricyclo[3.3.1.13,7]decane, 1-(3-bromo-4,4-dimethyl-1,2-pentadien-1-yl)-](/data/chemimg/3722300/2005527-22-8.png)
![Tricyclo[3.3.1.13,7]decane, 1-(3-bromo-4,4-dimethyl-1,2-pentadien-1-yl)-](/data/chemimg/3722300/2005527-22-8_b.png)




![Tricyclo[3.3.1.13,7]decane, 1-(1-bromo-4,4-dimethyl-1,2-pentadien-1-yl)-](/data/chemimg/3722300/2005527-25-1.png)
![Tricyclo[3.3.1.13,7]decane, 1-(1-bromo-4,4-dimethyl-1,2-pentadien-1-yl)-](/data/chemimg/3722300/2005527-25-1_b.png)








![6H-Dibenzo[d,f][1,3]diazepin-6-ylidene, 5,7-dihydro-5,7-dimethyl-](http://img.cochemist.com/ccimg/468800/468775-07-7.png)
![6H-Dibenzo[d,f][1,3]diazepin-6-ylidene, 5,7-dihydro-5,7-dimethyl-](http://img.cochemist.com/ccimg/468800/468775-07-7_b.png)




![1,6,8,13-Tetraoxa-7-azoniaspiro[6.6]trideca-2,4,9,11-tetraene](http://img.cochemist.com/ccimg/528900/528877-62-5.png)
![1,6,8,13-Tetraoxa-7-azoniaspiro[6.6]trideca-2,4,9,11-tetraene](http://img.cochemist.com/ccimg/528900/528877-62-5_b.png)


![Oxaziridine, 2-[(4-methylphenyl)sulfonyl]-3-(4-nitrophenyl)-](http://img.cochemist.com/ccimg/90700/90687-53-9.png)
![Oxaziridine, 2-[(4-methylphenyl)sulfonyl]-3-(4-nitrophenyl)-](http://img.cochemist.com/ccimg/90700/90687-53-9_b.png)
![1,6,8,13-Tetraaza-7-azoniaspiro[6.6]trideca-2,4,9,11-tetraene](http://img.cochemist.com/ccimg/528700/528609-44-1.png)
![1,6,8,13-Tetraaza-7-azoniaspiro[6.6]trideca-2,4,9,11-tetraene](http://img.cochemist.com/ccimg/528700/528609-44-1_b.png)
![Bicyclo[5.2.0]nona-1,3,4,6,8-pentaene](http://img.cochemist.com/ccimg/811900/811859-89-9.png)
![Bicyclo[5.2.0]nona-1,3,4,6,8-pentaene](http://img.cochemist.com/ccimg/811900/811859-89-9_b.png)














![2H-Pyran, 2-[(1-ethynylcyclohexyl)oxy]tetrahydro-](http://img.cochemist.com/ccimg/43000/42969-66-4.png)
![2H-Pyran, 2-[(1-ethynylcyclohexyl)oxy]tetrahydro-](http://img.cochemist.com/ccimg/43000/42969-66-4_b.png)
![CYCLOHEXANONE, 2-[(1R)-1-HYDROXY-2-METHYLPROPYL]-, (2S)-](http://img.cochemist.com/ccimg/562900/562837-03-0.png)
![CYCLOHEXANONE, 2-[(1R)-1-HYDROXY-2-METHYLPROPYL]-, (2S)-](http://img.cochemist.com/ccimg/562900/562837-03-0_b.png)
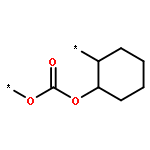

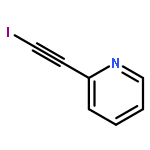
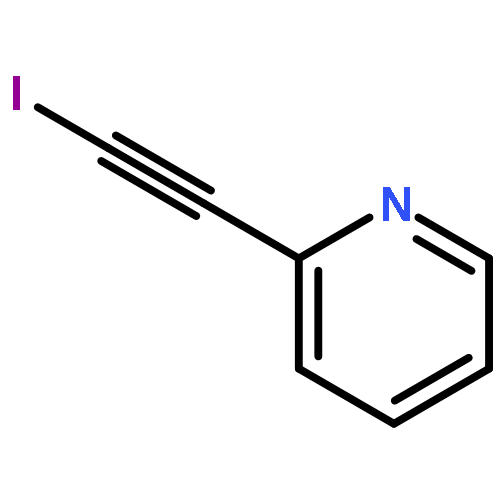








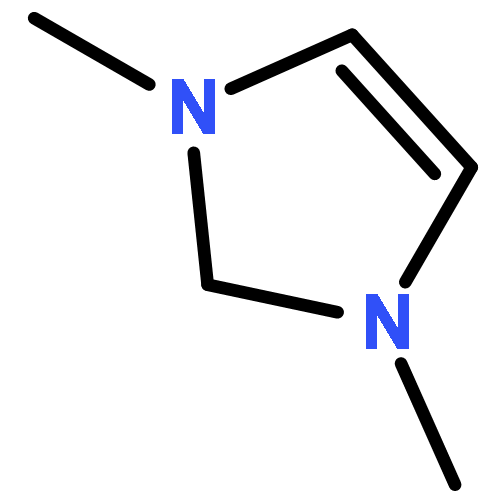
![CYCLOHEXANONE, 2-[(1S)-1-HYDROXY-2-METHYLPROPYL]-, (2S)-](http://img.cochemist.com/ccimg/324600/324535-32-2.png)
![CYCLOHEXANONE, 2-[(1S)-1-HYDROXY-2-METHYLPROPYL]-, (2S)-](http://img.cochemist.com/ccimg/324600/324535-32-2_b.png)


![Bicyclo[2.2.1]heptane, 2-chloro-1,7,7-trimethyl-, exo-](http://img.cochemist.com/ccimg/600/559-45-5.png)
![Bicyclo[2.2.1]heptane, 2-chloro-1,7,7-trimethyl-, exo-](http://img.cochemist.com/ccimg/600/559-45-5_b.png)









![(3R)-2,3,10,10-tetramethyl-2,3-dihydro-10H-3r,10ac-epidisulfano-pyrazino[1,2-a]indole-1,4-dione](http://img.cochemist.com/ccimg/59900/59888-49-2.png)
![(3R)-2,3,10,10-tetramethyl-2,3-dihydro-10H-3r,10ac-epidisulfano-pyrazino[1,2-a]indole-1,4-dione](http://img.cochemist.com/ccimg/59900/59888-49-2_b.png)
![2H-PYRAZINO[2,1-B]QUINAZOLINE-3,6(1H,4H)-DIONE, 2,4-DIMETHYL-, (4S)-](http://img.cochemist.com/ccimg/54800/54799-50-7.png)
![2H-PYRAZINO[2,1-B]QUINAZOLINE-3,6(1H,4H)-DIONE, 2,4-DIMETHYL-, (4S)-](http://img.cochemist.com/ccimg/54800/54799-50-7_b.png)
![1,3,5,7,9-DECAPENTAENE, 5,6-BIS[(R)-FLUOROIODOMETHYL]-, (3Z,5E,7Z)-](http://img.cochemist.com/ccimg/869400/869360-85-0.png)
![1,3,5,7,9-DECAPENTAENE, 5,6-BIS[(R)-FLUOROIODOMETHYL]-, (3Z,5E,7Z)-](http://img.cochemist.com/ccimg/869400/869360-85-0_b.png)


![5-HYDROXY-4-[(2S)-2-[(1R,2S,6R)-2-[(E)-3-HYDROXY-2-[(2R,3R,6S)-6-[(E, 3S)-3-[(3S,5R)-5-[(1S)-1-METHOXYETHYL]-3-METHYL-OXOLAN-2-YL]BUT-1-ENYL]-3-METHYL-OXAN-2-YL]PROP-1-ENYL]-6-METHYL-CYCLOHEXYL]PROPANOYL]FURAN-3-ONE](http://img.cochemist.com/ccimg/75200/75139-06-9.png)
![5-HYDROXY-4-[(2S)-2-[(1R,2S,6R)-2-[(E)-3-HYDROXY-2-[(2R,3R,6S)-6-[(E, 3S)-3-[(3S,5R)-5-[(1S)-1-METHOXYETHYL]-3-METHYL-OXOLAN-2-YL]BUT-1-ENYL]-3-METHYL-OXAN-2-YL]PROP-1-ENYL]-6-METHYL-CYCLOHEXYL]PROPANOYL]FURAN-3-ONE](http://img.cochemist.com/ccimg/75200/75139-06-9_b.png)

























![tricyclo[4.2.0.02,5]octa-3,7-diene, (1alpha-2alpha-5alpha-6alpha)-](http://img.cochemist.com/ccimg/20400/20380-31-8.png)
![tricyclo[4.2.0.02,5]octa-3,7-diene, (1alpha-2alpha-5alpha-6alpha)-](http://img.cochemist.com/ccimg/20400/20380-31-8_b.png)







![2-Oxabicyclo[2.2.0]hex-5-en-3-one](http://img.cochemist.com/ccimg/23000/22980-23-0.png)
![2-Oxabicyclo[2.2.0]hex-5-en-3-one](http://img.cochemist.com/ccimg/23000/22980-23-0_b.png)
![Tricyclo[4.2.0.02,5]octa-3,7-diene,(1R,2S,5R,6S)-rel-](http://img.cochemist.com/ccimg/20400/20380-30-7.png)
![Tricyclo[4.2.0.02,5]octa-3,7-diene,(1R,2S,5R,6S)-rel-](http://img.cochemist.com/ccimg/20400/20380-30-7_b.png)
![1,1'-Biphenyl,2-[(trifluoromethyl)thio]-](http://img.cochemist.com/ccimg/130000/129922-51-6.png)
![1,1'-Biphenyl,2-[(trifluoromethyl)thio]-](http://img.cochemist.com/ccimg/130000/129922-51-6_b.png)
![(1R,2R,5R,6R)-TRICYCLO[4.2.0.0<SUP>2,5</SUP>]OCTA-3,7-DIENE-1,5-DICARBALDEHYDE](http://img.cochemist.com/data/images/220338-69-2.png)
![(1R,2R,5R,6R)-TRICYCLO[4.2.0.0<SUP>2,5</SUP>]OCTA-3,7-DIENE-1,5-DICARBALDEHYDE](http://img.cochemist.com/data/images/220338-69-2_b.png)







![Ferrocene,1-[(S)-(dimethylamino)[2-(diphenylphosphino)phenyl]methyl]-2-(diphenylphosphino)-,(2R)- (9CI)](http://img.cochemist.com/ccimg/850500/850444-36-9.png)
![Ferrocene,1-[(S)-(dimethylamino)[2-(diphenylphosphino)phenyl]methyl]-2-(diphenylphosphino)-,(2R)- (9CI)](http://img.cochemist.com/ccimg/850500/850444-36-9_b.png)










![Ferrocene,1-[(1R)-1-(dicyclohexylphosphino)ethyl]-2-(diphenylphosphino)-, (2S)-](http://img.cochemist.com/ccimg/360100/360048-66-4.png)
![Ferrocene,1-[(1R)-1-(dicyclohexylphosphino)ethyl]-2-(diphenylphosphino)-, (2S)-](http://img.cochemist.com/ccimg/360100/360048-66-4_b.png)


![Poly[oxy(1-oxo-1,6-hexanediyl)]](http://img.cochemist.com/ccimg/25300/25248-42-4.png)
![Poly[oxy(1-oxo-1,6-hexanediyl)]](http://img.cochemist.com/ccimg/25300/25248-42-4_b.png)



![Indolo[3'',2'':4',5']pyrrolo[2',1':3,4]pyrazino[2,1-b]quinazoline-5,8(7H,9aH)-dione,10-acetyl-14b-(1,1-dimethyl-2-propen-1-yl)-10,14b,15,15a-tetrahydro-7-methyl-,(7R,9aR,14bR,15aS)-](http://img.cochemist.com/ccimg/148500/148441-26-3.png)
![Indolo[3'',2'':4',5']pyrrolo[2',1':3,4]pyrazino[2,1-b]quinazoline-5,8(7H,9aH)-dione,10-acetyl-14b-(1,1-dimethyl-2-propen-1-yl)-10,14b,15,15a-tetrahydro-7-methyl-,(7R,9aR,14bR,15aS)-](http://img.cochemist.com/ccimg/148500/148441-26-3_b.png)
![2,2'-Bi-1H-pyrrole,4-methoxy-5-[(5-methyl-4-pentyl-2H-pyrrol-2-ylidene)methyl]-](http://img.cochemist.com/ccimg/100/82-89-3.png)
![2,2'-Bi-1H-pyrrole,4-methoxy-5-[(5-methyl-4-pentyl-2H-pyrrol-2-ylidene)methyl]-](http://img.cochemist.com/ccimg/100/82-89-3_b.png)

![1R,5R,6R-(+)-1,6-BIS(DIPHENYLPHOSPHINOXY)SPIRO[4.4]NONANE](http://img.cochemist.com/ccimg/197200/197159-86-7.png)
![1R,5R,6R-(+)-1,6-BIS(DIPHENYLPHOSPHINOXY)SPIRO[4.4]NONANE](http://img.cochemist.com/ccimg/197200/197159-86-7_b.png)
![[3,3'-Biphenanthrene]-4,4'-diol,2,2'-diphenyl-, (3S)-](http://img.cochemist.com/ccimg/147800/147702-15-6.png)
![[3,3'-Biphenanthrene]-4,4'-diol,2,2'-diphenyl-, (3S)-](http://img.cochemist.com/ccimg/147800/147702-15-6_b.png)






![4-PHENYL-6,7-DIHYDROTHIENO[3,2-C]PYRIDINE](http://img.cochemist.com/ccimg/76400/76356-25-7.png)
![4-PHENYL-6,7-DIHYDROTHIENO[3,2-C]PYRIDINE](http://img.cochemist.com/ccimg/76400/76356-25-7_b.png)
![4-Phenyl-4,5,6,7-tetrahydrothieno[3,2-c]pyridine](http://img.cochemist.com/ccimg/91500/91477-84-8.png)
![4-Phenyl-4,5,6,7-tetrahydrothieno[3,2-c]pyridine](http://img.cochemist.com/ccimg/91500/91477-84-8_b.png)
























![Ferrocene,1,1'-bis[(2R,4R)-2,4-diethyl-1-phosphetanyl]-, stereoisomer (9CI)](http://img.cochemist.com/ccimg/268300/268220-91-3.png)
![Ferrocene,1,1'-bis[(2R,4R)-2,4-diethyl-1-phosphetanyl]-, stereoisomer (9CI)](http://img.cochemist.com/ccimg/268300/268220-91-3_b.png)


![(+)-1,2-BIS[(2S,5S)-2,5-DIMETHYLPHOSPHOLANO]BENZENE](http://img.cochemist.com/ccimg/136800/136735-95-0.png)
![(+)-1,2-BIS[(2S,5S)-2,5-DIMETHYLPHOSPHOLANO]BENZENE](http://img.cochemist.com/ccimg/136800/136735-95-0_b.png)




![[10b,10'b(11H,11'H)-Bi-3,11a-epidithio-11aH-pyrazino[1',2':1,5]pyrrolo[2,3-b]indole]-1,1',4,4'-tetrone,2,2',3,3',5a,5'a,6,6'-octahydro-3,3'-bis(hydroxymethyl)-2,2'-dimethyl-,(3S,3'S,5aR,5'aR,10bR,10'bR,11aS,11'aS)-](http://img.cochemist.com/ccimg/28100/28097-03-2.png)
![[10b,10'b(11H,11'H)-Bi-3,11a-epidithio-11aH-pyrazino[1',2':1,5]pyrrolo[2,3-b]indole]-1,1',4,4'-tetrone,2,2',3,3',5a,5'a,6,6'-octahydro-3,3'-bis(hydroxymethyl)-2,2'-dimethyl-,(3S,3'S,5aR,5'aR,10bR,10'bR,11aS,11'aS)-](http://img.cochemist.com/ccimg/28100/28097-03-2_b.png)
![Oxazole,2-[2-(diphenylphosphino)phenyl]-4,5-dihydro-4-(1-methylethyl)-, (4S)-](/data/chemimg/130100/148461-14-7.png)
![Oxazole,2-[2-(diphenylphosphino)phenyl]-4,5-dihydro-4-(1-methylethyl)-, (4S)-](/data/chemimg/130100/148461-14-7_b.png)















![Ferrocene,1-(diphenylphosphino)-2-[(1R)-1-[(diphenylphosphino)methylamino]ethyl]-, (1R)-](http://img.cochemist.com/ccimg/406700/406680-94-2.png)
![Ferrocene,1-(diphenylphosphino)-2-[(1R)-1-[(diphenylphosphino)methylamino]ethyl]-, (1R)-](http://img.cochemist.com/ccimg/406700/406680-94-2_b.png)
















![(SP,S'P)-1,1'-Bis(dicyclohexylphosphino)-2,2'-bis[(R)-alpha-(dimethylamino) benzyl]ferrocene](/data/chemimg/45300/494227-35-9.png)
![(SP,S'P)-1,1'-Bis(dicyclohexylphosphino)-2,2'-bis[(R)-alpha-(dimethylamino) benzyl]ferrocene](/data/chemimg/45300/494227-35-9_b.png)
![Dinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin,4-hydroxy-2,6-bis[2,4,6-tris(1-methylethyl)phenyl]-, 4-oxide, (11bR)-](/data/chemimg/115800/791616-63-2.png)
![Dinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin,4-hydroxy-2,6-bis[2,4,6-tris(1-methylethyl)phenyl]-, 4-oxide, (11bR)-](/data/chemimg/115800/791616-63-2_b.png)




![Dinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin,2,6-bis[3,5-bis(trifluoromethyl)phenyl]-4-hydroxy-, 4-oxide, (11bR)-](/data/chemimg/114800/791616-62-1.png)
![Dinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin,2,6-bis[3,5-bis(trifluoromethyl)phenyl]-4-hydroxy-, 4-oxide, (11bR)-](/data/chemimg/114800/791616-62-1_b.png)














![Cyclohexanone, 2-[(R)-hydroxyphenylmethyl]-, (2S)-](http://img.cochemist.com/ccimg/156100/156039-16-6.png)
![Cyclohexanone, 2-[(R)-hydroxyphenylmethyl]-, (2S)-](http://img.cochemist.com/ccimg/156100/156039-16-6_b.png)








![Phosphine,1,1'-[(1R)-5,5'-dichloro-6,6'-dimethoxy[1,1'-biphenyl]-2,2'-diyl]bis[1,1-diphenyl-](/data/chemimg/110700/185913-97-7.png)
![Phosphine,1,1'-[(1R)-5,5'-dichloro-6,6'-dimethoxy[1,1'-biphenyl]-2,2'-diyl]bis[1,1-diphenyl-](/data/chemimg/110700/185913-97-7_b.png)