Co-reporter:Masashi Kawasaki, Saki Kuroyanagi, Takuya Ito, Hiroyuki Morita, Yasuo Tanaka, Naoki Toyooka
Tetrahedron 2017 Volume 73, Issue 15(Issue 15) pp:
Publication Date(Web):13 April 2017
DOI:10.1016/j.tet.2017.02.055
Phantolide analogues 1a–1d were newly synthesized to evaluate their odor profiles. The enantiomers of 1a and 1b were also synthesized. Both (S) enantiomers of 1a and 1b had musk odor although weakly, and but neither of the (R) enantiomers 1a and 1b had musk odor. During the investigations, we encountered the undesirable racemization in Friedel-Crafts reaction of the intermediate (S)-5.Download high-res image (133KB)Download full-size image
Co-reporter:Hisashi Mori, Ryogo Wada, Satoyuki Takahara, Yoshikazu Horino, Hironori Izumi, Tetsuya Ishimoto, Tomoyuki Yoshida, Mineyuki Mizuguchi, Takayuki Obita, Hiroaki Gouda, Shuichi Hirono, Naoki Toyooka
Bioorganic & Medicinal Chemistry 2017 Volume 25, Issue 14(Issue 14) pp:
Publication Date(Web):15 July 2017
DOI:10.1016/j.bmc.2017.05.011
Serine racemase (SRR) is an enzyme that produces d-serine from l-serine. d-Serine acts as an endogenous coagonist of NMDA-type glutamate receptors (NMDARs), which regulate many physiological functions. Over-activation of NMDARs induces excitotoxicity, which is observed in many neurodegenerative disorders and epilepsy states. In our previous works on the generation of SRR gene knockout (Srr-KO) mice and its protective effects against NMDA- and Aβ peptide-induced neurodegeneration, we hypothesized that the regulation of NMDARs’ over-activation by inhibition of SRR activity is one such therapeutic strategy to combat these disease states. In the previous study, we performed in silico screening to identify four compounds with inhibitory activities against recombinant SRR. Here, we synthesized 21 derivatives of candidate 1, one of four hit compounds, and performed screening by in vitro evaluations. The derivative 13J showed a significantly lower IC50 value in vitro, and suppressed neuronal over-activation in vivo.Download high-res image (115KB)Download full-size image
Co-reporter:Daisuke Minehira, Takuya Okada, Ren Iwaki, Atsushi Kato, Isao Adachi, Naoki Toyooka
Tetrahedron Letters 2015 Volume 56(Issue 2) pp:331-334
Publication Date(Web):8 January 2015
DOI:10.1016/j.tetlet.2014.11.087
The enantiodivergent synthesis of polyhydroxylated pyrrolizidines has been achieved, starting from common intermediate 1. The synthesis involved the stereoselective construction of the pyrrolizidine core unit by using intramolecular Michael cyclization reaction as the key reaction. The synthesized eight isomers were evaluated for various glycosidase inhibition effects. Compound 15 showed a moderate inhibitory activity against α-l-fucosidase and a high selectivity compared from the other glycosidases.
Co-reporter:Hong-Yan Zhang, Yu-ichiro Yamakawa, Yuji Matsuya, Naoki Toyooka, Chihiro Tohda, Suresh Awale, Feng Li, Shigetoshi Kadota, Yasuhiro Tezuka
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 2) pp:604-608
Publication Date(Web):15 January 2014
DOI:10.1016/j.bmcl.2013.12.008
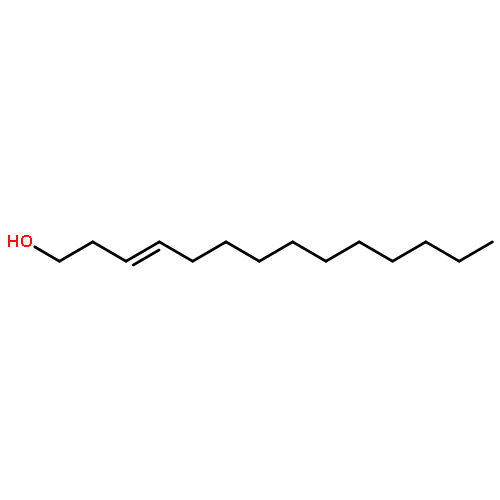
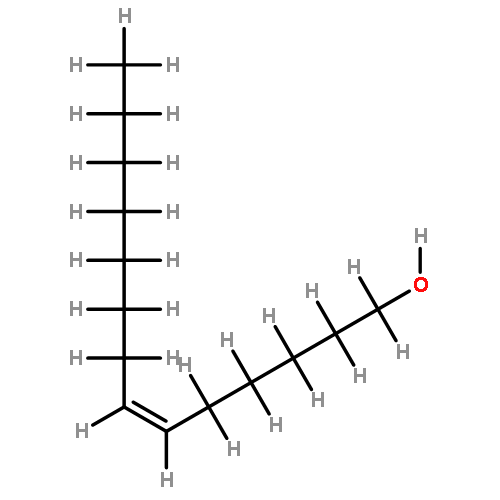
In order to develop new drugs for Alzheimer’s disease, we prepared 17 fatty acid derivatives with different chain lengths and different numbers and positions of double bonds by using Wittig reaction and stereospecific hydrogenation of triple bonds as key reactions. Among them, (4Z,15Z)-octadecadienoic acid (10) and (23Z,34Z)-heptatriacontadienoic acid (16) showed the most potent neurite outgrowth activities on Aβ(25–35)-treated rat cortical neurons, which activities were comparable to that of a positive control, NGF. Both fatty acids 10 and 16 possess two (Z)-double bonds at the n-3 and n-14 positions, which might be important for the neurite outgrowth activity.
Co-reporter:Yoshinori Ichihara, Ryohei Fujimura, Hiroshi Tsuneki, Tsutomu Wada, Kentaro Okamoto, Hiroaki Gouda, Shuichi Hirono, Kenji Sugimoto, Yuji Matsuya, Toshiyasu Sasaoka, Naoki Toyooka
European Journal of Medicinal Chemistry 2013 Volume 62() pp:649-660
Publication Date(Web):April 2013
DOI:10.1016/j.ejmech.2013.01.014
Novel 4-substituted 2-pyridin-2-ylamides were developed using in-silico ligand-based drug design (LBDD) in an attempt to identify inhibitors of SH2-containing 5′-inositol phosphatase 2 (SHIP2), which is implicated in insulin-resistant type 2 diabetes. Among the compounds synthesized, N-[4-(4-chlorobenzyloxy)pyridin-2-yl]-2-(2,6-difluorophenyl)- acetamide (CPDA, 4a) was identified as a potent SHIP2 inhibitor. CPDA was found to enhance in vitro insulin signaling through the Akt pathway more efficiently than the previously reported SHIP2 inhibitor AS1949490, and ameliorated abnormal glucose metabolism in diabetic (db/db) mice.Graphical abstractHighlights► Rational design of novel SHIP2 inhibitors using in-silico LBDD was reported. ► CPDA (4a) was found to enhance in vitro insulin signaling. ► CPDA (4a) was also found to improve the abnormal glucose metabolism in db/db mice.
Co-reporter:Atsushi Kato, Toru Okaki, Syohei Ifuku, Kasumi Sato, Yuki Hirokami, Ren Iwaki, Akiko Kamori, Shinpei Nakagawa, Isao Adachi, Peter G. Kiria, Osamu Onomura, Daishiro Minato, Kenji Sugimoto, Yuji Matsuya, Naoki Toyooka
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 21) pp:6565-6573
Publication Date(Web):1 November 2013
DOI:10.1016/j.bmc.2013.08.028
In this study we revealed that the addition of an N-phenylacetamide substituent to the C-1 position of 1-deoxyfuconojirimycin (DFJ) can lead to highly potent inhibitors of α-l-fucosidases. A structure–activity relationship study showed that a fluoro group on the phenyl ring greatly increased its potency and selectivity. In contrast the addition of two or three fluoro groups decreased their inhibition potency. Consequently, N-(2-fluorophenyl)-2β-DFJ acetamide (18j) was found to display very potent and selective inhibition of bovine kidney, rat epididymis, and human lysosome α-l-fucosidases, with IC50 value of 0.012, 0.044, and 0.0079 μM respectively. It is noteworthy that our designed N-phenyl-2β-DFJ acetamide derivative exhibited about 18-fold stronger effects on human lysosomal α-l-fucosidase than original DFJ and it occupied the active-site of this enzyme. We therefore expect that this compound may find applications in new therapeutic trials against genetic deficiency disorders.
Co-reporter:Toru Okaki;Ryohei Fujimura;Masataka Sekiguchi;Dejun Zhou;Kenji Sugimoto;Daishiro Minato;Yuji Matsuya;Atsushi Kato;Isao Adachi;Yasuhiro Tezuka;Ralph A. Saporito
European Journal of Organic Chemistry 2013 Volume 2013( Issue 14) pp:2841-2848
Publication Date(Web):
DOI:10.1002/ejoc.201201567
Abstract
Total synthesis of (–)-L-batzellasides A, B, and C has been achieved in 13 steps from known lactone 2 in 12.6, 13.2, and 13.8 % overall yields, respectively. The key steps in this synthesis were the stereoselective introduction of an allyl group at the C1 position by acyliminium chemistry and Brown's asymmetric allylation of the corresponding aldehydes to construct a stereocenter on the side chain.
Co-reporter:Xu Wang, Jie Li, Ralph A. Saporito, Naoki Toyooka
Tetrahedron 2013 69(48) pp: 10311-10315
Publication Date(Web):
DOI:10.1016/j.tet.2013.10.009
Co-reporter:Tomoki Saka, Toru Okaki, Shohei Ifuku, Yukiko Yamashita, Kasumi Sato, Shota Miyawaki, Akiko Kamori, Atsushi Kato, Isao Adachi, Yasuhiro Tezuka, Peter G. Kiria, Osamu Onomura, Daishiro Minato, Kenji Sugimoto, Yuji Matsuya, Naoki Toyooka
Tetrahedron 2013 69(49) pp: 10653-10661
Publication Date(Web):
DOI:10.1016/j.tet.2013.10.006
Co-reporter:Daisuke Minehira, Daisuke Takeda, Hirokazu Urata, Atsushi Kato, Isao Adachi, Xu Wang, Yuji Matsuya, Kenji Sugimoto, Mayuko Takemura, Satoshi Endo, Toshiyuki Matsunaga, Akira Hara, Jun Koseki, Kayo Narukawa, Shuichi Hirono, Naoki Toyooka
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 1) pp:356-367
Publication Date(Web):1 January 2012
DOI:10.1016/j.bmc.2011.10.073
New substituted (1-thioxo-1,2,3,4-tetrahydro-β-carbolin-9-yl)acetic acids were designed as the inhibitor of AKR1B1 based upon the structure of rhetsinine, a minor alkaloidal component of Evodia rutaecarpa, and twenty derivatives were synthesized and evaluated. The most active compound of the series was (2-benzyl-6-methoxy-1-thioxo-1,2,3,4-tetrahydro-β-carbolin-9-yl)acetic acid (7m), which showed comparable inhibitory activity for AKR1B1 (IC50 = 0.15 μM) with clinically used epalrestat (IC50 = 0.1 μM). In the view of activity and selectivity, the most potent compound was (2-benzyl-6-carboxy-1-thioxo-1,2,3,4-tetrahydro-β-carbolin-9-yl)acetic acid (7t), which showed strong inhibitory effect (IC50 = 0.17 μM) and very high selectivity for AKR1B1 against AKR1A1 (311:1) and AKR1B10 (253:1) compared with epalrestat.The binding conformation of the 7t (blue stick) for AKR1B1 (A) and for AKR1B10 (B), and the difference of binding pocket (C) between AKR1B1 (green solid surface) and AKR1B10 (red wireframe).

![(R)-(4-(5-Chlorobenzo[d]oxazol-2-yl)-7-methyl-1,4-diazepan-1-yl)(5-methyl-2-(2H-1,2,3-triazol-2-yl)phenyl)methanone](http://img.cochemist.com/ccimg/1030400/1030377-33-3.png)
![(R)-(4-(5-Chlorobenzo[d]oxazol-2-yl)-7-methyl-1,4-diazepan-1-yl)(5-methyl-2-(2H-1,2,3-triazol-2-yl)phenyl)methanone](http://img.cochemist.com/ccimg/1030400/1030377-33-3_b.png)



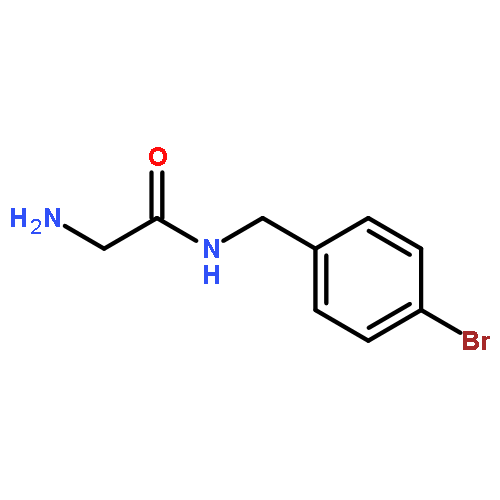

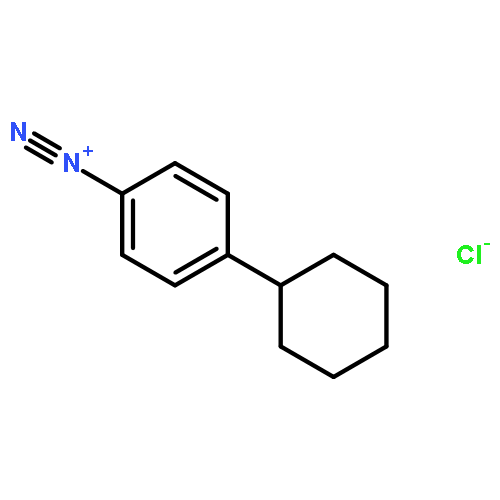
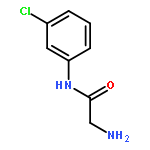
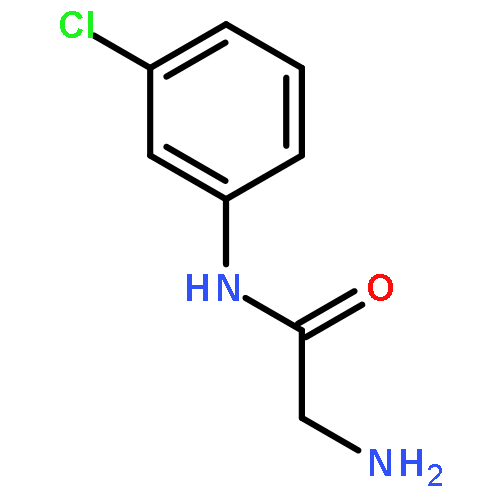
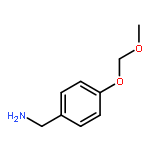
![[3-(METHOXYMETHOXY)PHENYL]METHANAMINE](http://img.cochemist.com/ccimg/553700/553611-73-7.png)
![[3-(METHOXYMETHOXY)PHENYL]METHANAMINE](http://img.cochemist.com/ccimg/553700/553611-73-7_b.png)

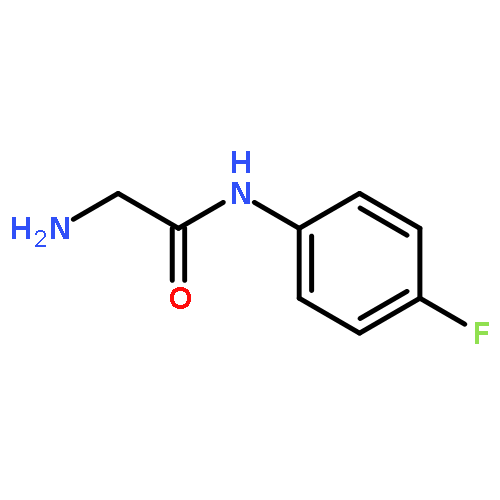
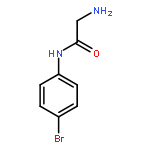

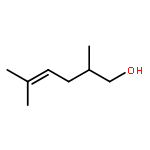
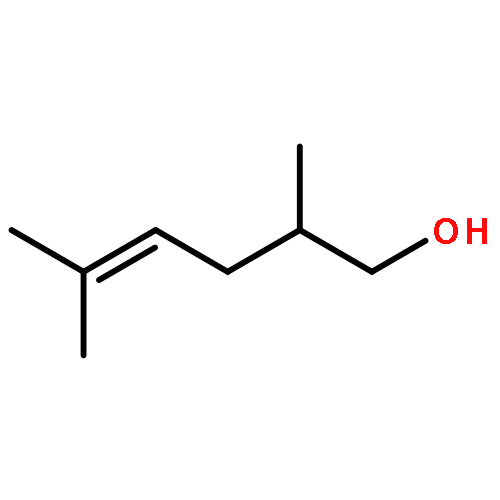
![Thiopyrano[3,4-b]indol-1(3H)-one, 4,9-dihydro-](http://img.cochemist.com/ccimg/180500/180403-45-6.png)
![Thiopyrano[3,4-b]indol-1(3H)-one, 4,9-dihydro-](http://img.cochemist.com/ccimg/180500/180403-45-6_b.png)






![Nonanal, 9-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-](http://img.cochemist.com/ccimg/139300/139258-86-9.png)
![Nonanal, 9-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-](http://img.cochemist.com/ccimg/139300/139258-86-9_b.png)
![1-Nonanol, 9-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-](http://img.cochemist.com/ccimg/139300/139258-85-8.png)
![1-Nonanol, 9-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-](http://img.cochemist.com/ccimg/139300/139258-85-8_b.png)
![Carbamic acid, [2-[(4-fluorophenyl)amino]-2-oxoethyl]-,1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/137900/137895-52-4.png)
![Carbamic acid, [2-[(4-fluorophenyl)amino]-2-oxoethyl]-,1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/137900/137895-52-4_b.png)
amino]-, methylester, (3R)-](http://img.cochemist.com/ccimg/134500/134430-97-0.png)
amino]-, methylester, (3R)-](http://img.cochemist.com/ccimg/134500/134430-97-0_b.png)
![1-Butanol, 4-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-](http://img.cochemist.com/ccimg/130400/130372-07-5.png)
![1-Butanol, 4-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-](http://img.cochemist.com/ccimg/130400/130372-07-5_b.png)
![Butanal, 4-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-](http://img.cochemist.com/ccimg/127800/127793-62-8.png)
![Butanal, 4-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-](http://img.cochemist.com/ccimg/127800/127793-62-8_b.png)

![4,9-dihydro-6-methyl-Pyrano[3,4-b]indol-1(3H)-one](http://img.cochemist.com/ccimg/112600/112585-23-6.png)
![4,9-dihydro-6-methyl-Pyrano[3,4-b]indol-1(3H)-one](http://img.cochemist.com/ccimg/112600/112585-23-6_b.png)
![1H-Pyrido[3,4-b]indol-1-one, 2,3,4,9-tetrahydro-2-(phenylmethyl)-](http://img.cochemist.com/ccimg/111500/111492-86-5.png)
![1H-Pyrido[3,4-b]indol-1-one, 2,3,4,9-tetrahydro-2-(phenylmethyl)-](http://img.cochemist.com/ccimg/111500/111492-86-5_b.png)
![Pyrano[3,4-b]indol-1(3H)-one, 6-chloro-4,9-dihydro-](http://img.cochemist.com/ccimg/111000/110977-89-4.png)
![Pyrano[3,4-b]indol-1(3H)-one, 6-chloro-4,9-dihydro-](http://img.cochemist.com/ccimg/111000/110977-89-4_b.png)



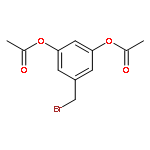
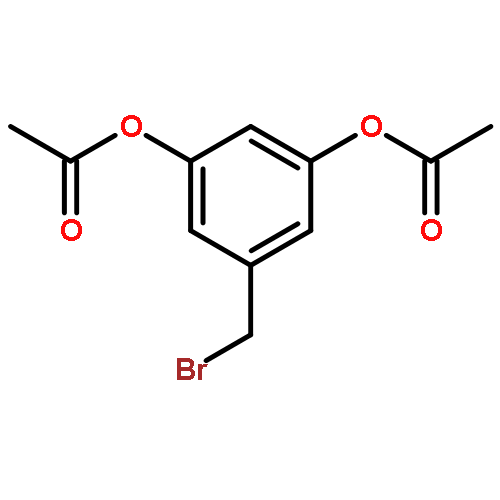
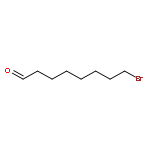
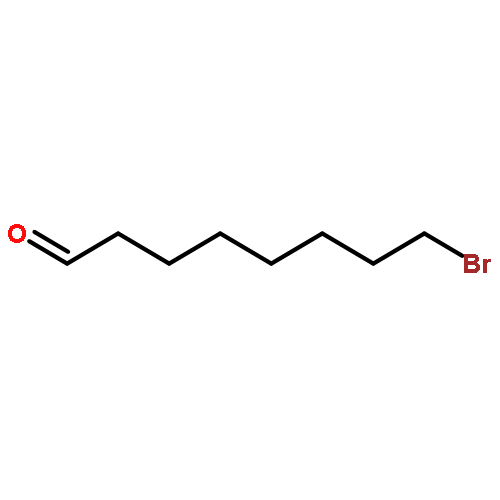

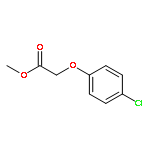
![Acetic acid, [4-(phenylmethoxy)phenoxy]-, methyl ester](http://img.cochemist.com/ccimg/81500/81466-35-5.png)
![Acetic acid, [4-(phenylmethoxy)phenoxy]-, methyl ester](http://img.cochemist.com/ccimg/81500/81466-35-5_b.png)







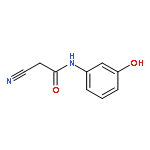
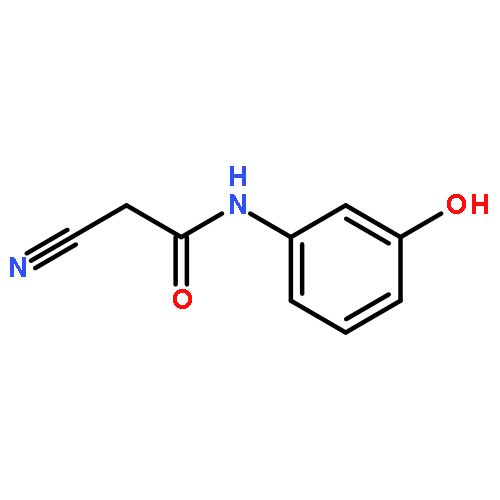
![Acetamide, N-[(3-chlorophenyl)methyl]-2-cyano-](http://img.cochemist.com/ccimg/64500/64488-08-0.png)
![Acetamide, N-[(3-chlorophenyl)methyl]-2-cyano-](http://img.cochemist.com/ccimg/64500/64488-08-0_b.png)
![Acetamide, N-[(2-chlorophenyl)methyl]-2-cyano-](http://img.cochemist.com/ccimg/64500/64488-07-9.png)
![Acetamide, N-[(2-chlorophenyl)methyl]-2-cyano-](http://img.cochemist.com/ccimg/64500/64488-07-9_b.png)
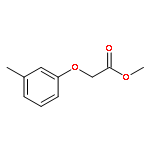
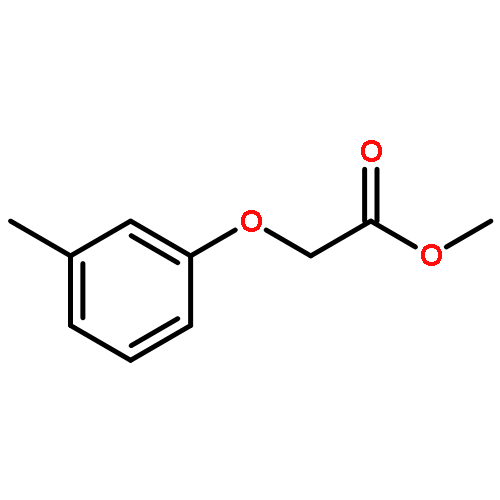


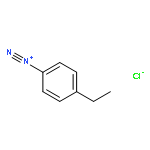
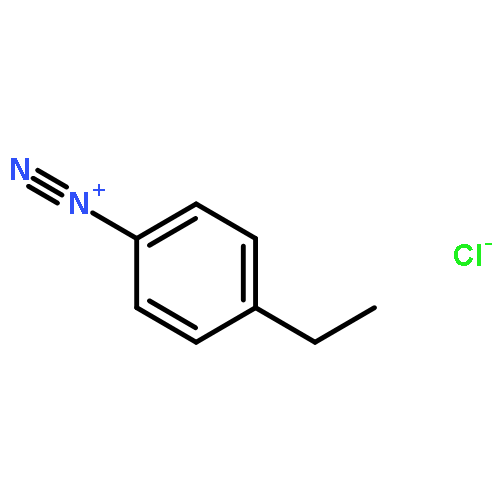
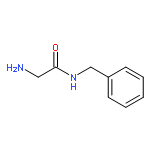
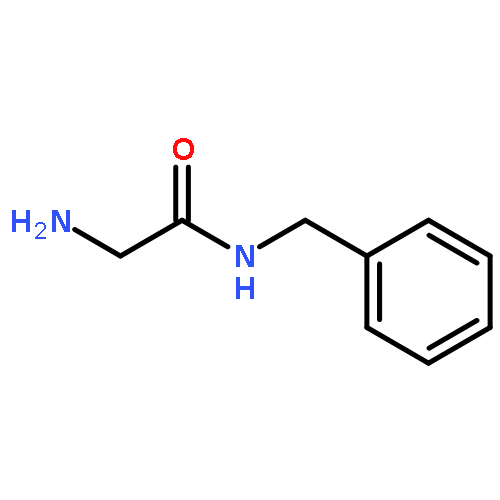
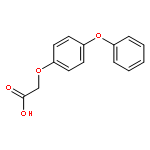








![Carbamic acid, N-[2-oxo-2-[(phenylmethyl)amino]ethyl]-, 1,1-dimethylethyl ester](/data/chemimg/3768200/19811-52-0.png)
![Carbamic acid, N-[2-oxo-2-[(phenylmethyl)amino]ethyl]-, 1,1-dimethylethyl ester](/data/chemimg/3768200/19811-52-0_b.png)
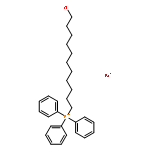
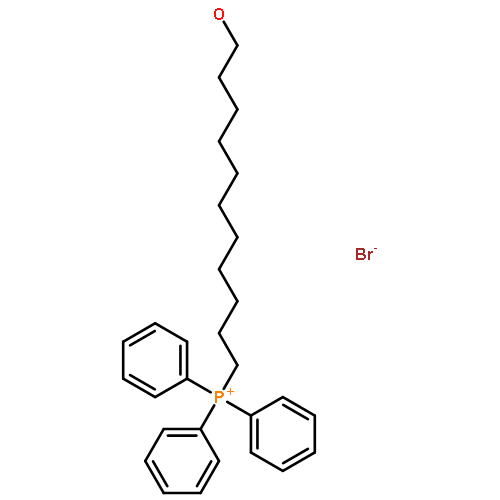
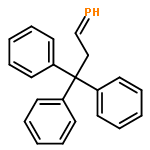
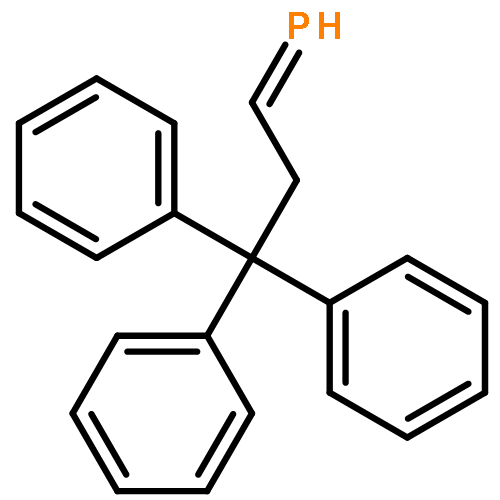
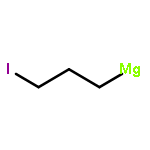
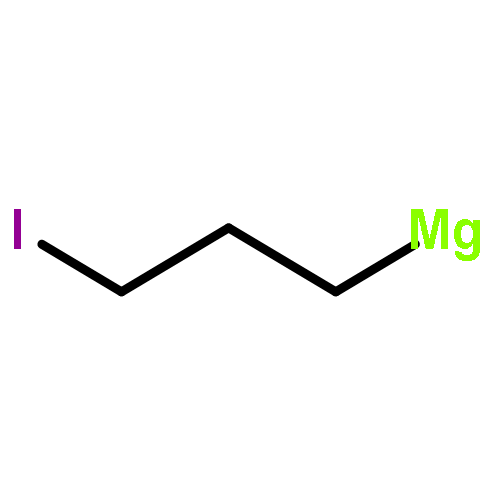



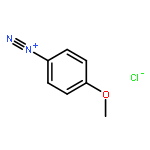
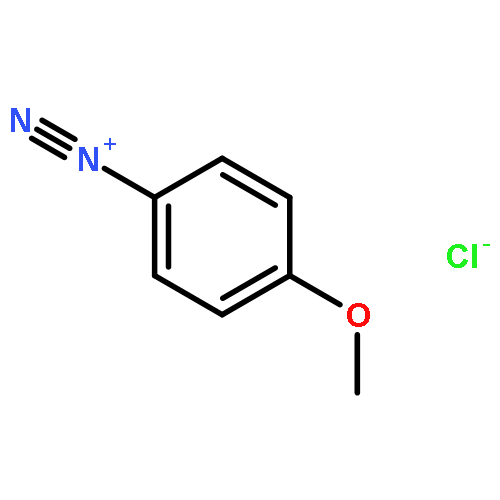
![[1,1'-Biphenyl]-4-diazonium, chloride](http://img.cochemist.com/ccimg/4200/4163-91-1.png)
![[1,1'-Biphenyl]-4-diazonium, chloride](http://img.cochemist.com/ccimg/4200/4163-91-1_b.png)














![4,9-dihydro-Pyrano[3,4-b]indol-1(3H)-one](http://img.cochemist.com/ccimg/6300/6250-88-0.png)
![4,9-dihydro-Pyrano[3,4-b]indol-1(3H)-one](http://img.cochemist.com/ccimg/6300/6250-88-0_b.png)