Co-reporter:Jianyu Zhang
Journal of the American Chemical Society 2016 Volume 138(Issue 29) pp:9158-9165
Publication Date(Web):June 29, 2016
DOI:10.1021/jacs.6b03462
Although an enormous and still growing number of biologically diverse methyltransferases have been reported and identified, a comprehensive understanding of the enzymatic methyl transfer mechanism is still lacking. Glycine N-methyltransferase (GNMT), a member of the family that acts on small metabolites as the substrate, catalyzes methyl transfer from S-adenosyl-l-methionine (AdoMet) to glycine to form S-adenosyl-l-homocysteine and sarcosine. We report primary carbon (12C/14C) and secondary (1H3/3H3) kinetic isotope effects at the transferred methyl group, together with 1H3/3H3 binding isotope effects for wild-type GNMT and a series of Tyr21 mutants. The data implicate a compaction effect in the methyl transfer step that is conferred by the protein structure. Furthermore, a remarkable similarity of properties is observed between GNMT and catechol O-methyltransferase, despite significant differences between these enzymes with regard to their active site structures and catalyzed reactions. We attribute these results to a catalytically relevant reduction in the methyl donor–acceptor distance that is dependent on a tyrosine side chain positioned behind the methyl-bearing sulfur of AdoMet.
Co-reporter:Adam R. Offenbacher, Hui Zhu, Judith P. Klinman
Tetrahedron Letters 2016 Volume 57(Issue 41) pp:4537-4540
Publication Date(Web):12 October 2016
DOI:10.1016/j.tetlet.2016.08.071
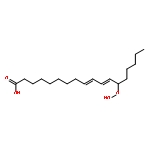
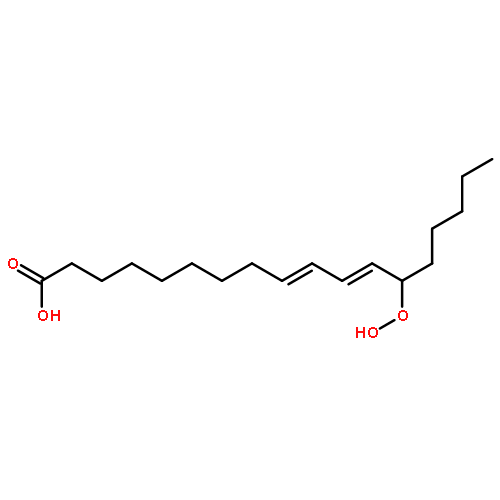
•Strategies for site-specific 13C isotope labeling of fatty acids.•Cu(I)-mediated cross coupling reactions.•Application of Corey–Fuchs aldehyde–alkyne conversion for selective 13C insertion.Soybean lipoxygenase-1 (SLO-1) catalyzes the C–H abstraction from the reactive carbon (C-11) in linoleic acid as the first and rate-determining step in the formation of alkylhydroperoxides. While previous labeling strategies have focused on deuterium labeling to ascertain the primary and secondary kinetic isotope effects for this reaction, there is an emerging interest and need for selectively enriched 13C isotopologues. In this Letter, we present synthetic strategies for site-specific 13C labeled linoleic acid substrates. We take advantage of a Corey–Fuchs formyl to terminal 13C-labeled alkyne conversion, using 13CBr4 as the labeling source, to reduce the number of steps from a previous fatty acid 13C synthetic labeling approach. The labeled linoleic acid substrates are useful as nuclear tunneling markers and for extracting active site geometries of the enzyme–substrate complex in lipoxygenase.
Co-reporter:Dr. Shenshen Hu;Jérôme Cattin-Ortolá;Dr. Jeffrey W. Munos;Dr. Judith P. Klinman
Angewandte Chemie International Edition 2016 Volume 55( Issue 32) pp:9361-9364
Publication Date(Web):
DOI:10.1002/anie.201603592
Abstract
The proposed contributions of distinct classes of local versus global protein motions during enzymatic bond making/breaking processes has been difficult to verify. We employed soybean lipoxygenase-1 as a model system to investigate the impact of high pressure at variable temperatures on the hydrogen-tunneling properties of the wild-type protein and three single-site mutants. For all variants, pressure dramatically elevates the enthalpies of activation for the C−H activation. In contrast, the primary kinetic isotope effects (KIEs) for C−H activation and their corresponding temperature dependencies remain unchanged up to ca. 700 bar. The differential impact of elevated hydrostatic pressure on the temperature dependencies of rate constants versus substrate KIEs provides direct evidence for two distinct classes of protein motions: local, isotope-dependent donor–acceptor distance-sampling modes, and a more global, isotope-independent search for productive protein conformational sub-states.
Co-reporter:Dr. Shenshen Hu;Jérôme Cattin-Ortolá;Dr. Jeffrey W. Munos;Dr. Judith P. Klinman
Angewandte Chemie 2016 Volume 128( Issue 32) pp:9507-9510
Publication Date(Web):
DOI:10.1002/ange.201603592
Abstract
The proposed contributions of distinct classes of local versus global protein motions during enzymatic bond making/breaking processes has been difficult to verify. We employed soybean lipoxygenase-1 as a model system to investigate the impact of high pressure at variable temperatures on the hydrogen-tunneling properties of the wild-type protein and three single-site mutants. For all variants, pressure dramatically elevates the enthalpies of activation for the C−H activation. In contrast, the primary kinetic isotope effects (KIEs) for C−H activation and their corresponding temperature dependencies remain unchanged up to ca. 700 bar. The differential impact of elevated hydrostatic pressure on the temperature dependencies of rate constants versus substrate KIEs provides direct evidence for two distinct classes of protein motions: local, isotope-dependent donor–acceptor distance-sampling modes, and a more global, isotope-independent search for productive protein conformational sub-states.
Co-reporter:Corey W. Meadows; Gurusamy Balakrishnan; Brandon L. Kier; Thomas G. Spiro
Journal of the American Chemical Society 2015 Volume 137(Issue 32) pp:10060-10063
Publication Date(Web):July 29, 2015
DOI:10.1021/jacs.5b04413
Protein dynamics on the microsecond (μs) time scale were investigated by temperature-jump fluorescence spectroscopy as a function of temperature in two variants of a thermophilic alcohol dehydrogenase: W87F and W87F:H43A. Both mutants exhibit a fast, temperature-independent μs decrease in fluorescence followed by a slower full recovery of the initial fluorescence. The results, which rule out an ionizing histidine as the origin of the fluorescence quenching, are discussed in the context of a Trp49-containing dimer interface that acts as a conduit for thermally activated structural change within the protein interior.
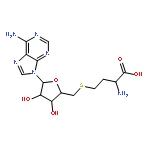
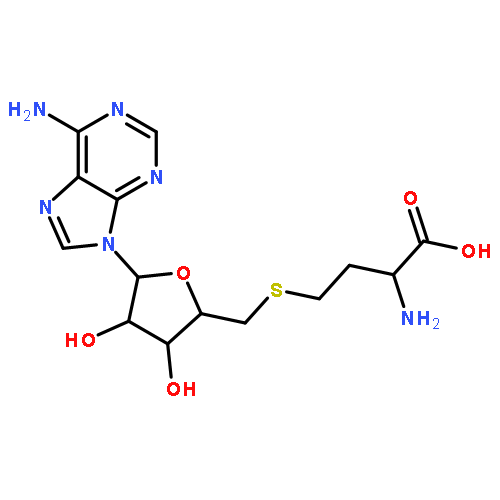
Co-reporter:Hui Zhu; Spencer C. Peck; Florence Bonnot; Wilfred A. van der Donk
Journal of the American Chemical Society 2015 Volume 137(Issue 33) pp:10448-10451
Publication Date(Web):August 12, 2015
DOI:10.1021/jacs.5b03907
Nonheme iron oxygenases that carry out four-electron oxidations of substrate have been proposed to employ iron(III) superoxide species to initiate this reaction [Paria, S.; Que, L.; Paine, T. K. Angew. Chem. Int. Ed. 2011, 50, 11129]. Here we report experimental evidence in support of this proposal. 18O KIEs were measured for two recently discovered mononuclear nonheme iron oxygenases: hydroxyethylphosphonate dioxygenase (HEPD) and methylphosphonate synthase (MPnS). Competitive 18O KIEs measured with deuterated substrates are larger than those measured with unlabeled substrates, which indicates that C–H cleavage must occur before an irreversible reductive step at molecular oxygen. A similar observation was previously used to implicate copper(II) superoxide in the H-abstraction reactions catalyzed by dopamine β-monooxygenase [Tian, G. C.; Klinman, J. P. J. Am. Chem. Soc. 1993, 115, 8891] and peptidylglycine α-hydroxylating monooxygenase [Francisco, W. A.; Blackburn, N. J.; Klinman, J. P. Biochemistry 2003, 42, 1813].
Co-reporter:Hui Zhu; Monika Sommerhalter; Andy K. L. Nguy
Journal of the American Chemical Society 2015 Volume 137(Issue 17) pp:5720-5729
Publication Date(Web):April 28, 2015
DOI:10.1021/ja512388n
Tyramine β-monooxygenase (TβM) belongs to a family of physiologically important dinuclear copper monooxygenases that function with a solvent-exposed active site. To accomplish each enzymatic turnover, an electron transfer (ET) must occur between two solvent-separated copper centers. In wild-type TβM, this event is too fast to be rate limiting. However, we have recently shown [Osborne, R. L.; et al. Biochemistry 2013, 52, 1179] that the Tyr216Ala variant of TβM leads to rate-limiting ET. In this study, we present a pH–rate profile study of Tyr216Ala, together with deuterium oxide solvent kinetic isotope effects (KIEs). A solvent KIE of 2 on kcat is found in a region where kcat is pH/pD independent. As a control, the variant Tyr216Trp, for which ET is not rate determining, displays a solvent KIE of unity. We conclude, therefore, that the observed solvent KIE arises from the rate-limiting ET step in the Tyr216Ala variant, and show how small solvent KIEs (ca. 2) can be fully accommodated from equilibrium effects within the Marcus equation. To gain insight into the role of the enzyme in the long-range ET step, a temperature dependence study was also pursued. The small enthalpic barrier of ET (Ea = 3.6 kcal/mol) implicates a significant entropic barrier, which is attributed to the requirement for extensive rearrangement of the inter-copper environment during PCET catalyzed by the Tyr216Ala variant. The data lead to the proposal of a distinct inter-domain pathway for PCET in the dinuclear copper monooxygenases.
Co-reporter:Sudhir C. Sharma and Judith P. Klinman
Biochemistry 2015 Volume 54(Issue 35) pp:
Publication Date(Web):July 8, 2015
DOI:10.1021/acs.biochem.5b00374
Soybean lipoxygenase-1 (SLO-1) is a paradigmatic enzyme system for studying the contribution of hydrogen tunneling to enzymatic proton-coupled electron transfer processes. In this study, the impact of pairs of double mutants on the properties of SLO-1 is presented. Steady-state rates and their deuterium kinetic isotope effects (KIEs) have been measured for the bimolecular reaction of enzyme with free substrate (kcat/Km) and compared to the unimolecular rate constant, kcat. A key kinetic finding is that the competitive KIEs on the second-order rate constant (kcat/Km) are all reduced from Dkcat and, despite large changes in rate and activation parameters, remain essentially unaltered under a variety of conditions. These data implicate a protein reaction coordinate that is orthogonal to the chemical reaction coordinate and controls the concentration of the active enzyme. This study introduces a new means to interrogate the alteration of conformational landscapes that can occur following site-specific mutagenesis.
Co-reporter:Jianyu Zhang, Judith P. Klinman
Analytical Biochemistry 2015 Volume 476() pp:81-83
Publication Date(Web):1 May 2015
DOI:10.1016/j.ab.2015.02.004
Abstract
S-adenosyl-l-methionine (AdoMet), an important biological cofactor, exists in two chiral forms, (S,S)- and (R,S)-, only the former of which is biologically active. Here, we have developed a chromatographic method to obtain pure (S,S)-AdoMet using a single C18 column.
Co-reporter:Jianyu Zhang;Heather J. Kulik;Todd J. Martinez
PNAS 2015 Volume 112 (Issue 26 ) pp:7954-7959
Publication Date(Web):2015-06-30
DOI:10.1073/pnas.1506792112
Enzymatic methyl transfer, catalyzed by catechol-O-methyltransferase (COMT), is investigated using binding isotope effects (BIEs), time-resolved fluorescence lifetimes, Stokes
shifts, and extended graphics processing unit (GPU)-based quantum mechanics/molecular mechanics (QM/MM) approaches. The WT
enzyme is compared with mutants at Tyr68, a conserved residue that is located behind the reactive sulfur of cofactor. Small
(>1) BIEs are observed for an S-adenosylmethionine (AdoMet)-binary and abortive ternary complex containing 8-hydroxyquinoline,
and contrast with previously reported inverse (<1) kinetic isotope effects (KIEs). Extended GPU-based computational studies
of a ternary complex containing catecholate show a clear trend in ground state structures, from noncanonical bond lengths
for WT toward solution values with mutants. Structural and dynamical differences that are sensitive to Tyr68 have also been
detected using time-resolved Stokes shift measurements and molecular dynamics. These experimental and computational results
are discussed in the context of active site compaction that requires an ionization of substrate within the enzyme ternary
complex.
Co-reporter:Judith P. Klinman
ACS Central Science 2015 Volume 1(Issue 3) pp:115
Publication Date(Web):June 5, 2015
DOI:10.1021/acscentsci.5b00215
Co-reporter:Judith P. Klinman
PNAS 2015 Volume 112 (Issue 34 ) pp:10568-10569
Publication Date(Web):2015-08-25
DOI:10.1073/pnas.1512507112
Co-reporter:Judith P. Klinman and Florence Bonnot
Chemical Reviews 2014 Volume 114(Issue 8) pp:4343
Publication Date(Web):December 18, 2013
DOI:10.1021/cr400475g
Co-reporter:Corey W. Meadows ; Jonathan E. Tsang
Journal of the American Chemical Society 2014 Volume 136(Issue 42) pp:14821-14833
Publication Date(Web):October 14, 2014
DOI:10.1021/ja506667k
Time-resolved fluorescence dynamics are investigated in two mutants of a thermophilic alcohol dehydrogenase (ht-ADH): Y25A (at the dimer interface) and V260A (at the cofactor-binding domain). These residues, ca. 32 Å apart, are shown to exhibit opposing low-temperature effects on the hydride tunneling step. Using single-tryptophan constructs at the active site (Trp87) and a remote, surface-exposed site (Trp167), time-dependent Stokes shifts and collisional quenching data allow an analysis of intra-protein dynamical communication. A double mutant, Y25A:V260A, was also inserted into each single-Trp construct and analyzed accordingly. None of the mutations affect fluorescence lifetimes, Stokes shift relaxation rates, and quenching data for the surface-exposed Trp167 to an appreciable extent. By contrast, fluorescent probes of the active-site tryptophan 87 reveal distinctive forms of dynamical communication. Stokes shifts show that the distal Y25A increases active-site flexibility, V260A introduces a temperature-dependent equilibration process not previously reported by such measurements, and the double mutant (Y25A:V260A) eliminates the temperature-dependent transition sensed by the active-site tryptophan in the presence of V260A. Collisional quenching data at Trp87 further show a structural change in the active-site environment/solvation for V260A. In the aggregate, the temperature dependencies of the fluorescence data are distinct from the breaks in behavior previously reported for catalysis and hydrogen/deuterium exchange, attributed to time scales for the interconversion of protein conformational substates that are slower and more global than the local motions monitored within. An extended network of dynamical communication between the protein dimer surface and substrate- and cofactor-binding domains emerges from the flourescent data.
Co-reporter:Shenshen Hu ; Sudhir C. Sharma ; Alexander D. Scouras ; Alexander V. Soudackov ; Cody A. Marcus Carr ; Sharon Hammes-Schiffer ; Tom Alber
Journal of the American Chemical Society 2014 Volume 136(Issue 23) pp:8157-8160
Publication Date(Web):June 2, 2014
DOI:10.1021/ja502726s
The enzyme soybean lipoxygenase (SLO) has served as a prototype for hydrogen-tunneling reactions, as a result of its unusual kinetic isotope effects (KIEs) and their temperature dependencies. Using a synergy of kinetic, structural, and theoretical studies, we show how the interplay between donor–acceptor distance and active-site flexibility leads to catalytic behavior previously predicted by quantum tunneling theory. Modification of the size of two hydrophobic residues by site-specific mutagenesis in SLO reduces the reaction rate 104-fold and is accompanied by an enormous and unprecedented room-temperature KIE. Fitting of the kinetic data to a non-adiabatic model implicates an expansion of the active site that cannot be compensated by donor–acceptor distance sampling. A 1.7 Å resolution X-ray structure of the double mutant further indicates an unaltered backbone conformation, almost identical side-chain conformations, and a significantly enlarged active-site cavity. These findings show the compelling property of room-temperature hydrogen tunneling within a biological context and demonstrate the very high sensitivity of such tunneling to barrier width.
Co-reporter:Cody A. Marcus Carr and Judith P. Klinman
Biochemistry 2014 Volume 53(Issue 14) pp:
Publication Date(Web):March 18, 2014
DOI:10.1021/bi500070q
A bacterial lipoxygenase (LOX) shows a deuterium kinetic isotope effect (KIE) that is similar in magnitude and temperature dependence to the very large KIE of eukaryotic LOXs. This occurs despite the evolutionary distance, an ∼25% smaller catalytic domain, and an increase in Ea of ∼11 kcal/mol. Site-specific mutagenesis leads to a protein variant with an Ea similar to that of the prototypic plant LOX, providing possible insight into the origin of evolutionary differences. These findings, which extend the phenomenon of hydrogen tunneling to a prokaryotic LOX, are discussed in the context of a role for protein size and/or flexibility in enzymatic hydrogen tunneling.
Co-reporter:Corey W. Meadows, Ryan Ou, and Judith P. Klinman
The Journal of Physical Chemistry B 2014 Volume 118(Issue 23) pp:6049-6061
Publication Date(Web):June 3, 2014
DOI:10.1021/jp500825x
Two single-tryptophan variants were generated in a thermophilic alcohol dehydrogenase with the goal of correlating temperature-dependent changes in local fluorescence with the previously demonstrated catalytic break at ca. 30 °C (Kohen et al., Nature 1999, 399, 496). One tryptophan variant, W87in, resides at the active site within van der Waals contact of bound alcohol substrate; the other variant, W167in, is a remote-site surface reporter located >25 Å from the active site. Picosecond-resolved fluorescence measurements were used to analyze fluorescence lifetimes, time-dependent Stokes shifts, and the extent of collisional quenching at Trp87 and Trp167 as a function of temperature. A subnanosecond fluorescence decay rate constant has been detected for W87in that is ascribed to the proximity of the active site Zn2+ and shows a break in behavior at 30 °C. For the remainder of the reported lifetime measurements, there is no detectable break between 10 and 50 °C, in contrast with previously reported hydrogen/deuterium exchange experiments that revealed a temperature-dependent break analogous to catalysis (Liang et al., Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 9556). We conclude that the motions that lead to the rigidification of ht-ADH below 30 °C are likely to be dominated by global processes slower than the picosecond to nanosecond motions measured herein. In the case of collisional quenching of fluorescence by acrylamide, W87in and W167in behave in a similar manner that resembles free tryptophan in water. Stokes shift measurements, by contrast, show distinctive behaviors in which the active-site tryptophan relaxation is highly temperature-dependent, whereas the solvent-exposed tryptophan’s dynamics are temperature-independent. These data are concluded to reflect a significantly constrained environment surrounding the active site Trp87 that both increases the magnitude of the Stokes shift and its temperature-dependence. The results are discussed in the context of spatially distinct differences in enthalpic barriers for protein conformational sampling that may be related to catalysis.
Co-reporter:Robert L. Osborne, Hui Zhu, Anthony T. Iavarone, Ninian J. Blackburn, and Judith P. Klinman
Biochemistry 2013 Volume 52(Issue 7) pp:
Publication Date(Web):January 15, 2013
DOI:10.1021/bi3013609
The enzyme tyramine β-monooxygenase (TβM) belongs to a small eukaryotic family of physiologically important mononuclear dicopper monooxygenases. The properties of this family include noncoupled mononuclear copper centers ∼11 Å apart, with CuM performing C–H and O2 activation and CuH functioning as an electron storage site [Klinman, J. P. (2006) J. Biol. Chem. 281, 3013–3016]. A conserved tyrosine (Y216 in TβM) is positioned between the copper domains and is associated with CuH (through an interaction with a CuH-coordinating histidine). Mutations at Y216 (to W, I, and A) indicate little or no difference in electron paramagnetic resonance spectra, while X-ray absorption spectroscopy studies show only a very small decrease in distance between CuM and its Met471 ligand in reduced enzyme. High-performance liquid chromatography assays demonstrate that turnover of substrate is complete with Y216W and Y216I, whereas Y216A undergoes a secondary inactivation that is linked to oxidation of ligands at CuM. Steady-state kinetic and isotope effect measurements were investigated. The significantly elevated Km,Tyr for Y216A, together with a very large D(kcat/Km,Tyr) of ∼12, indicates a major impact on the binding of substrate at the CuM site. The kinetic and isotopic parameters lead to estimated rate constants for C–H bond cleavage, dissociation of substrate from the CuM site, and, in the case of Y216A, the rate of electron transfer (ET) from CuH to CuM. These studies uncover a rate-limiting ET within the solvent-filled interface and lead to a paradigm shift in our understanding of the mononuclear dicopper monooxygenases.
Co-reporter:Judith P. Klinman
Biochemistry 2013 Volume 52(Issue 12) pp:
Publication Date(Web):February 1, 2013
DOI:10.1021/bi301504m
Quantum tunneling and protein dynamics have emerged as important components of enzyme function. This review focuses on soybean lipoxygenase-1, to illustrate how the properties of enzymatic C–H bond activation link protein motions to the fundamental bond making–breaking processes.
Co-reporter:Florence Bonnot, Anthony T. Iavarone, and Judith P. Klinman
Biochemistry 2013 Volume 52(Issue 27) pp:
Publication Date(Web):May 29, 2013
DOI:10.1021/bi4003315
The final step of the biosynthesis of prokaryotic cofactor PQQ is catalyzed by PqqC, a cofactorless oxidase that brings about a ring closure and overall eight-electron oxidation of its substrate. Time-dependent acid quenching and subsequent high-performance liquid chromatography separation and mass spectrometric analyses of reaction mixtures were performed to correlate the structures of intermediates with previously observed UV–visible signatures. The reaction is composed of four stepwise oxidations: three steps use O2 as the two-electron acceptor, and the fourth uses hydrogen peroxide (H2O2). The chemical nature of the intermediates, the stoichiometry of the reaction, and their dependence on the oxygen concentration indicate that the third oxidation uses the product, H2O2, from the preceding step to produce water. The last oxidation step can also be studied separately and is a reaction between O2 and PQQH2 trapped in the active site. This oxidation is approximately 10 times slower than the reoxidation of PQQH2 in solution. From the order of the four oxidation steps and their sensitivity to O2 concentration, we propose a progressive closure of the active site as the enzyme proceeds through its catalytic cycle.
Co-reporter:Yao-Qing Shen, Florence Bonnot, Erin M. Imsand, Jordan M. RoseFigura, Kimmen Sjölander, and Judith P. Klinman
Biochemistry 2012 Volume 51(Issue 11) pp:
Publication Date(Web):February 10, 2012
DOI:10.1021/bi201763d
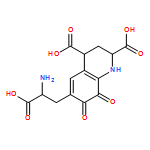
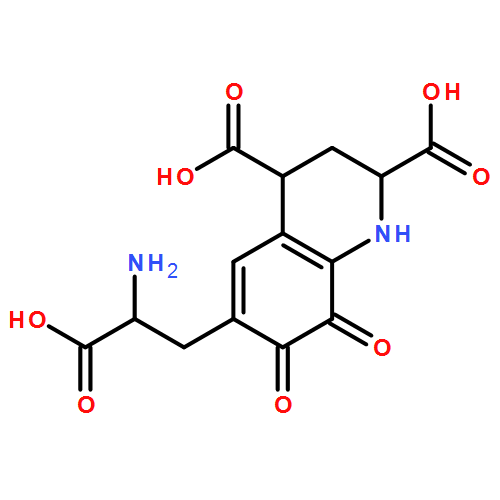
Pyrroloquinoline quinone (PQQ) is a small, redox active molecule that serves as a cofactor for several bacterial dehydrogenases, introducing pathways for carbon utilization that confer a growth advantage. Early studies had implicated a ribosomally translated peptide as the substrate for PQQ production. This study presents a sequence- and structure-based analysis of the components of the pqq operon. We find the necessary components for PQQ production are present in 126 prokaryotes, most of which are Gram-negative and a number of which are pathogens. A total of five gene products, PqqA, PqqB, PqqC, PqqD, and PqqE, are identified as being obligatory for PQQ production. Three of the gene products in the pqq operon, PqqB, PqqC, and PqqE, are members of large protein superfamilies. By combining evolutionary conservation patterns with information from three-dimensional structures, we are able to differentiate the gene products involved in PQQ biosynthesis from those with divergent functions. The observed persistence of a conserved gene order within analyzed operons strongly suggests a role for protein–protein interactions in the course of cofactor biosynthesis. These studies propose previously unidentified roles for several of the gene products, as well as identifying possible new targets for antibiotic design and application.
Co-reporter:Zachary D. Nagel, Corey W. Meadows, Ming Dong, Brian J. Bahnson, and Judith P. Klinman
Biochemistry 2012 Volume 51(Issue 20) pp:4147-4156
Publication Date(Web):May 8, 2012
DOI:10.1021/bi3001352
A growing body of data suggests that protein motion plays an important role in enzyme catalysis. Two highly conserved hydrophobic active site residues in the cofactor-binding pocket of ht-ADH (Leu176 and V260) have been mutated to a series of hydrophobic side chains of smaller size, as well as one deletion mutant, L176Δ. Mutations decrease kcat and increase KM(NAD+). Most of the observed decreases in effects on kcat at pH 7.0 are due to an upward shift in the optimal pH for catalysis; a simple electrostatic model is invoked that relates the change in pKa to the distance between the positively charged nicotinamide ring and bound substrate. Structural modeling of the L176Δ and V260A variants indicates the development of a cavity behind the nicotinamide ring without any significant perturbation of the secondary structure of the enzyme relative to that of the wild type. Primary kinetic isotope effects (KIEs) are modestly increased for all mutants. Above the dynamical transition at 30 °C for ht-ADH [Kohen, A., et al. (1999) Nature399, 496], the temperature dependence of the KIE is seen to increase with a decrease in side chain volume at positions 176 and 260. Additionally, the relative trends in the temperature dependence of the KIE above and below 30 °C appear to be reversed for the cofactor-binding pocket mutants in relation to wild-type protein. The aggregate results are interpreted in the context of a full tunneling model of enzymatic hydride transfer that incorporates both protein conformational sampling (preorganization) and active site optimization of tunneling (reorganization). The reduced temperature dependence of the KIE in the mutants below 30 °C indicates that at low temperatures, the enzyme adopts conformations refractory to donor–acceptor distance sampling.
Co-reporter:Robert L. Osborne, Hui Zhu, Anthony T. Iavarone, Corinna R. Hess, and Judith P. Klinman
Biochemistry 2012 Volume 51(Issue 38) pp:
Publication Date(Web):August 14, 2012
DOI:10.1021/bi300456f
Tyramine β-monooxygenase (TβM), the insect homologue of dopamine β-monooxygenase, is a neuroregulatory enzyme that catalyzes the β-hydroxylation of tyramine to yield octopamine. Mutation of the methionine (Met) ligand to CuM of TβM, Met471Cys, yielded a form of TβM that is catalytically active but susceptible to inactivation during turnover [Hess, C. R., Wu, Z., Ng, A., Gray, E. E., McGuirl, M. M., and Klinman, J. P. (2008) J. Am. Chem. Soc. 130, 11939–11944]. Further, although the wild-type (WT) enzyme undergoes coordination of Met471 to CuM in its reduced form, the generation of Met471Cys almost completely eliminates this interaction [Hess, C. R., Klinman, J. P., and Blackburn, N. J. (2010) J. Biol. Inorg. Chem. 15, 1195–1207]. The aim of this study is to identify the chemical consequence of the poor ability of Cys to coordinate CuM. We show that Met471Cys TβM is ∼5-fold more susceptible to inactivation than the WT enzyme in the presence of the cosubstrate/reductant ascorbate and that this process is not facilitated by the substrate tyramine. The resulting 50-fold smaller ratio for turnover to inactivation in the case of Met471Cys prevents full turnover of the substrate under all conditions examined. Liquid chromatography–tandem mass spectrometry analysis of proteolytic digests of inactivated Met471Cys TβM leads to the identification of cysteic acid at position 471. While both Met and Cys side chains are expected to be similarly subject to oxidative damage in proteins, the enhanced reactivity of Met471Cys toward solution oxidants in TβM is attributed to its weaker interaction with Cu(I)M.
Co-reporter:Jianyu Zhang
Journal of the American Chemical Society 2011 Volume 133(Issue 43) pp:17134-17137
Publication Date(Web):September 29, 2011
DOI:10.1021/ja207467d
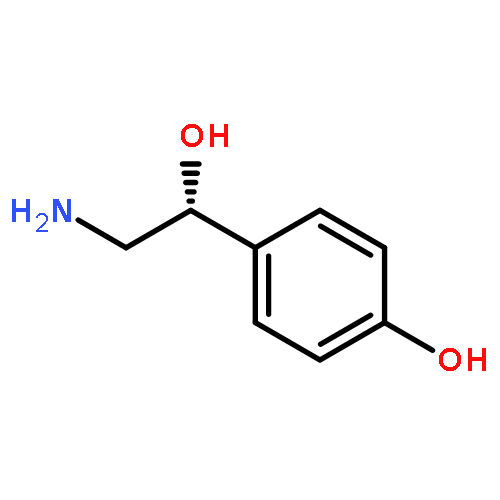
Human catechol-O-methyltransferase (COMT) catalyzes a methyl transfer from S-adenosylmethionine (AdoMet) to dopamine. Site-specific mutants at three positions (Tyr68, Trp38, and Val108) have been characterized with regard to product distribution, catalytic efficiency, and secondary kinetic isotope effects. The series of mutations at Tyr68 within wild-type protein and the common polymorphic variant (Val108Met) yields a linear correlation between the catalytic efficiency and the size of the secondary kinetic isotope effect. We conclude that active site compaction in COMT is modulated by a proximal side chain residing behind the sulfur-bearing methyl group of AdoMet. These findings are discussed in the context of the active site compression that has been postulated to accompany enzyme-supported hydrogen tunneling.
Co-reporter:Olayinka A. Oyeyemi, Kevin M. Sours, Thomas Lee, Amnon Kohen, Katheryn A. Resing, Natalie G. Ahn, and Judith P. Klinman
Biochemistry 2011 Volume 50(Issue 38) pp:
Publication Date(Web):August 22, 2011
DOI:10.1021/bi200640s
The technique of hydrogen–deuterium exchange coupled to mass spectrometry (HDX-MS) has been applied to a mesophilic (E. coli) dihydrofolate reductase under conditions that allow direct comparison to a thermophilic (B. stearothermophilus) ortholog, Ec-DHFR and Bs-DHFR, respectively. The analysis of hydrogen–deuterium exchange patterns within proteolytically derived peptides allows spatial resolution, while requiring a series of controls to compare orthologous proteins with only ca. 40% sequence identity. These controls include the determination of primary structure effects on intrinsic rate constants for HDX as well as the use of existing 3-dimensional structures to evaluate the distance of each backbone amide hydrogen to the protein surface. Only a single peptide from the Ec-DHFR is found to be substantially more flexible than the Bs-DHFR at 25 °C in a region located within the protein interior at the intersection of the cofactor and substrate-binding sites. The surrounding regions of the enzyme are either unchanged or more flexible in the thermophilic DHFR from B. stearothermophilus. The region with increased flexibility in Ec-DHFR corresponds to one of two regions previously proposed to control the enthalpic barrier for hydride transfer in Bs-DHFR [Oyeyemi et al. (2010) Proc. Natl. Acad. Sci. U.S.A.107, 10074].
Co-reporter:
Biochemistry 2011 Volume 50(Issue 9) pp:1556-1566
Publication Date(Web):December 14, 2010
DOI:10.1021/bi1015474
PQQ is an exogenous, tricyclic, quino-cofactor for a number of bacterial dehydrogenases. The final step of PQQ formation is catalyzed by PqqC, a cofactorless oxidase. This study focuses on the activation of molecular oxygen in an enzyme active site without metal or cofactor and has identified a specific oxygen binding and activating pocket in PqqC. The active site variants H154N, Y175F,S, and R179S were studied with the goal of defining the site of O2 binding and activation. Using apo-glucose dehydrogenase to assay for PQQ production, none of the mutants in this “O2 core” are capable of PQQ/PQQH2 formation. Spectrophotometric assays give insight into the incomplete reactions being catalyzed by these mutants. Active site variants Y175F, H154N, and R179S form a quinoid intermediate (Figure 1) anaerobically. Y175S is capable of proceeding further from quinoid to quinol, whereas Y175F, H154N, and R179S require O2 to produce the quinol species. None of the mutations precludes substrate/product binding or oxygen binding. Assays for the oxidation of PQQH2 to PQQ show that these O2 core mutants are incapable of catalyzing a rate increase over the reaction in buffer, whereas H154N can catalyze the oxidation of PQQH2 to PQQ in the presence of H2O2 as an electron acceptor. Taken together, these data indicate that none of the targeted mutants can react fully to form quinone even in the presence of bound O2. The data indicate a successful separation of oxidative chemistry from O2 binding. The residues H154, Y175, and R179 are proposed to form a core O2 binding structure that is essential for efficient O2 activation.
Co-reporter:Brian J. Bahnson;Zachary D. Nagel;Ming Dong
PNAS 2011 Volume 108 (Issue 26 ) pp:10520-10525
Publication Date(Web):2011-06-28
DOI:10.1073/pnas.1104989108
A growing body of data supports a role for protein motion in enzyme catalysis. In particular, the ability of enzymes to sample
catalytically relevant conformational substates has been invoked to model kinetic and spectroscopic data. However, direct
experimental links between rapidly interconverting conformations and the chemical steps of catalysis remain rare. We report
here on the kinetic analysis and characterization of the hydride transfer step catalyzed by a series of mutant thermophilic
alcohol dehydrogenases (ht-ADH), presenting evidence for Arrhenius prefactor values that become enormously elevated above
an expected value of approximately 1013 s-1 when the enzyme operates below its optimal temperature range. Restoration of normal Arrhenius behavior in the ht-ADH reaction
occurs at elevated temperatures. A simple model, in which reduced temperature alters the ability of the ht-ADH variants to
sample the catalytically relevant region of conformational space, can reproduce the available data. These findings indicate
an impaired landscape that has been generated by the combined condition of reduced temperature and mutation at a single, active-site
hydrophobic side chain. The broader implication is that optimal enzyme function requires the maintenance of a relatively smooth
landscape that minimizes low energy traps.
Co-reporter:Zachary D. Nagel and Judith P. Klinman
Chemical Reviews 2010 Volume 110(Issue 12) pp:PR41-PR67
Publication Date(Web):December 8, 2010
DOI:10.1021/cr1001035
Co-reporter:Kevin P. McCusker
Journal of the American Chemical Society 2010 Volume 132(Issue 14) pp:5114-5120
Publication Date(Web):March 19, 2010
DOI:10.1021/ja909416z
Enzymes that cleave C−H bonds are often found to depend on well-packed hydrophobic cores that influence the distance between the hydrogen donor and acceptor. Residue F159 in taurine α-ketoglutarate dioxygenase (TauD) is demonstrated to play an important role in the binding and orientation of its substrate, which undergoes a hydrogen atom transfer to the active site Fe(IV)═O. Mutation of F159 to smaller hydrophobic side chains (L, V, A) leads to substantially reduced rates for substrate binding and for C−H bond cleavage, as well as increased contribution of the chemical step to kcat under steady-state turnover conditions. The greater sensitivity of these substrate-dependent processes to mutation at position 159 than observed for the oxygen activation process supports a previous conclusion of modularity of function within the active site of TauD (McCusker, K. P.; Klinman, J. P. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 19791−19795). Extraction of intrinsic deuterium kinetic isotope effects (KIEs) using single turnover transients shows 2- to 4-fold increase in the size of the KIE for F159V in relation to wild-type and F159L. It appears that there is a break in behavior following removal of a single methylene from the side chain of F159L to generate F159V, whereby the protein active site loses its ability to restore the internuclear distance between substrate and Fe(IV)═O that supports optimal hydrogenic wave function overlap.
Co-reporter:Matthew P. Meyer
Journal of the American Chemical Society 2010 Volume 133(Issue 3) pp:430-439
Publication Date(Web):December 30, 2010
DOI:10.1021/ja1050742
This work describes the application of NMR to the measurement of secondary deuterium (2° 2H) and carbon-13 (13C) kinetic isotope effects (KIEs) at positions 9−13 within the substrate linoleic acid (LA) of soybean lipoxygenase-1. The KIEs have been measured using LA labeled with either protium (11,11-h2-LA) or deuterium (11,11-d2-LA) at the reactive C11 position, which has been previously shown to yield a primary deuterium isotope effect of ca. 80. The conditions of measurement yield the intrinsic 2° 2H and 13C KIEs on kcat/Km directly for 11,11-d2-LA, whereas the values for the 2° 2H KIEs for 11,11-h2-LA are obtained after correction for a kinetic commitment. The pattern of the resulting 2° 2H and 13C isotope effects reveals values that lie far above those predicted from changes in local force constants. Additionally, many of the experimental values cannot be modeled by electronic effects, torsional strain, or the simple inclusion of a tunneling correction to the rate. Although previous studies have shown the importance of extensive tunneling for cleavage of the primary hydrogen at C11 of LA, the present findings can only be interpreted by extending the conclusion of nonclassical behavior to the secondary hydrogens and carbons that flank the position undergoing C−H bond cleavage. A quantum mechanical method introduced by Buhks et al. [J. Phys. Chem. 1981, 85, 3763] to model the inner-sphere reorganization that accompanies electron transfer has been shown to be able to reproduce the scale of the 2° 2H KIEs.
Co-reporter:Stephen R. Wecksler, Stefan Stoll, Anthony T. Iavarone, Erin M. Imsand, Ha Tran, R. David Britt and Judith P. Klinman
Chemical Communications 2010 vol. 46(Issue 37) pp:7031-7033
Publication Date(Web):25 Aug 2010
DOI:10.1039/C0CC00968G
pqqD is one of six genes required for PQQ production in Klebsiella pneumoniae. Herein, we demonstrate that PqqD interacts specifically with the radical SAM enzyme PqqE, causing a perturbation in the electronic environment around the [4Fe–4S]+ clusters. This interaction redirects the role for PqqD in PQQ biosynthesis.
Co-reporter:Cindy M. Chang, Valerie J. Klema, Bryan J. Johnson, Minae Mure, Judith P. Klinman and Carrie M. Wilmot
Biochemistry 2010 Volume 49(Issue 11) pp:
Publication Date(Web):February 15, 2010
DOI:10.1021/bi901933d
The structural underpinnings of enzyme substrate specificity are investigated in a pair of copper amine oxidases (CAOs) from Hansenula polymorpha (HPAO-1 and HPAO-2). The X-ray crystal structure (to 2.0 Å resolution) and steady state kinetic data of the second copper amine oxidase (HPAO-2) are presented for comparison to those of HPAO-1. Despite 34% sequence identity and superimposable active site residues implicated in catalysis, the enzymes vary considerably in their substrate entry channel. The previously studied CAO, HPAO-1, has a narrow substrate channel. In contrast, HPAO-2 has a wide funnel-shaped substrate channel, which also contains a side chamber. In addition, there are a number of amino acid changes within the channels of HPAO-2 and HPAO-1 that may sterically impact the ability of substrates to form covalent Schiff base catalytic intermediates and to initiate chemistry. These differences can partially explain the greatly different substrate specificities as characterized by kcat/Km value differences. In HPAO-1, the kcat/Km for methylamine is 330-fold greater than for benzylamine, whereas in HPAO-2, it is benzylamine that is the better substrate by 750-fold. In HPAO-2, an inflated Dkcat/Km(methylamine) in relation to Dkcat/Km(benzylamine) indicates that proton abstraction has been impeded more than substrate release. In HPAO-1, Dkcat/Km(S) changes little with the slow substrate and indicates a similar increase in the energy barriers that control both substrate binding and subsequent catalysis. In neither case is kcat/Km for the second substrate, O2, significantly altered. These results reinforce the modular nature of the active sites of CAOs and show that multiple factors contribute to substrate specificity and catalytic efficiency. In HPAO-1, the enzyme with the smaller substrate binding pocket, both initial substrate binding and proton loss are affected by an increase in substrate size, while in HPAO-2, the enzyme with the larger substrate binding pocket, the rate of proton loss is differentially affected when a phenyl substituent in the substrate is reduced to the size of a methyl group.
Co-reporter:Zhi-wei Chen, Saumen Datta, Jennifer L. DuBois, Judith P. Klinman and F. Scott Mathews
Biochemistry 2010 Volume 49(Issue 34) pp:
Publication Date(Web):July 13, 2010
DOI:10.1021/bi100643y
The copper amine oxidases carry out two copper-dependent processes: production of their own redox-active cofactor (2,4,5-trihydroxyphenylalanine quinone, TPQ) and the subsequent oxidative deamination of substrate amines. Because the same active site pocket must facilitate both reactions, individual active site residues may serve multiple roles. We have examined the roles of a strictly conserved active site tyrosine Y305 in the copper amine oxidase from Hansenula polymorpha kinetically, spetroscopically (Dubois and Klinman (2006) Biochemistry 45, 3178), and, in the present work, structurally. While the Y305A enzyme is almost identical to the wild type, a novel, highly oxygenated species replaces TPQ in the Y305F active sites. This new structure not only provides the first direct detection of peroxy intermediates in cofactor biogenesis but also indicates the critical control of oxidation chemistry that can be conferred by a single active site residue.
Co-reporter:Judith P. Klinman
Journal of Physical Organic Chemistry 2010 Volume 23( Issue 7) pp:606-612
Publication Date(Web):
DOI:10.1002/poc.1661
Abstract
Measurements of kinetic hydrogen isotope effects in enzymatic CH bond cleavage reactions have generated a body of data that is inconsistent with either a semi-classical formalism for the origin of the isotope effects or a semi-classical formalism together with a modest degree of hydrogen tunneling. A model that treats the movement of hydrogen as a wave, and whose behavior is dictated by the heavy atom environment, is presented and discussed. A list of unanswered questions and challenges is given at the end of this paper. Copyright © 2010 John Wiley & Sons, Ltd.
Co-reporter:Olayinka A. Oyeyemi;Kevin M. Sours;Thomas Lee;Katheryn A. Resing;Natalie G. Ahn;
Proceedings of the National Academy of Sciences 2010 107(22) pp:10074-10079
Publication Date(Web):May 13, 2010
DOI:10.1073/pnas.1003678107
We report hydrogen deuterium exchange by mass spectrometry (HDX-MS) as a function of temperature in a thermophilic dihydrofolate
reductase from Bacillus stearothermophilus (Bs-DHFR). Protein stability, probed with circular dichroism, established an accessible temperature range of 10 °C to 55 °C
for the interrogation of HDX-MS. Although both the rate and extent of HDX are sensitive to temperature, the majority of peptides
showed rapid kinetics of exchange, allowing us to focus on plateau values for the maximal extent of exchange at each temperature.
Arrhenius plots of the ratio of hydrogens exchanged at 5 h normalized to the number of exchangeable hydrogens vs. 1/T provides
an estimate for the apparent enthalpic change of local unfolding, ΔH°unf(avg). Most regions in the enzyme show ΔH°unf(avg) ≤ 2.0 kcal/mol, close to the value of kT; by contrast, significantly elevated values for ΔH°unf(avg) are observed in regions within the core of protein that contain the cofactor and substrate-binding sites. Our technique introduces
a new strategy for probing the temperature dependence of local protein unfolding within native proteins. These findings are
discussed in the context of the demonstrated role for nuclear tunneling in hydride transfer from NADPH to dihydrofolate, and
relate the observed enthalpic changes to two classes of motion, preorganization and reorganization, that have been proposed
to control the efficiency of hydrogenic wave function overlap. Our findings suggest that the enthalpic contribution to the
heavy atom environmental reorganizations controlling the hydrogenic wave function overlap will be dominated by regions of
the protein proximal to the bound cofactor and substrate.
Co-reporter:Stephen R. Wecksler, Stefan Stoll, Ha Tran, Olafur T. Magnusson, Shu-pao Wu, David King, R. David Britt and Judith P. Klinman
Biochemistry 2009 Volume 48(Issue 42) pp:
Publication Date(Web):September 11, 2009
DOI:10.1021/bi900918b
Biogenesis of pyrroloquinoline quinone (PQQ) in Klebsiella pneumoniae requires the expression of six genes (pqqA−F). One of these genes (pqqE) encodes a 43 kDa protein (PqqE) that plays a role in the initial steps in PQQ formation [Veletrop, J. S., et al. (1995) J. Bacteriol. 177, 5088−5098]. PqqE contains two highly conserved cysteine motifs at the N- and C-termini, with the N-terminal motif comprised of a CX3CX2C consensus sequence that is unique to a family of proteins known as radical S-adenosyl-l-methionine (SAM) enzymes [Sofia, H. J., et al. (2001) Nucleic Acids Res. 29, 1097−1106]. PqqE from K. pneumoniae was cloned into Escherichia coli and expressed as the native protein and with an N-terminal His6 tag. Anaerobic expression and purification of the His6-tagged PqqE results in an enzyme with a brownish-red hue indicative of Fe−S cluster formation. Spectroscopic and physical analyses indicate that PqqE contains a mixture of Fe−S clusters, with the predominant form of the enzyme containing two [4Fe-4S] clusters. PqqE isolated anaerobically yields an active enzyme capable of cleaving SAM to methionine and 5′-deoxyadenosine in an uncoupled reaction (kobs = 0.011 ± 0.001 min−1). In this reaction, the 5′-deoxyadenosyl radical either abstracts a hydrogen atom from a solvent accessible position in the enzyme or obtains a proton and electron from buffer. The putative PQQ substrate PqqA has not yet been shown to be modified by PqqE, implying that PqqA must be modified before becoming the substrate for PqqE and/or that another protein in the biosynthetic pathway is critical for the initial steps in PQQ biogenesis.
Co-reporter:Judith P. Klinman
Chemical Physics Letters 2009 Volume 471(4–6) pp:179-193
Publication Date(Web):26 March 2009
DOI:10.1016/j.cplett.2009.01.038
Abstract
The origins of the enormous rate accelerations brought about by enzymes are discussed. The focus is on enzymatic C–H activation, which has been shown to take place via tunneling. Four enzyme systems illustrate the impact of site-specific mutagenesis, changes in temperature or changes in protein solvation on the tunneling properties. A model emerges in which conformational sampling is required to access a subset of protein conformers where the H-donor and acceptor undergo a close approach. The evidence for an inverse relationship between protein flexibility and active site compression is likely to extend to all classes of enzyme catalysts.
Co-reporter:Kevin P. McCusker
PNAS 2009 Volume 106 (Issue 47 ) pp:19791-19795
Publication Date(Web):2009-11-24
DOI:10.1073/pnas.0910660106
Taurine α-ketoglutarate dioxygenase (tauD) is one of the best-studied α-ketoglutarate (αKG)-dependent nonheme iron oxygenases.
As with all oxygenases, a fine balance must be struck between generating a species sufficiently reactive for the required
chemistry and controlling that species to prevent undesirable side reactions [Klinman JP (2007) Accts Chem Res 40:325–333]. In the case of tauD, the substrate oxidizing species has been shown to be a ferryl-oxo, and the introduction
of deuterium at the reactive position of substrate results in an enormous kinetic isotope effect together with a partial uncoupling
of oxygen activation from substrate oxidation [Price JC, Barr EW, Glass TE, Krebs C, Bollinger JM (2003) J Am Chem Soc 125:13008–13009]. We have generated a series of site-specific variants at a position that resides directly behind bound substrate
(F159 to L, V, A, and G). Decreasing side-chain bulk diminishes the coupling of oxygen activation to C-H cleavage, which is
further reduced by substrate deuteration. Despite this impact, oxygen activation remains completely coupled to the oxidative
decarboxylation of αKG. The concentration of bis-Tris buffer impacts the extent of coupling of oxygen activation to C-H cleavage,
implicating the buffer in the uncoupling pathway. These data indicate a critical role for residue 159 in substrate positioning
and reaction in tauD and show that minor active-site perturbations in these enzymes could allow for changes in substrate reactivity
while maintaining substrate triggering and oxygen binding/activation.
Co-reporter:Kevin P. McCusker, Judith P. Klinman
Tetrahedron Letters 2009 50(6) pp: 611-613
Publication Date(Web):
DOI:10.1016/j.tetlet.2008.11.063
Co-reporter:Liviu M. Mirica
PNAS 2008 Volume 105 (Issue 6 ) pp:1814-1819
Publication Date(Web):2008-02-12
DOI:10.1073/pnas.0711626105
Ethylene is a plant hormone important in many aspects of plant growth and development such as germination, fruit ripening,
and senescence. 1-Aminocyclopropane-1-carboxylic acid (ACC) oxidase (ACCO), an O2-activating ascorbate-dependent nonheme iron enzyme, catalyzes the last step in ethylene biosynthesis. The O2 activation process by ACCO was investigated using steady-state kinetics, solvent isotope effects (SIEs), and competitive
oxygen kinetic isotope effects (18O KIEs) to provide insights into the nature of the activated oxygen species formed at the active-site iron center and its
dependence on ascorbic acid. The observed large 18O KIE of 1.0215 ± 0.0005 strongly supports a rate-determining step formation of an FeIV
O species, which acts as the reactive intermediate in substrate oxidation. The large SIE on k
cat/K
m(O2) of 5.0 ± 0.9 suggests that formation of this FeIV
O species is linked to a rate-limiting proton or hydrogen atom transfer step. Based on the observed decrease in SIE and 18O KIE values for ACCO at limiting ascorbate concentrations, ascorbate is proposed to bind in a random manner, depending on
its concentration. We conclude that ascorbate is not essential for initial O2 binding and activation but is required for rapid FeIV
O formation under catalytic turnover. Similar studies can be performed for other nonheme iron enzymes, with the 18O KIEs providing a kinetic probe into the chemical nature of Fe/O2 intermediates formed in the first irreversible step of the O2 activation.
Co-reporter:Matthew P. Meyer;Diana R. Tomchick
PNAS 2008 Volume 105 (Issue 4 ) pp:1146-1151
Publication Date(Web):2008-01-29
DOI:10.1073/pnas.0710643105
This study examines the impact of a series of mutations at position 553 on the kinetic and structural properties of soybean
lipoxygenase-1 (SLO-1). The previously uncharacterized mutants reported herein are I553L, I553V, and I553G. High-resolution
x-ray studies of these mutants, together with the earlier studied I553A, show almost no structural change in relation to the
WT-enzyme. By contrast, a progression in kinetic behavior occurs in which the decrease in the size of the side chain at position
553 leads to an increased importance of donor–acceptor distance sampling in the course of the hydrogen transfer process. These
dynamical changes in behavior are interpreted in the context of two general classes of protein motions, preorganization and
reorganization, with the latter including the distance sampling modes [Klinman JP (2006) Philos Trans R Soc London Ser B 361:1323–1331; Nagel Z, Klinman JP (2006) Chem Rev 106:3095–3118]. The aggregate data for SLO-1 show how judicious placement of hydrophobic side chains can influence enzyme
catalysis via enhanced donor–acceptor hydrogenic wave function overlap.
Co-reporter:Judith P. Klinman
Accounts of Chemical Research 2007 Volume 40(Issue 5) pp:325
Publication Date(Web):May 3, 2007
DOI:10.1021/ar6000507
Detailed analyses of the oxidative half-reactions of glucose oxidase and soybean lipoxygenase provide insight into Nature’s solution to the “trouble with oxygen”. Coupled with studies of other O2-activating enzymes, two key features emerge. The first is the predominance of a rate-limiting transfer of the first electron transfer to O2, with subsequent electron and proton transfers occurring in rapid steps. The second feature is the identification of non-metal binding sites and channels for O2. These permit a controlled reactivity of oxygen to generate the desired regio- and stereochemical products, while minimizing deleterious side reactions.
Co-reporter:Stephen R. Wecksler, Stefan Stoll, Anthony T. Iavarone, Erin M. Imsand, Ha Tran, R. David Britt and Judith P. Klinman
Chemical Communications 2010 - vol. 46(Issue 37) pp:NaN7033-7033
Publication Date(Web):2010/08/25
DOI:10.1039/C0CC00968G
pqqD is one of six genes required for PQQ production in Klebsiella pneumoniae. Herein, we demonstrate that PqqD interacts specifically with the radical SAM enzyme PqqE, causing a perturbation in the electronic environment around the [4Fe–4S]+ clusters. This interaction redirects the role for PqqD in PQQ biosynthesis.
.jpg)
![1-Nonanol, 9-[(tetrahydro-2H-pyran-2-yl)oxy]-](http://img.cochemist.com/ccimg/98900/98847-00-8.png)
![1-Nonanol, 9-[(tetrahydro-2H-pyran-2-yl)oxy]-](http://img.cochemist.com/ccimg/98900/98847-00-8_b.png)




![[13C,2H]paraformaldehyde](http://img.cochemist.com/ccimg/63200/63101-50-8.png)
![[13C,2H]paraformaldehyde](http://img.cochemist.com/ccimg/63200/63101-50-8_b.png)







![5-HYDROXY-4-OXO-1,6-DIHYDROPYRROLO[2,3-F]QUINOLINE-2,7,9-TRICARBOXYLIC ACID](http://img.cochemist.com/ccimg/79200/79127-57-4.png)
![5-HYDROXY-4-OXO-1,6-DIHYDROPYRROLO[2,3-F]QUINOLINE-2,7,9-TRICARBOXYLIC ACID](http://img.cochemist.com/ccimg/79200/79127-57-4_b.png)











![L-Glutamic acid,N-[4-[[(2-amino-3,4,7,8-tetrahydro-4-oxo-6-pteridinyl)methyl]amino]benzoyl]-](http://img.cochemist.com/ccimg/4100/4033-27-6.png)
![L-Glutamic acid,N-[4-[[(2-amino-3,4,7,8-tetrahydro-4-oxo-6-pteridinyl)methyl]amino]benzoyl]-](http://img.cochemist.com/ccimg/4100/4033-27-6_b.png)




![[2,4'-Bi-1H-indole]-3,3'-dipropanoicacid, a3,a3'-diamino-6',7'-dihydro-6',7'-dioxo-, (a3S,a3'S)-](http://img.cochemist.com/ccimg/134700/134645-25-3.png)
![[2,4'-Bi-1H-indole]-3,3'-dipropanoicacid, a3,a3'-diamino-6',7'-dihydro-6',7'-dioxo-, (a3S,a3'S)-](http://img.cochemist.com/ccimg/134700/134645-25-3_b.png)

![4,5-Dioxo-4,5-dihydro-1H-pyrrolo[2,3-f]quinoline-2,7,9-tricarboxylic acid](/data/chemimg/654800/72909-34-3.png)
![4,5-Dioxo-4,5-dihydro-1H-pyrrolo[2,3-f]quinoline-2,7,9-tricarboxylic acid](/data/chemimg/654800/72909-34-3_b.png)