Co-reporter:Yi-pu Zhao;Wei-liang Ye;Dao-zhou Liu;Han Cui;Ying Cheng;Miao Liu;Bang-le Zhang;Qi-bing Mei
Nanoscale (2009-Present) 2017 vol. 9(Issue 19) pp:6264-6277
Publication Date(Web):2017/05/18
DOI:10.1039/C7NR00962C
Bone is an especially prone metastatic site for breast cancer, and to block the vicious cycle between bone resorption and tumor growth is an important strategy for the treatment of breast cancer bone metastasis. In this paper, pH- and redox-sensitive as well as breast cancer bone metastasis-targeting nanoparticles (DOX@ALN-(HA-PASP)CL) were prepared, and also their anti-tumor activity and anti-bone resorption effect were investigated in detail. The in vitro experimental results indicated that DOX released from DOX@ALN-(HA-PASP)CL exhibited a GSH-, DTT- and pH-dependent manner. Moreover, in an in vitro 3D breast cancer bone metastasis model, DOX@ALN-(HA-PASP)CL decreased bone resorption through inhibiting the proliferation of human breast cancer cells (MDA-MB-231 cells) and reducing the activity of osteoclasts. The in vivo experimental results indicated that a large amount of DOX was delivered to a breast cancer bone metastasis site after tumor-bearing mice were treated with DOX@ALN-(HA-PASP)CL; meanwhile, DOX@ALN-(HA-PASP)CL significantly decreased the tumor volume and bone resorption in tumor-bearing mice without causing obvious systemic toxicity. In conclusion, the in vitro and in vivo experimental results indicate that DOX@ALN-(HA-PASP)CL has great potential in the treatment of breast cancer bone metastasis.
Co-reporter:Han Cui, Meng-lei Huan, Wei-liang Ye, Dao-zhou Liu, Zeng-hui Teng, Qi-Bing Mei, and Si-yuan Zhou
Molecular Pharmaceutics 2017 Volume 14(Issue 3) pp:
Publication Date(Web):February 1, 2017
DOI:10.1021/acs.molpharmaceut.6b01016
Doxorubicin (DOX) is a broad-spectrum chemotherapy drug to treat tumors. However, severe side effects and development of DOX resistance hinder its clinical application. In order to overcome DOX resistance, DOX/TPP–DOX@Pasp-hyd-PEG-FA micelles were prepared by using newly synthesized comb-like amphiphilic material Pasp-hyd-PEG-FA. Drug released in vitro from micelles showed a pH-dependent manner. DOX/TPP–DOX@Pasp-hyd-PEG-FA induced more apoptosis in KB cell and MCF-7/ADR cell than DOX@Pasp-hyd-PEG-FA. Confocal laser scanning microscopy experiment indicated that DOX/TPP–DOX@Pasp-hyd-PEG-FA delivered TPP–DOX and DOX to the nucleus and mitochondria of the tumor cell simultaneously. Thus, DOX/TPP–DOX@Pasp-hyd-PEG-FA could significantly damage the mitochondrial membrane potential. DOX/TPP–DOX@Pasp-hyd-PEG-FA markedly shrinked the tumor volume in tumor-bearing nude mice grafted with MCF-7/ADR cell as compared with the same dose of free DOX. DOX was mainly accumulated in tumor tissue after DOX/TPP–DOX@Pasp-hyd-PEG-FA was injected to tumor-bearing nude mice by tail vein. After free DOX was injected to tumor-bearing nude mice by tail vein, DOX widely distributed through the whole body. Therefore, mitochondria and nucleus dual delivery system has potential in overcoming DOX resistance.Keywords: (3-carboxypropyl)triphenylphosphonium; doxorubicin; mitochondria; pH-sensitive micelle;
Co-reporter:Jiang-bo Du; Ying Cheng; Zeng-hui Teng; Meng-lei Huan; Miao Liu; Han Cui; Bang-le Zhang
Molecular Pharmaceutics 2016 Volume 13(Issue 5) pp:1711-1722
Publication Date(Web):March 21, 2016
DOI:10.1021/acs.molpharmaceut.6b00158
PLGA nanoparticles are widely used in tumor targeting drug delivery systems. However, the naked PLGA nanoparticles (NNPs) not only have low drug loading but also can be rapidly removed from blood circulation by the immune system. The aim of this study was to prepare pH-triggered surface charge reversed lipid hybrid PLGA nanoparticles (LNPs) to enhance drug loading and drug delivery efficiency. CHO-Arg-His-OMe and FA-PEG-DSPE were synthesized to modify PLGA nanoparticles to prepare LNPs. The drug loading and encapsulation rate of LNPs were greatly improved as compared with NNPs. In pH 7.4 medium, doxorubicin (DOX)-loaded LNPs showed negative charge and released DOX slowly. In pH 5.0 medium, DOX-loaded LNPs exhibited positive charge and released DOX quickly. DOX-loaded LNPs delivered more DOX to the nucleus of KB cells and MBA-MD-231/ADR cells than did free DOX. In addition, DOX-loaded LNPs significantly inhibited the proliferation of KB cells and MBA-MD-231/ADR cells. Compared with free DOX, the same dose of the DOX-loaded LNPs delivered more DOX to tumor tissue. Thus, DOX-loaded LNPs significantly inhibited the growth of tumor in tumor-bearing nude mice and obviously reduced the systemic toxicity of DOX. In conclusion, pH-triggered surface charge reversed DOX-loaded LNPs significantly enhanced the antitumor activity of DOX in vitro and in vivo. DOX-loaded LNPs had great potential in tumor targeted chemotherapy.
Co-reporter:Wei-Liang Ye;Yi-Pu Zhao;Ren Na;Fei Li;Qi-Bing Mei;Ming-Gao Zhao
Journal of Pharmaceutical Sciences 2015 Volume 104( Issue 7) pp:2293-2303
Publication Date(Web):
DOI:10.1002/jps.24476
Alendronate-monoethyl adipate-(hydrazone)-doxorubicin conjugate (ALN-MA-hyd-DOX) was synthesized to specifically deliver doxorubicin (DOX) to bone tumor tissue. The binding kinetics of ALN-MA-hyd-DOX with hydroxyapatite (HA) and natural bone were detected by using spectrophotometer. Cytotoxicity of ALN-MA-hyd-DOX on tumor cells was determined by MTT [3-(4,5-dimethylthiaol-2-yl)-2,5-diphenyl-tetrazolium bromide] method. The cellular uptake of ALN-MA-hyd-DOX was observed by using fluorescence microscopy. The in vivo antitumor activity of ALN-MA-hyd-DOX was investigated by using tumor-bearing nude mice model. The results indicated that ALN-MA-hyd-DOX was able to quickly bind with HA and natural bone. ALN-MA-hyd-DOX immobilized on the natural bone released more DOX in pH 5.0 medium than that in pH 6.0 or 7.4 medium. The cytotoxicity of ALN-MA-hyd-DOX toward A549 cells and MDA-MB-231/ADR cells was greater than DOX. ALN-MA-hyd-DOX was rapidly uptaken by A549 cells and MDA-MB-231/ADR cells. Compared with the same dose of free DOX, ALN-MA-hyd-DOX significantly decreased tumor volume of tumor-bearing nude mice. DOX mainly distributed in bone tumor tissue after ALN-MA-hyd-DOX was intravenously administered to tumor-bearing nude mice, whereas DOX distributed through the whole body after DOX was intravenously administered to tumor-bearing nude mice. These findings implied that the ALN-MA-hyd-DOX was a promising bone-targeted conjugate for treating bone neoplasms. © 2015 Wiley Periodicals, Inc. and the American Pharmacists Association J Pharm Sci 104:2293–2303, 2015
Co-reporter:Chun-hui Liang, Wei-liang Ye, Chun-lai Zhu, Ren Na, Ying Cheng, Han Cui, Dao-zhou Liu, Zhi-fu Yang, and Si-yuan Zhou
Molecular Pharmaceutics 2014 Volume 11(Issue 5) pp:1378-1390
Publication Date(Web):March 31, 2014
DOI:10.1021/mp4004139
Doxorubicin (DOX) is a broad-spectrum antitumor drug used in the clinic. However, it can cause serious heart toxicity. To increase the therapeutic index of DOX and to attenuate its toxicity toward normal tissues, we conjugated DOX with either α-linolenic acid (LNA) or palmitic acid (PA) by a hydrazone or an amide bond to produce DOX-hyd-LNA, DOX-ami-LNA, DOX-hyd-PA, and DOX-ami-PA. The cytotoxicity of DOX-hyd-LNA on HepG2, MCF-7, and MDA-231 cells was higher compared to that of DOX, DOX-ami-LNA, DOX-hyd-PA, and DOX-ami-PA. The cytotoxicity of DOX-hyd-LNA on HUVECs was lower than that of DOX. DOX-hyd-LNA released significantly more DOX in pH 5.0 medium than it did in pH 7.4 medium. DOX-hyd-LNA induced more apoptosis in MCF-7 and HepG2 cells than DOX or DOX-ami-LNA. Significantly more DOX was released from DOX-hyd-LNA in both MCF-7 and HepG2 cells compared with DOX-ami-LNA. Compared to free DOX, a biodistribution study showed that DOX-hyd-LNA greatly increased the content of DOX in tumor tissue and decreased the content of DOX in heart tissue after it was intravenously administered. DOX-hyd-LNA improved the survival rate, prolonged the life span, and slowed the growth of the tumor in tumor-bearing nude mice. These results indicate that DOX-hyd-LNA improved the therapeutic index of DOX. Therefore, DOX-hyd-LNA is a potential compound for use as a cancer-targeting therapy.Keywords: doxorubicin; palmitic acid; pH sensitive; tumor targeting; α-linolenic acid;
Co-reporter:Wei-Liang Ye;Zeng-Hui Teng;Dao-Zhou Liu;Han Cui;Miao Liu;Ying Cheng;Tie-Hong Yang;Qi-Bing Mei
Journal of Pharmaceutical Sciences 2013 Volume 102( Issue 2) pp:530-540
Publication Date(Web):
DOI:10.1002/jps.23381
Abstract
Folate–aminocaproic acid–doxorubicin (FA–AMA–DOX) was synthesized and characterized by H NMR spectroscopy and mass spectrometry. Cytotoxicity and cellular uptake experiments were performed in KB and HepG2 cells, which express folic acid receptor, and the cell line A549, which does not express folic acid receptor. Cytotoxicity was determined using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay, and cellular uptake was monitored using fluorescence microscopy. The amount of DOX released from FA–AMA–DOX was much greater at pH 5.0 than that at pH 6.5 or 7.4. The cytotoxicity of FA–AMA–DOX toward KB and HepG2 cells was greater than that of DOX or AMA–DOX at the same concentrations, and cytotoxicity could be attenuated by FA in a dose-dependent manner. On the contrary, the cytotoxicity of FA–AMA–DOX and AMA–DOX toward A549 cells was lower than that of DOX at the same concentration, and cytotoxicity could not be reduced by FA. Compared with FA–AMA, FA–AMA–DOX increased the intracellular accumulation of DOX in KB cells. These results suggested that FA–AMA–DOX have suitable attributes for the active targeting of folate-receptor-positive tumor cells and for releasing the chemotherapeutic agent, DOX, in situ; it therefore has potential as a novel cancer therapeutic. © 2012 Wiley Periodicals, Inc. and the American Pharmacists Association J Pharm Sci 102:530–540, 2013
Co-reporter:Bang-le Zhang, Wei He, Xin Shi, Meng-lei Huan, Qiu-ju Huang, Si-yuan Zhou
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 3) pp:805-808
Publication Date(Web):1 February 2010
DOI:10.1016/j.bmcl.2009.12.104
An efficient total synthesis of (R) and (S)-3-methyl 5-pentyl 2,6-dimethyl-4-(3-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate in high optical purities is reported. The useful step is the resolution of racemic 2, 6-dimethyl-5-methoxycarbonyl-4-(3-nitrophenyl)-1,4-dihydropyridine-3-carboxylic acid by using commercially available Cinchona alkaloids cinchonidine and quinidine as the resolving agents. Under the optimum conditions, the optical purities for R- and S-enantiomers are extremely high (ee >99.5%). The further dihydropyridine receptor binding activity assay shows that the S-enantiomer is more potent than R-enantiomer both in rat cardiac (approximately 19 times) and cerebral cortex membrane (12 times).The synthesis of the calcium modulator (R) and (S)-1 in extremely high optical purities (ee >99.5%) is reported. (S)-1 was defined as active isomer which is more potent than (R)-1 both in rat cardiac and cerebral cortex membrane.
Co-reporter:Wei-Dong Zhang, Xiao-Juan Wang, Si-Yuan Zhou, Yi Gu, Rong Wang, Tao-Li Zhang, Hong-Quan Gan
Journal of Chromatography B 2010 Volume 878(Issue 23) pp:2137-2140
Publication Date(Web):1 August 2010
DOI:10.1016/j.jchromb.2010.06.002
Flavanoid kaempferol is mainly present as glucuronides and sulfates in rat plasma, and small amounts of the intact aglycone are also detected. In the this study, a rapid, specific and sensitive liquid chromatography–electrospray ionization-tandem mass spectrometry method (HPLC–MS/MS) was developed and validated for determination of kaempferol and its major metabolite glucuronidated kaempferol in rat plasma. A liquid–liquid extraction with acetic ether was involved for the extraction of kaempferol and internal standard. Analytes were separated on a C18 column (150 mm × 2.1 mm, 4.5 μm, Waters Corp.) with isocratic elution at a flow-rate of 0.3 ml min−1. The mobile phase was consisted of 0.5% formic acid and acetonitrile (50:50, v/v). The Quattro Premier HPLC–MS/MS was operated under the multiple reaction-monitoring mode (MRM) using the electrospray ionization technique. The method was validated according to the FDA guidelines for validation of bioanalytical method. The validated method was successfully applied to the study of the pharmacokinetics in rats after oral administration of kaempferol with different doses.
Co-reporter:Wei-Liang Ye, Yi-Pu Zhao, Ren Na, Fei Li, ... Si-Yuan Zhou
Journal of Pharmaceutical Sciences (July 2015) Volume 104(Issue 7) pp:2293-2303
Publication Date(Web):1 July 2015
DOI:10.1002/jps.24476
Alendronate-monoethyl adipate-(hydrazone)-doxorubicin conjugate (ALN-MA-hyd-DOX) was synthesized to specifically deliver doxorubicin (DOX) to bone tumor tissue. The binding kinetics of ALN-MA-hyd-DOX with hydroxyapatite (HA) and natural bone were detected by using spectrophotometer. Cytotoxicity of ALN-MA-hyd-DOX on tumor cells was determined by MTT [3-(4,5-dimethylthiaol-2-yl)-2,5-diphenyl-tetrazolium bromide] method. The cellular uptake of ALN-MA-hyd-DOX was observed by using fluorescence microscopy. The in vivo antitumor activity of ALN-MA-hyd-DOX was investigated by using tumor-bearing nude mice model. The results indicated that ALN-MA-hyd-DOX was able to quickly bind with HA and natural bone. ALN-MA-hyd-DOX immobilized on the natural bone released more DOX in pH 5.0 medium than that in pH 6.0 or 7.4 medium. The cytotoxicity of ALN-MA-hyd-DOX toward A549 cells and MDA-MB-231/ADR cells was greater than DOX. ALN-MA-hyd-DOX was rapidly uptaken by A549 cells and MDA-MB-231/ADR cells. Compared with the same dose of free DOX, ALN-MA-hyd-DOX significantly decreased tumor volume of tumor-bearing nude mice. DOX mainly distributed in bone tumor tissue after ALN-MA-hyd-DOX was intravenously administered to tumor-bearing nude mice, whereas DOX distributed through the whole body after DOX was intravenously administered to tumor-bearing nude mice. These findings implied that the ALN-MA-hyd-DOX was a promising bone-targeted conjugate for treating bone neoplasms.
Co-reporter:Wei-Liang Ye, Zeng-Hui Teng, Dao-Zhou Liu, Han Cui, ... Si-Yuan Zhou
Journal of Pharmaceutical Sciences (February 2013) Volume 102(Issue 2) pp:530-540
Publication Date(Web):1 February 2013
DOI:10.1002/jps.23381
Folate–aminocaproic acid–doxorubicin (FA–AMA–DOX) was synthesized and characterized by H NMR spectroscopy and mass spectrometry. Cytotoxicity and cellular uptake experiments were performed in KB and HepG2 cells, which express folic acid receptor, and the cell line A549, which does not express folic acid receptor. Cytotoxicity was determined using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay, and cellular uptake was monitored using fluorescence microscopy. The amount of DOX released from FA–AMA–DOX was much greater at pH 5.0 than that at pH 6.5 or 7.4. The cytotoxicity of FA–AMA–DOX toward KB and HepG2 cells was greater than that of DOX or AMA–DOX at the same concentrations, and cytotoxicity could be attenuated by FA in a dose-dependent manner. On the contrary, the cytotoxicity of FA–AMA–DOX and AMA–DOX toward A549 cells was lower than that of DOX at the same concentration, and cytotoxicity could not be reduced by FA. Compared with FA–AMA, FA–AMA–DOX increased the intracellular accumulation of DOX in KB cells. These results suggested that FA–AMA–DOX have suitable attributes for the active targeting of folate-receptor-positive tumor cells and for releasing the chemotherapeutic agent, DOX, in situ; it therefore has potential as a novel cancer therapeutic. © 2012 Wiley Periodicals, Inc. and the American Pharmacists Association J Pharm Sci 102:530–540, 2013

![N-[4-[[(2-amino-3,4-dihydro-4-oxo-6-pteridinyl)methyl]amino]benzoyl]-L-Glutamic acid 5-(2,5-dioxo-1-pyrrolidinyl) ester](http://img.cochemist.com/ccimg/153500/153445-05-7.png)
![N-[4-[[(2-amino-3,4-dihydro-4-oxo-6-pteridinyl)methyl]amino]benzoyl]-L-Glutamic acid 5-(2,5-dioxo-1-pyrrolidinyl) ester](http://img.cochemist.com/ccimg/153500/153445-05-7_b.png)
![[3-[HYDROXY(2-HYDROXYETHOXY)PHOSPHORYL]OXY-2-[(E)-OCTADEC-9-ENOYL]OXYPROPYL] (E)-OCTADEC-9-ENOATE](http://img.cochemist.com/ccimg/76400/76391-83-8.png)
![[3-[HYDROXY(2-HYDROXYETHOXY)PHOSPHORYL]OXY-2-[(E)-OCTADEC-9-ENOYL]OXYPROPYL] (E)-OCTADEC-9-ENOATE](http://img.cochemist.com/ccimg/76400/76391-83-8_b.png)











![1-Hexanol, 6-[[(3b)-cholest-5-en-3-yl]oxy]-](http://img.cochemist.com/ccimg/68400/68354-84-7.png)
![1-Hexanol, 6-[[(3b)-cholest-5-en-3-yl]oxy]-](http://img.cochemist.com/ccimg/68400/68354-84-7_b.png)
![Poly[imino[(1S)-1-(carboxymethyl)-2-oxo-1,2-ethanediyl]]](http://img.cochemist.com/ccimg/26100/26063-13-8.png)
![Poly[imino[(1S)-1-(carboxymethyl)-2-oxo-1,2-ethanediyl]]](http://img.cochemist.com/ccimg/26100/26063-13-8_b.png)
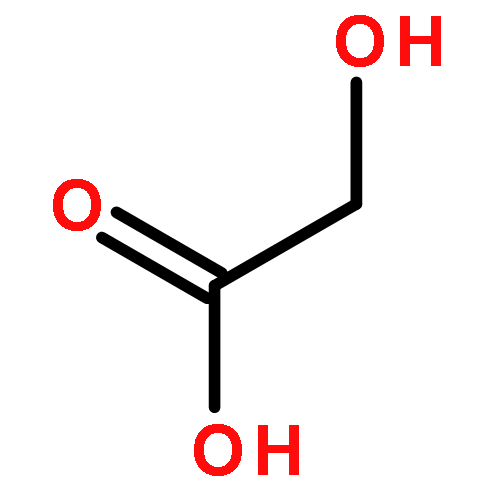
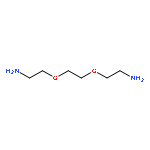
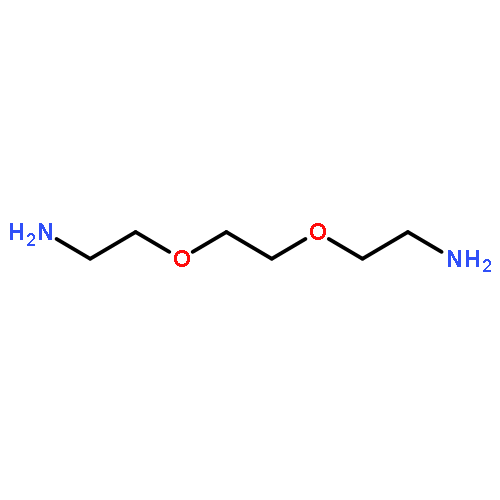
![Hexanoic acid, 6-[(4-methoxybenzoyl)amino]-](http://img.cochemist.com/ccimg/22900/22834-47-5.png)
![Hexanoic acid, 6-[(4-methoxybenzoyl)amino]-](http://img.cochemist.com/ccimg/22900/22834-47-5_b.png)