Co-reporter:Sicheng Zhang, Jihong Lan, Zhuqi Chen, Guochuan Yin, and Guangxing Li
ACS Sustainable Chemistry & Engineering October 2, 2017 Volume 5(Issue 10) pp:9360-9360
Publication Date(Web):August 16, 2017
DOI:10.1021/acssuschemeng.7b02396
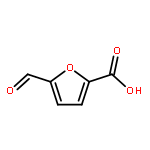
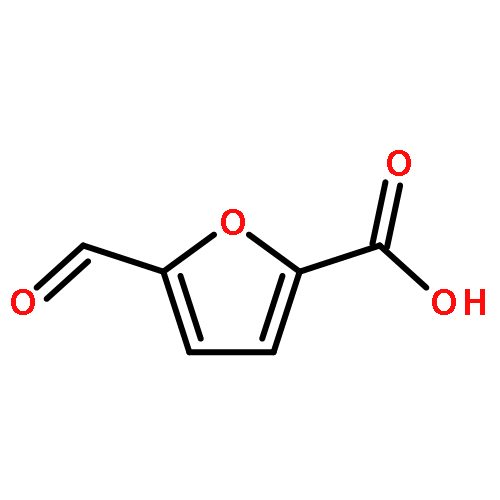
2,5-Furandicarboxylic acid (2,5-FDCA), a renewable alternative to p-phthalic acid, is the most promising subproduct from 5-hydroxymethylfurfural (HMF). However, HMF is currently synthesized from mono- and polysaccharides like glucose and fructose with limited volume, which apparently blocks the utilization of 2,5-FDCA to replace p-phthalic acid in the polymer industry. Here, we presented a novel route to 2,5-FDCA originating from C5-based furfural which is industrially produced from bulky raw biomaterials, and is not competitive with food for humans. The starting chemical of this synthesis is furoic acid which is currently produced from furfural. Furoic acid can be feasibly transformed to 2,5-FDCA through consecutive bromination, esterification, carbonylation, and hydrolysis with 65% total yield in four steps and above 80% isolated yield in each step. In particular, the key step, palladium-catalyzed carbonylation of ethyl 5-bromo-furan-2-carboxylate, retains 90% isolated yield in the scale-up synthesis. The route introduced here has offered a promising opportunity to access HMF products from furfural derivatives with a large market; meanwhile it offers one of the key C1 resources, that is, CO, a promising utilization in industry.Keywords: 2,5-Furandicarboxylic acid; Biomass; C5 to C6 platform transformation; Noncompetitive with food; Pd-catalyzed carbonylation;
Co-reporter:Ahmed M. Senan, Sicheng Zhang, Miao Zeng, Zhuqi Chen, and Guochuan Yin
Journal of Agricultural and Food Chemistry August 16, 2017 Volume 65(Issue 32) pp:6912-6912
Publication Date(Web):July 18, 2017
DOI:10.1021/acs.jafc.7b02017
Utilization of renewable biomass to partly replace the fossil resources in industrial applications has attracted attention due to the limited fossil feedstock with the increased environmental concerns. This work introduced a modified Wacker-type oxidation for transformation of unsaturated fatty acids/esters to the corresponding keto fatty acids/esters, in which Cu2+ cation was replaced with common nonredox metal ions, that is, a novel Pd(II)/Lewis acid (LA) catalyst. It was found that adding nonredox metal ions can effectively promote Pd(II)-catalyzed oxidation of unsaturated fatty acids/esters to the corresponding keto fatty acids/esters, even much better than Cu2+, and the promotional effect is highly dependent on the Lewis acidity of added nonredox metal ions. The improved catalytic efficiency is attributed to the formation of heterobimetallic Pd(II)/LA species, and the oxidation mechanism of this Pd(II)/LA catalyst is also briefly discussed.Keywords: biomass valorization; Lewis acid; oxidation; palladium(II); unsaturated fatty acid;
Co-reporter:Jisheng Zhang, Hang Yang, Tingting Sun, Zhuqi Chen, and Guochuan Yin
Inorganic Chemistry 2017 Volume 56(Issue 2) pp:
Publication Date(Web):January 5, 2017
DOI:10.1021/acs.inorgchem.6b02277
Dioxygen activation toward efficient catalysis at ambient temperature is still a big challenge for industrial oxidations, while it proceeds smoothly in nature. This work presents an example of that adding nonredox metal ions as Lewis acid can enhance dioxygen activation by oxidovanadium(IV) complex, [VIV(O)Cl(TPA)]PF6 (where TPA is tris-[(2-pyridy)methyl]amine), which leads to efficient hydrogen abstraction at ambient temperature, whereas, in the absence of a Lewis acid, the catalytic hydrogen abstraction of the oxidovanadium(IV) complex is very sluggish. Ultraviolet-visible light (UV-vis), electron paramagnetic resonance (EPR), mass, and nuclear magnetic resonance (NMR) studies have provided informative clues to indicate the interaction between the Lewis acid and vanadium complexes, including assisting the dissociation of the chloride from the oxidovanadium(IV) complex, interacting with the vanadium oxido group, and stabilizing the vanadium(V) superoxo species. These interactions enhanced the dioxgyen activation efficiency of oxidovanadium(IV) complex, and improved the hydrogen abstraction ability of vanadium(V) oxido species, which leads to efficient hydrogen abstraction in a catalytic process. A brief mechanism has also been proposed for dioxygen activation toward hydrogen abstraction by an oxidovanadium(IV) complex.
Co-reporter:Sicheng Zhang;Haosheng Xu;Chenlin Lou;Ahmed M. Senan;Zhuqi Chen
European Journal of Organic Chemistry 2017 Volume 2017(Issue 14) pp:1870-1875
Publication Date(Web):2017/04/10
DOI:10.1002/ejoc.201601495
Transition-metal-catalyzed nitrile hydration is an atom-economic method for the synthesis of various amides. This work demonstrates for the first time that the addition of non-redox metal ions like Sc3+ dramatically accelerate the hydration of various nitriles to amides at ambient temperature with the simple Pd(OAc)2 salt as catalyst, whereas the reactions with Pd(OAc)2 alone were very sluggish. The formation of a heterobimetallic PdII/ScIII species has been proposed as the key species for the hydration that demonstrates a bimetallic synergistic effect in this process.
Co-reporter:Cholho Choe, Zhanao Lv, Yunfeng Wu, Zhuqi Chen, Tingting Sun, Haibin Wang, Guangxing Li, Guochuan Yin
Molecular Catalysis 2017 Volume 438(Volume 438) pp:
Publication Date(Web):1 September 2017
DOI:10.1016/j.mcat.2017.05.030
•Lewis and Brønsted acids can sharply improve the oxygen transfer efficiency of a manganese(II) catalyst with non-heme ligand.•The catalytic activity improvement is acidity strength or net charge dependent.•The promotional effect by either Lewis acid or Brønsted acid was originated from the dissociating sluggish di-μ-oxo core.•Distinctions of reactive intermediate were also demonstrated for Lewis acid or Brønsted acid.This work demonstrates that certain Lewis and Brønsted acids can sharply improve the oxygen transfer efficiency of a manganese(II) catalyst bearing non-heme ligand. In the absence of Lewis and Brønsted acids, oxidation of manganese(II) complex will generate di-μ-oxo-bridged dinuclear Mn2(III,IV) core which is very sluggish for olefin epoxidation. Adding non-redox metal ions as Lewis acid or Brønsted acid will both improve the catalytic epoxidation of olefin, and this improvement is dependent on the pKa of Brønsted acid, or the net charge of non-redox metals of Lewis acid. Mechanism study revealed that similar promotional effect by either Lewis or Brønsted acids was originated from a similar reaction pathway by dissociating aforementioned sluggish di-μ-oxo core. However, distinctions of reactive intermediate were also demonstrated for Lewis or Brønsted acids.Download full-size image
Co-reporter:Ahmed M. Senan;Sicheng Zhang;Shuhao Qin
Journal of the American Oil Chemists' Society 2017 Volume 94( Issue 12) pp:1481-1489
Publication Date(Web):26 October 2017
DOI:10.1007/s11746-017-3052-5
With the rapid depletion of fossil resources, the exploitation of biomass to partly replace fossil resources as the source of carbon in the chemical industry constitutes a promising alternative for the near future. This work introduces catalytic transformation of vegetable oil, i.e., methyl linoleate, to its conjugated esters by a simple Pd(OAc)2/Sc(OTf)3 catalyst, which has extensive applications in industry. It was found that adding non-redox metal ions like Sc(III) to a simple Pd(OAc)2 catalyst can effectively improve its isomerization activity in toluene/t-BuOH solvent, whereas Pd(OAc)2 alone is inactive. Preliminary mechanistic investigations together with previous studies suggested that the in situ-generated heterobimetallic Pd(II)/Sc(III) dimer serves as the key species for methyl linoleate isomerization, and the reaction proceeds by [1,3]-hydrogen shift mechanism involving a formal Pd(II)/Pd(IV) cycle.
Co-reporter:Ahmed M. Senan, Shuhao Qin, Sicheng Zhang, Chenling Lou, Zhuqi Chen, Rong-Zhen Liao, and Guochuan Yin
ACS Catalysis 2016 Volume 6(Issue 7) pp:4144
Publication Date(Web):May 27, 2016
DOI:10.1021/acscatal.6b01061
Redox metal-ion-catalyzed olefin isomerization represents one of the important chemical processes. This work illustrates that nonredox metal ions can sharply accelerate Pd(II)-catalyzed olefin isomerization, while Pd(II) alone is very sluggish. Nuclear magnetic resonance (NMR) and ultraviolet–visible light (UV-vis) characterizations disclosed that the acceleration effect originates from the formation of heterobimetallic Pd(II) species with added nonredox metal ions, which improves the C–H activation capability of the Pd(II) moiety. Density functional theory (DFT) calculations further confirmed the sharp decrease of the energy barrier in C–H activation by the heterobimetallic Pd(II)/Al(III) species.Keywords: density functional theory; energy barrier; isomerization; nonredox metal ions; olefins
Co-reporter:Sicheng Zhang, Zhuqi Chen, Shuhao Qin, Chenlin Lou, Ahmed M. Senan, Rong-Zhen Liao and Guochuan Yin
Organic & Biomolecular Chemistry 2016 vol. 14(Issue 17) pp:4146-4157
Publication Date(Web):05 Apr 2016
DOI:10.1039/C6OB00401F
Developing new catalytic technologies through C–H bond activation to synthesize versatile pharmaceuticals has attracted much attention in recent decades. This work introduces a new strategy in catalyst design for Pd(II)-catalyzed C–H bond activation in which non-redox metal ions serving as Lewis acids play significant roles. In the oxidative coupling of indoles with olefins using dioxygen, it was found that Pd(OAc)2 alone as the catalyst is very sluggish at ambient temperature which provided a low yield of the olefination product, whereas adding non-redox metal ions to Pd(OAc)2 substantially improves its catalytic efficiency. In particular, it provided bis(indolyl)methane derivatives as the dominant product, a category of pharmacological molecules which could not be synthesized by Pd(II)-catalyzed oxidative coupling previously. Detailed investigations revealed that the reaction proceeds by heterobimetallic Pd(II)/Sc(III)-catalyzed oxidative coupling of an indole with an olefin followed by Sc(III)-catalyzed addition with a second indole molecule. DFT calculations disclosed that the formation of heterobimetallic Pd(II)/Sc(III) species substantially decreases the C–H bond activation energy barrier, and shifts the rate determining step from C–H bond activation of indole to the olefination step. This non-redox metal ion promoted Pd(II)-catalyzed C–H bond activation may offer a new opportunity for catalyst design in organic synthesis, which has not been fully recognized yet.
Co-reporter:Zhanao Lv, Wenrui Zheng, Zhuqi Chen, Zhiming Tang, Wanling Mo and Guochuan Yin
Dalton Transactions 2016 vol. 45(Issue 28) pp:11369-11383
Publication Date(Web):07 Jun 2016
DOI:10.1039/C6DT01077F
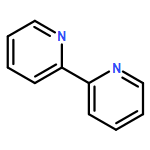
Non-redox metal ions can affect the reactivity of active redox metal ions in versatile biological and heterogeneous oxidation processes; however, the intrinsic roles of these non-redox ions still remain elusive. This work demonstrates the first example of the use of non-redox metal ions as Lewis acids to sharply improve the catalytic oxygen atom transfer efficiency of a ruthenium complex bearing the classic 2,2′-bipyridine ligand. In the absence of Lewis acid, the oxidation of ruthenium(II) complex by PhI(OAc)2 generates the Ru(IV)O species, which is very sluggish for olefin epoxidation. When Ru(bpy)2Cl2 was tested as a catalyst alone, only 21.2% of cyclooctene was converted, and the yield of 1,2-epoxycyclooctane was only 6.7%. As evidenced by electronic absorption spectra and EPR studies, both the oxidation of Ru(II) by PhI(OAc)2 and the reduction of Ru(IV)O by olefin are kinetically slow. However, adding non-redox metal ions such as Al(III) can sharply improve the oxygen transfer efficiency of the catalyst to 100% conversion with 89.9% yield of epoxide under identical conditions. Through various spectroscopic characterizations, an adduct of Ru(IV)O with Al(III), Ru(IV)O/Al(III), was proposed to serve as the active species for epoxidation, which in turn generated a Ru(III)–O–Ru(III) dimer as the reduced form. In particular, both the oxygen transfer from Ru(IV)O/Al(III) to olefin and the oxidation of Ru(III)–O–Ru(III) back to the active Ru(IV)O/Al(III) species in the catalytic cycle can be remarkably accelerated by adding a non-redox metal, such as Al(III). These results have important implications for the role played by non-redox metal ions in catalytic oxidation at redox metal centers as well as for the understanding of the redox mechanism of ruthenium catalysts in the oxygen atom transfer reaction.
Co-reporter:Ali Jawad, Yibing Li, Lianshuang Guo, Aimal Khan, Zhuqi Chen, Jingyu Wang, Jiakuan Yang, Weidong Liu and Guochuan Yin
RSC Advances 2016 vol. 6(Issue 76) pp:72643-72653
Publication Date(Web):04 Jul 2016
DOI:10.1039/C6RA10402A
Catalytic wastewater treatment is confronted by varied challenges like catalyst stability and efficiency in aqueous media due to the complex chemistry during organic compound degradation. Herein, we attempt to address this challenge by creating a synergistically stable and active bimetallic CuCoOx–LDH catalyst via facile copper ion hydrothermal impregnation in a CoOx–LDH catalyst. Different instrumental techniques like BET, XRD, FTIR, SEM, XPS and electrochemical studies etc. were conducted to investigate the properties of the catalyst before and after impregnation of copper ions. It was found that the changes in the electrochemistry and redox properties of the CuCoOx–LDH catalyst based on cyclic voltammetry (CV), electrochemical impedance spectroscopy (EIS), and X-ray photoelectron spectroscopy (XPS) appeared in the form of enhanced activity and excellent stability. In the bicarbonate activation of hydrogen peroxide (BAP) system, the synthesized CuCoOx–LDH catalyst can efficiently degrade 200 ppm 4-chlorophenol (4-CP) with 84% COD and 78% TOC removal in less than 40 minutes, and even 1000 ppm of 4-CP in hours, while the CoOx–LDH and CuOx–LDH catalysts or their physical mixtures are apparently sluggish. This catalyst can also effectively degrade various substituted phenols including 2,4-dichlorophenol (DCP), 2,4,4-trichlorophenol (TCP), 2-chlorophenol (2-CP), phenol, and chlorobenzene with significant COD removal. The findings from fluorescence, scavengers, electron paramagnetic resonance (EPR), XPS, and electrochemical studies suggest collectively the generation of ˙OH, 1O2, and ˙O2− species and that the regeneration of active sites may be part of the degradation process. This approach based on CV, EIS and XPS studies has provided novel knowledge about the intrinsic origins of synergetic acceleration of catalyst activity.
Co-reporter:Zhuqi Chen and Guochuan Yin
Chemical Society Reviews 2015 vol. 44(Issue 5) pp:1083-1100
Publication Date(Web):07 Jan 2015
DOI:10.1039/C4CS00244J
While the significance of the redox metal oxo moieties has been fully acknowledged in versatile oxidation processes, active metal hydroxo moieties are gradually realized to play the key roles in certain biological oxidation events, and their reactivity has also been evidenced by related biomimic models. However, compared with the metal oxo moieties, the significance of the metal hydroxo moieties has not been fully recognized, and their relationships in oxidations remain elusive until recently. This review summarizes the reactivity of the metal oxo and hydroxo moieties in different oxidation processes including hydrogen atom transfer, oxygen atom transfer and electron transfer, and their reactivity similarities and differences have been discussed as well. Particularly, how the physicochemical properties like metal–oxygen bond order, net charge and potential of a redox metal ion affect its reactivity has also been presented based on available data. We hope that this review may provide new clues to understand the origins of the enzyme's choice on them in a specific event, to explain the elusive phenomena occurring in those enzymatic, homogeneous and heterogeneous oxidations, to design selective redox catalysts and control their reactivity.
Co-reporter:Jihong Lan, Jinchi Lin, Zhuqi Chen, and Guochuan Yin
ACS Catalysis 2015 Volume 5(Issue 4) pp:2035
Publication Date(Web):February 18, 2015
DOI:10.1021/cs501776n
Developing novel technologies to utilize renewable biomass as the source of energy and carbon to partially replace the fossil resources has been well recognized by the global governments and both industrial and academic communities. This work explored a catalytic transformation of biomass-based 5-hydroxymethylfurfural to maleic anhydride and maleic acid through aerobic oxidation with vanadium-substituted heteropolyacid. Under the optimized conditions, total yields of 64% for maleic anhydride and maleic acid could be achieved. Mechanistic studies with control experiments excluded 2, 5-furandicarboxylic acid, 2, 5-diformylfuran, 5-formyl-2-furancarboxylic acid, and 5-hydroxymethyl-2-furancarboxylic acid as the intermediates in the pathway of 5-hydroxymethylfurfural oxidation to maleic anhydride. Alternatively, a new mechanism initialized by the C–C bond cleavage between the hydroxymethyl group and furan sketch of HMF by heteropolyacid has been proposed for MA formation, in which several intermediates have been identified through GC-MS analysis.Keywords: 5-hydroxymethylfurfural; biomass utilization; catalytic oxidation; heteropolyacid; maleic anhydride
Co-reporter:Zhuqi Chen, Ling Yang, Cholho Choe, Zhanao Lv and Guochuan Yin
Chemical Communications 2015 vol. 51(Issue 10) pp:1874-1877
Publication Date(Web):15 Dec 2014
DOI:10.1039/C4CC07981G
This work demonstrates that non-redox metal ions as Lewis acids can sharply improve the oxygen transfer efficiency of a manganese(II) catalyst having a non-heme ligand. In the absence of Lewis acid, oxidation of a manganese(II) complex will generate the known di-μ-oxo-bridged dinuclear Mn2(III,IV) core which is very sluggish for olefin epoxidation. Adding non-redox metal ions causes the dissociation of the dinuclear core, leading to sharp improvement in its oxygen transfer efficiency.
Co-reporter:Donald G. Jones, Kevin R. Wilson, Desiray J. Cannon-Smith, Anthony D. Shircliff, Zhan Zhang, Zhuqi Chen, Timothy J. Prior, Guochuan Yin, and Timothy J. Hubin
Inorganic Chemistry 2015 Volume 54(Issue 5) pp:2221-2234
Publication Date(Web):February 11, 2015
DOI:10.1021/ic502699m
The first 2-pyridylmethyl pendant-armed ethylene cross-bridged cyclam ligand has been synthesized and successfully complexed to Mn2+, Fe2+, Co2+, Ni2+, Cu2+, and Zn2+ cations. X-ray crystal structures were obtained for all six complexes and demonstrate pentadentate binding of the ligand with the requisite cis-V configuration of the cross-bridged cyclam ring in all cases, leaving a potential labile binding site cis to the pyridine donor for interaction of the complex with oxidants and/or substrates. The electronic properties of the complexes were evaluated using solid-state magnetic moment determination and acetonitrile solution electronic spectroscopy, which both agree with the crystal structure determination of high-spin divalent metal complexes in all cases. Cyclic voltammetry in acetonitrile revealed reversible redox processes in all but the Ni2+ complex, suggesting that catalytic reactivity involving electron-transfer processes is possible for complexes of this ligand. Kinetic studies of the dissociation of the ligand from the copper(II) complex under strongly acidic conditions and elevated temperatures revealed that the pyridine pendant arm actually destabilizes the complex compared to the parent cross-bridged cyclam complex. Screening for oxidation catalysis using hydrogen peroxide as the terminal oxidant for the most biologically relevant Mn2+, Fe2+, and Cu2+ complexes identified the Mn2+ complex as a potential mild oxidation catalyst worthy of continued development.
Co-reporter:Shuhao Qin, Lei Dong, Zhuqi Chen, Sicheng Zhang and Guochuan Yin
Dalton Transactions 2015 vol. 44(Issue 40) pp:17508-17515
Publication Date(Web):04 Sep 2015
DOI:10.1039/C5DT02612A
In Wacker oxidation and inspired Pd(II)/Cu(II)-catalyzed C–H activations, copper(II) is believed to serve in re-oxidizing of Pd(0) in the catalytic cycle. Herein we report that non-redox metal ions like Sc(III) can promote Wacker-type oxidations even better than Cu(II); both Sc(III) and Cu(II) can greatly promote Pd(II)-catalyzed olefin isomerization in which the redox properties of Cu(II) are not essential, indicating that the Lewis acid properties of Cu(II) can play a significant role in Pd(II)-catalyzed C–H activations in addition to its redox properties. Characterization of catalysts using UV-Vis and NMR indicated that adding Sc(OTf)3 to the acetonitrile solution of Pd(OAc)2 generates a new Pd(II)/Sc(III) bimetallic complex having a diacetate bridge which serves as the key active species for Wacker-type oxidation and olefin isomerization. Linkage of trivalent Sc(III) to the Pd(II) species makes it more electron-deficient, thus facilitating the coordination of olefin to the Pd(II) cation. Due to the improved electron transfer from olefin to the Pd(II) cation, it benefits the nucleophilic attack of water on the olefinic double bond, leading to efficient olefin oxidation. The presence of excess Sc(III) prevents the palladium(0) black formation, which has been rationalized by the formation of the Sc(III)⋯H–Pd(II) intermediate. This intermediate inhibits the reductive elimination of the H–Pd(II) bond, and facilitates the oxygen insertion to form the HOO–Pd(II) intermediate, and thus avoids the formation of the inactive palladium(0) black. The Lewis acid promoted Wacker-type oxidation and olefin isomerization demonstrated here may open up a new opportunity in catalyst design for versatile C–H activations.
Co-reporter:Cholho Choe, Ling Yang, Zhanao Lv, Wanling Mo, Zhuqi Chen, Guangxin Li and Guochuan Yin
Dalton Transactions 2015 vol. 44(Issue 19) pp:9182-9192
Publication Date(Web):13 Apr 2015
DOI:10.1039/C4DT03993A
Redox-inactive metal ions can modulate the reactivity of redox-active metal ions in a variety of biological and chemical oxidations. Many synthetic models have been developed to help address the elusive roles of these redox-inactive metal ions. Using a non-heme manganese(II) complex as the model, the influence of redox-inactive metal ions as a Lewis acid on its catalytic efficiency in oxygen atom transfer was investigated. In the absence of redox-inactive metal ions, the manganese(II) catalyst is very sluggish, for example, in cyclooctene epoxidation, providing only 9.9% conversion with 4.1% yield of epoxide. However, addition of 2 equiv. of Al3+ to the manganese(II) catalyst sharply improves the epoxidation, providing up to 97.8% conversion with 91.4% yield of epoxide. EPR studies of the manganese(II) catalyst in the presence of an oxidant reveal a 16-line hyperfine structure centered at g = 2.0, clearly indicating the formation of a mixed valent di-μ-oxo-bridged diamond core, MnIII-(μ-O)2-MnIV. The presence of a Lewis acid like Al3+ causes the dissociation of this diamond MnIII-(μ-O)2-MnIV core to form monomeric manganese(IV) species which is responsible for improved epoxidation efficiency. This promotional effect has also been observed in other manganese complexes bearing various non-heme ligands. The findings presented here have provided a promising strategy to explore the catalytic reactivity of some di-μ-oxo-bridged complexes by adding non-redox metal ions to in situ dissociate those dimeric cores and may also provide clues to understand the mechanism of methane monooxygenase which has a similar diiron diamond core as the intermediate.
Co-reporter:Jisheng Zhang, Yujuan Wang, Nengchao Luo, Zhuqi Chen, Kangbing Wu and Guochuan Yin
Dalton Transactions 2015 vol. 44(Issue 21) pp:9847-9859
Publication Date(Web):20 Apr 2015
DOI:10.1039/C5DT00804B
Utilization of dioxygen as the terminal oxidant at ambient temperature is always a challenge in redox chemistry, because it is hard to oxidize a stable redox metal ion like iron(III) to its high oxidation state to initialize the catalytic cycle. Inspired by the dioxygenation and co-oxidase activity of lipoxygenases, herein, we introduce an alternative protocol to activate the sluggish iron(III) species with non-redox metal ions, which can promote its oxidizing power to facilitate substrate oxidation with dioxygen, thus initializing the catalytic cycle. In oxidations of N,N-dimethylaniline and its analogues, adding Zn(OTf)2 to the [Fe(TPA)Cl2]Cl catalyst can trigger the amine oxidation with dioxygen, whereas [Fe(TPA)Cl2]Cl alone is very sluggish. In stoichiometric oxidations, it has also been confirmed that the presence of Zn(OTf)2 can apparently improve the electron transfer capability of the [Fe(TPA)Cl2]Cl complex. Experiments using different types of substrates as trapping reagents disclosed that the iron(IV) species does not occur in the catalytic cycle, suggesting that oxidation of amines is initialized by electron transfer rather than hydrogen abstraction. Combined experiments from UV-Vis, high resolution mass spectrometry, electrochemistry, EPR and oxidation kinetics support that the improved electron transfer ability of iron(III) species originates from its interaction with added Lewis acids like Zn2+ through a plausible chloride or OTf− bridge, which has promoted the redox potential of iron(III) species. The amine oxidation mechanism was also discussed based on the available data, which resembles the co-oxidase activity of lipoxygenases in oxidative dealkylation of xenobiotic metabolisms where an external electron donor is not essential for dioxygen activation.
Co-reporter:Jihong Lan, Zhuqi Chen, Jinchi Lin and Guochuan Yin
Green Chemistry 2014 vol. 16(Issue 10) pp:4604-4604
Publication Date(Web):10 Sep 2014
DOI:10.1039/C4GC90039A
Correction for ‘Catalytic aerobic oxidation of renewable furfural to maleic anhydride and furanone derivatives with their mechanistic studies’ by Jihong Lan et al., Green Chem., 2014, 16, 4351–4358.
Co-reporter:Jihong Lan, Zhuqi Chen, Jinchi Lin and Guochuan Yin
Green Chemistry 2014 vol. 16(Issue 9) pp:4351-4358
Publication Date(Web):10 Jul 2014
DOI:10.1039/C4GC00829D
Catalytic transformation of biomass-based furfural to value-added chemicals is an alternative route to the on-going fossil feedstock-based processes. This work describes catalytic aerobic oxidation of furfural to maleic anhydride, an important polymer starting material having a large market with H5PV2Mo10O40 and Cu(CF3SO3)2 catalysts. Under the optimized conditions, 54.0% yield of maleic anhydride can be achieved with about 7.5% yield of 5-acetoxyl-2(5H)-furanone formation. Notably, 5-acetoxyl-2(5H)-furanone is a highly value-added, biologically important intermediate that has been applied in pharmaceutical synthesis. The catalytic mechanism for furfural oxidation to maleic anhydride and 5-acetoxyl-2(5H)-furanone has been investigated in detail with identification of several key intermediates.
Co-reporter:Zhan Zhang, Katherine L. Coats, Zhuqi Chen, Timothy J. Hubin, and Guochuan Yin
Inorganic Chemistry 2014 Volume 53(Issue 22) pp:11937-11947
Publication Date(Web):November 6, 2014
DOI:10.1021/ic501342c
Available data from different laboratories have confirmed that both Ca2+ and Cl– are crucial for water oxidation in Photosystem II. However, their roles are still elusive. Using a manganese(II) complex having a cross-bridged cyclen ligand as a model, the influence of Ca2+ on the oxidative reactivity of the manganese(II) complex and its corresponding manganese(IV) analogue were investigated. It has been found that adding Ca2+ can significantly improve the oxygenation efficiency of the manganese(II) complex in sulfide oxidation and further accelerate the oxidation of sulfoxide to sulfone. Similar improvements have also been observed for Mg2+, Sr2+, and Ba2+. A new monomeric manganese(IV) complex having two cis-hydroxide ligands has also been isolated through oxidation of the corresponding manganese(II) complex with H2O2 in the presence of NH4PF6. This rare cis-dihydroxomanganese(IV) species has been well characterized by X-ray crystallography, electrochemistry, electron paramagnetic resonance, and UV–vis spectroscopy. Notably, using the manganese(IV) complex as a catalyst demonstrates higher activity than the corresponding manganese(II) complex, and adding Ca2+ further improves its catalytic efficiency. However, adding Cl– decreases its catalytic activity. In electrochemical studies of manganese(IV) complexes with no chloride ligand present, adding Ca2+ positively shifted the redox potential of the MnIV/MnIII couple but negatively shifted its MnV/MnIV couple. In the manganese(II) complex having a chloride ligand, adding Ca2+ shifted both the MnIV/MnIII and MnV/MnIV couples in the negative direction. The revealed oxidative reactivity and redox properties of the manganese species affected by Ca2+ and Cl– may provide new clues to understanding their roles in the water oxidation process of Photosystem II.
Co-reporter:Ali Jawad, Xiaoyan Lu, Zhuqi Chen, and Guochuan Yin
The Journal of Physical Chemistry A 2014 Volume 118(Issue 43) pp:10028-10035
Publication Date(Web):October 6, 2014
DOI:10.1021/jp5085313
Toxic and bioresistant compounds have attracted researchers to develop more efficient and cost-effective technologies for degradation of organic compounds in wastewater. This work demonstrates the degradation of 4-chlorophenol, 2,4-dichlorophenol, 2,4,6-trichlorophenol, and phenol as model compounds using bicarbonate-activated H2O2 oxidation system in the presence of supported catalysts. The catalytic activity of the catalyst was investigated in term of degradation of target compounds, chemical oxygen demand (COD), and total organic carbon (TOC) removals both for batch mode and in fixed bed reactor using CoMgAl-HTs and CoMgAl-SHTs, respectively. The leaching of cobalt ion was efficiently prohibited because of the presence of a weakly basic medium provided by bicarbonate, and the CoMgAl-SHTs catalyst was found to retain its stability and good catalytic activity in fixed bed reactor for over 300 h. Extensive chemical probing, fluorescence, and electron paired resonance (EPR) studies were conducted to identify the actual reactive species in the degradation pathway, which revealed that the reaction proceeds through generation of superoxide, hydroxyl radical along with carbonate radical.
Co-reporter:Li Zhou, Wei Song, Zhuqi Chen, and Guochuan Yin
Environmental Science & Technology 2013 Volume 47(Issue 8) pp:3833
Publication Date(Web):March 14, 2013
DOI:10.1021/es400101f
Developing novel technologies to cleanup wastewater has attracted attention for a long while in academic and industrial communities not only for environmental issues but also for recycling water sources. This work demonstrates that bicarbonate-activated H2O2 can be applied as a novel oxidant source in pollutant degradation. Using a supported cobalt catalyst, bicarbonate-activated H2O2 can efficiently degrade various dyes and phenol at ambient temperature. Because the reaction media remains weakly basic during degradation, the cobalt leaching from the solid catalyst has been efficiently avoided and the lifetime of the catalyst can be extended to above 180 h without significant activity loss in a fixed-bed test. Different scavengers, including ascorbic acid, t-butanol, sodium azide, benzoquinone, and tiron, have been tested to identify the active species, which may be involved in pollutant degradation, and it was found that singlet oxygen and the carbonate radical may play a key role in the degradation process.
Co-reporter:Lei Dong, Yujuan Wang, Yanzong Lv, Zhuqi Chen, Fuming Mei, Hui Xiong, and Guochuan Yin
Inorganic Chemistry 2013 Volume 52(Issue 9) pp:5418-5427
Publication Date(Web):April 19, 2013
DOI:10.1021/ic400361s
Redox-inactive metal ions have been recognized to be able to participate in redox metal-ion-mediated biological and chemical oxidative events; however, their roles are still elusive. This work presents how the redox-inactive metal ions affect the oxidative reactivity of a well-investigated manganese(II) with its corresponding manganese(IV) complexes having cross-bridged cyclam ligand. In dry acetone, the presence of these metal ions can greatly accelerate stoichiometric oxidations of triphenylphosphine and sulfides by the manganese(IV) complexes through electron transfer or catalytic sulfoxidations by the corresponding manganese(II) complexes with PhIO. Significantly, the rate enhancements are highly Lewis-acid strength dependent on added metal ions. These metal ions like Al3+ can also promote the thermodynamic driving force of the MnIV–OH moiety to facilitate its hydrogen abstraction from ethylbenzene having a BDECH value of 85 kcal/mol, while it is experimentally limited to 80 kcal/mol for MnIV–OH alone. Adding Al3+ may also improve the manganese(II)-catalyzed olefin epoxidation with PhIO. However, compared with those in electron transfer, improvements in hydrogen abstraction and electron transfer are minor. The existence of the interaction between Lewis acid and the manganese(IV) species was evidenced by the blue shift of the characteristic absorbance of the manganese(IV) species from 554 to 537 nm and by converting its EPR signal at g = 2.01 into a hyperfine 6-line signal upon adding Al3+ (I = 5/2). Cyclic voltammograms of the manganese(IV) complexes reveal that adding Lewis acid would substantially shift its potential to the positive direction, thus enhancing its oxidizing capability.
Co-reporter:Yujuan Wang, Song Shi, Huajun Wang, Dajian Zhu and Guochuan Yin
Chemical Communications 2012 vol. 48(Issue 63) pp:7832-7834
Publication Date(Web):02 Jul 2012
DOI:10.1039/C2CC33615D
The kinetics of hydrogen abstraction by manganese(IV) species having hydroxo or oxo group reveals that they have very similar reactive characters in the transition state of hydrogen abstraction.
Co-reporter:Yujuan Wang, Song Shi, Dajian Zhu and Guochuan Yin
Dalton Transactions 2012 vol. 41(Issue 9) pp:2612-2619
Publication Date(Web):05 Jan 2012
DOI:10.1039/C2DT11814A
Clear elucidation of the oxidative relationships of the active metal hydroperoxide moiety with its corresponding metal oxo and hydroxo intermediates would help the understanding of the different roles they may play in redox metalloenzymes and oxidation chemistry. Using an Mn(Me2EBC)Cl2 complex, it was found that, in t-butanol–water (4:1) with excess H2O2 at pH 1.5, the MnIV-OOH moiety may exist in the catalytic solution with a mass signal of m/z = 358.1, which provides a particular chance to investigate its oxidative properties. In catalytic oxidations, the MnIV-OOH moiety demonstrates a relatively poor activity in hydrogen abstraction from diphenyl methane and ethylbenzene with TOF of only 1.2 h−1 and 1.1 h−1 at 50 °C, whereas it can efficiently oxygenate diphenyl sulfide, methyl phenyl sulfide and benzyl phenyl sulfide with TOF of 13.8 h−1, 15.4 h−1 and 17.8 h−1, respectively. In mechanistic studies using H218O and H218O2, it was found that, in the MnIV-OOH moiety mediated hydrogen abstraction and sulfide oxygenations, the reaction proceeds by two parallel pathways: one by direct oxygen insertion/transfer, and the other by plausible electron transfer. Together with a good understanding of the corresponding manganese(IV) oxo and hydroxo intermediates, this work provides the first chance to compare the reactivity differences and similarities of the active metal oxo, hydroxo and hydroperoxide intermediates. The available evidence reveals that the MnIV-OOH moiety has a much more powerful oxidizing capability than the corresponding MnIVO and MnIV–OH functional groups in both hydrogen abstraction and oxygenation.
Co-reporter:Yujuan Wang, Jiayi Sheng, Song Shi, Dajian Zhu, and Guochuan Yin
The Journal of Physical Chemistry C 2012 Volume 116(Issue 24) pp:13231-13239
Publication Date(Web):June 1, 2012
DOI:10.1021/jp303281z
Clarifying how versatile physicochemical parameters of an active metal intermediate affect its reactivity would help to understand its roles in chemical and enzymatic oxidations. The influence of the net charge on electron transfer and hydrogen abstraction reactions of a manganese(IV) species having hydroxide ligand has been investigated here. It was found that increasing one unit of the positive net charge from 2+ to 3+ would accelerate its electron-transfer rate by 10–20 fold in oxygenation of tris(4-methoxyphenyl)phosphine. In contrast, the hydrogen abstraction rate is insensitive to its net charge change, and the insensitivity has been attributed to the compensation effect between the redox potential and pKa, which determine the hydrogen abstraction capability of a metal ion. Similar net-charge-promoted electron transfer but not hydrogen abstraction has also been observed in intramolecular electron transfer and hydrogen abstraction reactions when using thioxanthene as substrate. Together with the previous understanding of the reactivity of the identical manganese(IV) species having MnIV–OH or MnIV=O functional groups, the relationships of the oxidative reactivity of an active metal intermediate with its physicochemical parameters such as the net charge, the redox potential and the metal–oxygen bond order (M–O versus M═O) have been discussed with this manganese(IV) model.
Co-reporter:Huajun Guo;Wei-Dong Liu
Applied Organometallic Chemistry 2011 Volume 25( Issue 11) pp:836-842
Publication Date(Web):
DOI:10.1002/aoc.1848
A simple, efficient method for oxidation of primary and secondary alcohols to the corresponding aldehydes and ketones has been developed. Using RuCl3/Et3N as catalyst, the oxidation of benzyl alcohol with oxygen could be achieved with 332 h−1 turnover frequency in the absence of solvent. The influence of versatile N-containing additives on the catalytic efficiency has been discussed. The presence of minor water would substantially promote the catalytic efficiency, and its role in catalysis has been investigated in detail. The insensitive Hammett correlations of the substituted benzyl alcohols, the normal substrate isotope effect (kH/kD = 3.5 at 335 K), and the linear relationship between O2 pressure and turnover frequency imply that the reoxidation of the Ru(III) hydride intermediate to the active species shares the rate-determining step with the hydride transfer in the catalytic cycle. Copyright © 2011 John Wiley & Sons, Ltd.
Co-reporter:Huajun Guo and Guochuan Yin
The Journal of Physical Chemistry C 2011 Volume 115(Issue 35) pp:17516-17522
Publication Date(Web):August 3, 2011
DOI:10.1021/jp2054712
Developing new technologies to obtain chemicals from biomass in place of the fossil feedstock have attracted attention in academic and industrial communities. In this work, using renewable furfural as the feedstock, catalytic aerobic oxidation of furfural to maleic acid was investigated with phosphomolybdic acid catalyst in the aqueous/organic biphase system. The oxidation happens in the aqueous phase, and the organic phase serves as the reservoir to release the substrate gradually through phase equilibrium. Under the optimized conditions, 34.5% yield of maleic acid could be obtained with 68.6% of selectivity, and the conversion of furfural is 50.4%. Because furfural and maleic acid dominantly exist in two different phases, the product separation and reactant recycle would be very simple in its potential application. The FT-IR and 31P NMR technologies were applied to characterize the phosphomolybdic acid catalyst, and the pathway of maleic acid formation was also discussed based on obtained mechanistic information. This work demonstrates an alternative, renewable route to maleic acid, and the mechanistic information from this study also provides clues to improve the catalyst for efficient oxidation of furfural to maleic acid.
Co-reporter:Guochuan Yin
Coordination Chemistry Reviews 2010 Volume 254(15–16) pp:1826-1842
Publication Date(Web):August 2010
DOI:10.1016/j.ccr.2010.01.016
The high oxidation state transition metal oxo moieties in redox enzymes and their models are generally recognized to serve as the key active intermediates in a series of hydrogen abstraction, oxygen transfer, and electron transfer processes. New evidence suggests that certain transition metal hydroxo moieties also play key roles in oxidative processes in biological and chemical systems. Clarifying the structure and reactivity similarities and differences between the metal oxo functionality and its corresponding metal hydroxo form will help promote understanding of their complementary roles in oxidation processes and aid in the rational design of selective oxidation catalysts to match different requirements. This review summarizes the structure and reactivity similarities and differences of the reported redox enzymes and their models in which the metal oxo and/or corresponding metal hydroxo moieties have demonstrated their activity in oxidation processes. Those enzymes include heme enzymes, lipoxygenases, sulfite oxidases and xanthine oxidases, because the heme enzymes and lipoxygenases would provide the platform to compare the iron oxo with its corresponding hydroxo species, and the sulfite oxidases and xanthine oxidases provide the platform for molybdenum oxo and hydroxo species.
Co-reporter:Zhanao Lv, Haibin Wang, Zhuqi Chen, Shuhua Zou, Shuaishuai Zhu, Chenlin Lou, Guochuan Yin
Molecular Catalysis (May 2017) Volume 432() pp:259-266
Publication Date(Web):1 May 2017
DOI:10.1016/j.mcat.2016.12.027
•Synergetic effect led to remarkably improved performances in various dehydrogenation reactions.•The catalytic activity improvement is Lewis acidity strength dependent.•Dioxygen was used as the solely terminal oxidant under mild conditions.•An adduct of Ru(IV) = O/Sc(III) was proposed as the key active species to improve the catalytic efficiency.Dioxygen activation as the solely terminal oxidant in organic synthesis and catalytic oxidation is particularly attractive from the point of economic and environmental view. In our previous study, we have displayed that the introducing of non-redox metal ions can sharply improve the olefin epoxidation catalyzed by ruthenium complex with PhI(OAc)2 as the organic oxidation. Inspired by the successful strategy and dioxygen activation, in this study, we demonstrate an alternative protocol that adding non-redox metal ions to cis-Ru(bpy)2Cl2 catalyst can remarkably improve the oxidation from Ru(II) to Ru(IV) = O with dioxygen as the oxidant and promote oxidative dehydrogenation of saturated C–C bond whereas Ru(bpy)2Cl2 alone is very sluggish. Through UV–Vis, NMR, cyclic voltammogram and EPR characterization, it has been testified that oxidation from Ru(II) to Ru(IV) = O can be realized under the condition of 1 atm of dioxygen and 323 K within 25 min while extreme tough condition of 20 atm of dioxygen, 343 K and 8 h is necessary for Ru(bpy)2Cl2 alone. Combined the reaction data with characterization results, an adduct of Ru(IV) = O/Sc(III) is proposed as the key active species to improve the catalytic efficiency. The activity improvement in oxidative dehydrogenation illustrated a novel strategy of that adding non-redox metal ions to sluggish catalysts can remarkably improve its efficiency for dehydrogenation of saturated C–C bond, especially through dioxygen as ultimate oxidant.Download full-size image
Co-reporter:Chenlin Lou, Shuhao Qin, Sicheng Zhang, Zhanao Lv, Ahmed M. Senan, Zhuqi Chen, Guochuan Yin
Catalysis Communications (February 2017) Volume 90() pp:5-9
Publication Date(Web):1 February 2017
DOI:10.1016/j.catcom.2016.11.007
•Lewis acid can promote dehydrogenation of saturated CC bond with Pd(OAc)2.•The promotional effect is Lewis acidity dependent.•The plausible Pd(II)/Lewis acid dimer may play the key role in dehydrogenation.Adding non-redox metal ions to simple Pd(OAc)2 catalyst can remarkably promote oxidative dehydrogenation of saturated CC bond, and the activity improvement is Lewis acidity strength dependent. Through UV–vis and NMR characterizations of the catalyst, it was proposed that in-situ generated heteronuclear Pd(II)/Zn(II) dimer is the key active species for dehydrogenation.Adding non-redox metal ions to simple Pd(OAc)2 catalyst can sharply improve its efficiency in oxidative dehydrogenation reaction.Download high-res image (134KB)Download full-size image
Co-reporter:Song Shi, Huajun Guo, Guochuan Yin
Catalysis Communications (31 March 2011) Volume 12(Issue 8) pp:731-733
Publication Date(Web):31 March 2011
DOI:10.1016/j.catcom.2010.12.033
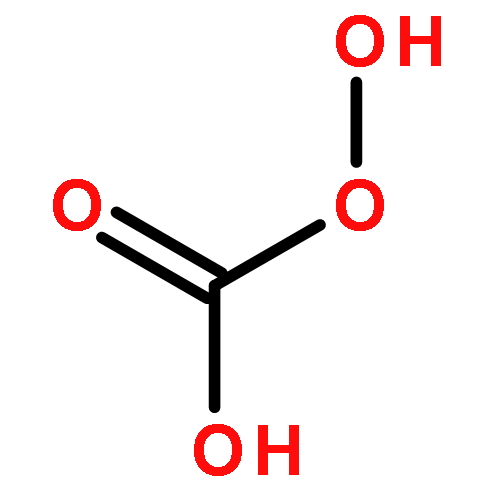
Developing novel technologies to obtain fuel and organic chemicals from renewable resources has been the immediate issue in academic and industrial communities. The present work introduces a new route to synthesize maleic acid from the renewable furfural. The current data reveal that, using dioxygen as oxidant, the simple copper salts can catalyze oxidation of furfural to maleic acid in aqueous solution. The combination of copper nitrate with phosphomolybdic acid could achieve a 49.2% yield of maleic acid with selectivity of 51.7%. The major challenge for this route is how to avoid the polymerization of furfural to resins under oxidative conditions.Download full-size imageResearch Highlights►A novel route for maleic acid synthesis from renewable furfural has been explored. ►Copper nitrate displays high efficiency in maleic acid formation. ►Copper nitrate plus phosphomolybdic acid can provide maleic acid with a 49.2% yield.
Co-reporter:Ali Jawad, Yibing Li, Xiaoyan Lu, Zhuqi Chen, Weidong Liu, Guochuan Yin
Journal of Hazardous Materials (30 May 2015) Volume 289() pp:165-173
Publication Date(Web):30 May 2015
DOI:10.1016/j.jhazmat.2015.02.056
•Degradation of organic dyes was performed with supported Co–LDH catalyst.•H2O2was activated with bicarbonate and supported catalyst.•No leaching of metal ions was monitored with enhanced degradation in basic buffer condition.•Fixed bed catalyst demonstrated good stability for over 300 h.The effluents from industries are commonly non-biodegradable and produce various hazardous intermediate products by chemical reactions that have direct impact on environment. In the present investigation, a series of Co–Mg/AL ternary LDH catalysts with fixed Mg/Al ratio were prepared by co-precipitation method. The effect of Co on the activity of the catalyst was monitored on the degradation of methylene blue (MB) as model compound at batch level using bicarbonate activation of H2O2 (BAP) system. On bench level, the best CoMgAl-4 catalyst can completely decolorize both methylene blue (MB) and methylene orange (MO) in short time, while in fixed bed, the catalyst was found stable for over 300 h with nearly 100% decolorization and excellent chemical oxygen demand (COD) removal. No leaching of Co was detected for the entire fixed experiment which may be accounted for long life stability and good activity of the catalyst. The ternary LDH catalysts were characterized by AES, XRD, FTIR, BET, and SEM for its compositional, phase structure, optical properties, textural, and surface morphology respectively. The XRD analysis confirmed characteristic pattern of hydrotalcite like structures without impurity phases. The formation of superoxide and hydroxyl radical as ROS was proposed with CoMgAl-4 by radical’s scavengers.Download full-size image
Co-reporter:Yibing Li, Lianshuang Guo, Dekang Huang, Ali Jawad, Zhuqi Chen, Jiakuan Yang, Weidong Liu, Yan Shen, Mingkui Wang, Guochuan Yin
Journal of Hazardous Materials (15 April 2017) Volume 328() pp:56-62
Publication Date(Web):15 April 2017
DOI:10.1016/j.jhazmat.2016.12.063
•The active species formation for degradation is controlled by supporting material.•The Cu(III) species based degradation displays better efficiency than OH in base.•Redox metal oxide catalyst leaching can be controlled to a very low level in base.Redox metal ions play the crucial role in versatile advanced oxidation technologies, in which controlling the active species formation through catalyst design is one of the key challenges in oxidant utilization. This work describes an example of different active species formations in CuO-mediated degradation just because of supporting material differences. Although three CuO catalysts were prepared by similar procedures, it was found that CuO-MgO catalyst demonstrated high efficiency in phenol degradation with bicarbonate activated H2O2, in which the superoxide radical is crucial, while hydroxyl radical and singlet oxygen are ignorable. For the CuO-MgO-Al2O3 and CuO-Al2O3 catalysts, the degradation proceeds by popular hydroxyl radical based process, however, the efficiency was poor. The EPR experiments also confirmed the absence of hydroxyl radical in CuO-MgO system but its presence in CuO-MgO-Al2O3 and CuO-Al2O3 system. The high catalytic efficiency with ignorable hydroxyl radical in the CuO-MgO system leads us to propose that an alternative Cu(III) species dominates the degradation. The basic MgO support may facilitate the formation of the Cu(III) species, whereas the neutral MgO-Al2O3 and acidic Al2O3 supports are unable to stabilize the high valent Cu(III) species, leading to the common hydroxyl radical mechanism with low efficiency of H2O2 in alkaline conditions.Download high-res image (105KB)Download full-size image
Co-reporter:Shuhao Qin, Lei Dong, Zhuqi Chen, Sicheng Zhang and Guochuan Yin
Dalton Transactions 2015 - vol. 44(Issue 40) pp:NaN17515-17515
Publication Date(Web):2015/09/04
DOI:10.1039/C5DT02612A
In Wacker oxidation and inspired Pd(II)/Cu(II)-catalyzed C–H activations, copper(II) is believed to serve in re-oxidizing of Pd(0) in the catalytic cycle. Herein we report that non-redox metal ions like Sc(III) can promote Wacker-type oxidations even better than Cu(II); both Sc(III) and Cu(II) can greatly promote Pd(II)-catalyzed olefin isomerization in which the redox properties of Cu(II) are not essential, indicating that the Lewis acid properties of Cu(II) can play a significant role in Pd(II)-catalyzed C–H activations in addition to its redox properties. Characterization of catalysts using UV-Vis and NMR indicated that adding Sc(OTf)3 to the acetonitrile solution of Pd(OAc)2 generates a new Pd(II)/Sc(III) bimetallic complex having a diacetate bridge which serves as the key active species for Wacker-type oxidation and olefin isomerization. Linkage of trivalent Sc(III) to the Pd(II) species makes it more electron-deficient, thus facilitating the coordination of olefin to the Pd(II) cation. Due to the improved electron transfer from olefin to the Pd(II) cation, it benefits the nucleophilic attack of water on the olefinic double bond, leading to efficient olefin oxidation. The presence of excess Sc(III) prevents the palladium(0) black formation, which has been rationalized by the formation of the Sc(III)⋯H–Pd(II) intermediate. This intermediate inhibits the reductive elimination of the H–Pd(II) bond, and facilitates the oxygen insertion to form the HOO–Pd(II) intermediate, and thus avoids the formation of the inactive palladium(0) black. The Lewis acid promoted Wacker-type oxidation and olefin isomerization demonstrated here may open up a new opportunity in catalyst design for versatile C–H activations.
Co-reporter:Yujuan Wang, Song Shi, Dajian Zhu and Guochuan Yin
Dalton Transactions 2012 - vol. 41(Issue 9) pp:NaN2619-2619
Publication Date(Web):2012/01/05
DOI:10.1039/C2DT11814A
Clear elucidation of the oxidative relationships of the active metal hydroperoxide moiety with its corresponding metal oxo and hydroxo intermediates would help the understanding of the different roles they may play in redox metalloenzymes and oxidation chemistry. Using an Mn(Me2EBC)Cl2 complex, it was found that, in t-butanol–water (4:1) with excess H2O2 at pH 1.5, the MnIV-OOH moiety may exist in the catalytic solution with a mass signal of m/z = 358.1, which provides a particular chance to investigate its oxidative properties. In catalytic oxidations, the MnIV-OOH moiety demonstrates a relatively poor activity in hydrogen abstraction from diphenyl methane and ethylbenzene with TOF of only 1.2 h−1 and 1.1 h−1 at 50 °C, whereas it can efficiently oxygenate diphenyl sulfide, methyl phenyl sulfide and benzyl phenyl sulfide with TOF of 13.8 h−1, 15.4 h−1 and 17.8 h−1, respectively. In mechanistic studies using H218O and H218O2, it was found that, in the MnIV-OOH moiety mediated hydrogen abstraction and sulfide oxygenations, the reaction proceeds by two parallel pathways: one by direct oxygen insertion/transfer, and the other by plausible electron transfer. Together with a good understanding of the corresponding manganese(IV) oxo and hydroxo intermediates, this work provides the first chance to compare the reactivity differences and similarities of the active metal oxo, hydroxo and hydroperoxide intermediates. The available evidence reveals that the MnIV-OOH moiety has a much more powerful oxidizing capability than the corresponding MnIVO and MnIV–OH functional groups in both hydrogen abstraction and oxygenation.
Co-reporter:Zhuqi Chen, Ling Yang, Cholho Choe, Zhanao Lv and Guochuan Yin
Chemical Communications 2015 - vol. 51(Issue 10) pp:NaN1877-1877
Publication Date(Web):2014/12/15
DOI:10.1039/C4CC07981G
This work demonstrates that non-redox metal ions as Lewis acids can sharply improve the oxygen transfer efficiency of a manganese(II) catalyst having a non-heme ligand. In the absence of Lewis acid, oxidation of a manganese(II) complex will generate the known di-μ-oxo-bridged dinuclear Mn2(III,IV) core which is very sluggish for olefin epoxidation. Adding non-redox metal ions causes the dissociation of the dinuclear core, leading to sharp improvement in its oxygen transfer efficiency.
Co-reporter:Zhanao Lv, Wenrui Zheng, Zhuqi Chen, Zhiming Tang, Wanling Mo and Guochuan Yin
Dalton Transactions 2016 - vol. 45(Issue 28) pp:NaN11383-11383
Publication Date(Web):2016/06/07
DOI:10.1039/C6DT01077F
Non-redox metal ions can affect the reactivity of active redox metal ions in versatile biological and heterogeneous oxidation processes; however, the intrinsic roles of these non-redox ions still remain elusive. This work demonstrates the first example of the use of non-redox metal ions as Lewis acids to sharply improve the catalytic oxygen atom transfer efficiency of a ruthenium complex bearing the classic 2,2′-bipyridine ligand. In the absence of Lewis acid, the oxidation of ruthenium(II) complex by PhI(OAc)2 generates the Ru(IV)O species, which is very sluggish for olefin epoxidation. When Ru(bpy)2Cl2 was tested as a catalyst alone, only 21.2% of cyclooctene was converted, and the yield of 1,2-epoxycyclooctane was only 6.7%. As evidenced by electronic absorption spectra and EPR studies, both the oxidation of Ru(II) by PhI(OAc)2 and the reduction of Ru(IV)O by olefin are kinetically slow. However, adding non-redox metal ions such as Al(III) can sharply improve the oxygen transfer efficiency of the catalyst to 100% conversion with 89.9% yield of epoxide under identical conditions. Through various spectroscopic characterizations, an adduct of Ru(IV)O with Al(III), Ru(IV)O/Al(III), was proposed to serve as the active species for epoxidation, which in turn generated a Ru(III)–O–Ru(III) dimer as the reduced form. In particular, both the oxygen transfer from Ru(IV)O/Al(III) to olefin and the oxidation of Ru(III)–O–Ru(III) back to the active Ru(IV)O/Al(III) species in the catalytic cycle can be remarkably accelerated by adding a non-redox metal, such as Al(III). These results have important implications for the role played by non-redox metal ions in catalytic oxidation at redox metal centers as well as for the understanding of the redox mechanism of ruthenium catalysts in the oxygen atom transfer reaction.
Co-reporter:Cholho Choe, Ling Yang, Zhanao Lv, Wanling Mo, Zhuqi Chen, Guangxin Li and Guochuan Yin
Dalton Transactions 2015 - vol. 44(Issue 19) pp:NaN9192-9192
Publication Date(Web):2015/04/13
DOI:10.1039/C4DT03993A
Redox-inactive metal ions can modulate the reactivity of redox-active metal ions in a variety of biological and chemical oxidations. Many synthetic models have been developed to help address the elusive roles of these redox-inactive metal ions. Using a non-heme manganese(II) complex as the model, the influence of redox-inactive metal ions as a Lewis acid on its catalytic efficiency in oxygen atom transfer was investigated. In the absence of redox-inactive metal ions, the manganese(II) catalyst is very sluggish, for example, in cyclooctene epoxidation, providing only 9.9% conversion with 4.1% yield of epoxide. However, addition of 2 equiv. of Al3+ to the manganese(II) catalyst sharply improves the epoxidation, providing up to 97.8% conversion with 91.4% yield of epoxide. EPR studies of the manganese(II) catalyst in the presence of an oxidant reveal a 16-line hyperfine structure centered at g = 2.0, clearly indicating the formation of a mixed valent di-μ-oxo-bridged diamond core, MnIII-(μ-O)2-MnIV. The presence of a Lewis acid like Al3+ causes the dissociation of this diamond MnIII-(μ-O)2-MnIV core to form monomeric manganese(IV) species which is responsible for improved epoxidation efficiency. This promotional effect has also been observed in other manganese complexes bearing various non-heme ligands. The findings presented here have provided a promising strategy to explore the catalytic reactivity of some di-μ-oxo-bridged complexes by adding non-redox metal ions to in situ dissociate those dimeric cores and may also provide clues to understand the mechanism of methane monooxygenase which has a similar diiron diamond core as the intermediate.
Co-reporter:Zhuqi Chen and Guochuan Yin
Chemical Society Reviews 2015 - vol. 44(Issue 5) pp:NaN1100-1100
Publication Date(Web):2015/01/07
DOI:10.1039/C4CS00244J
While the significance of the redox metal oxo moieties has been fully acknowledged in versatile oxidation processes, active metal hydroxo moieties are gradually realized to play the key roles in certain biological oxidation events, and their reactivity has also been evidenced by related biomimic models. However, compared with the metal oxo moieties, the significance of the metal hydroxo moieties has not been fully recognized, and their relationships in oxidations remain elusive until recently. This review summarizes the reactivity of the metal oxo and hydroxo moieties in different oxidation processes including hydrogen atom transfer, oxygen atom transfer and electron transfer, and their reactivity similarities and differences have been discussed as well. Particularly, how the physicochemical properties like metal–oxygen bond order, net charge and potential of a redox metal ion affect its reactivity has also been presented based on available data. We hope that this review may provide new clues to understand the origins of the enzyme's choice on them in a specific event, to explain the elusive phenomena occurring in those enzymatic, homogeneous and heterogeneous oxidations, to design selective redox catalysts and control their reactivity.
Co-reporter:Sicheng Zhang, Zhuqi Chen, Shuhao Qin, Chenlin Lou, Ahmed M. Senan, Rong-Zhen Liao and Guochuan Yin
Organic & Biomolecular Chemistry 2016 - vol. 14(Issue 17) pp:NaN4157-4157
Publication Date(Web):2016/04/05
DOI:10.1039/C6OB00401F
Developing new catalytic technologies through C–H bond activation to synthesize versatile pharmaceuticals has attracted much attention in recent decades. This work introduces a new strategy in catalyst design for Pd(II)-catalyzed C–H bond activation in which non-redox metal ions serving as Lewis acids play significant roles. In the oxidative coupling of indoles with olefins using dioxygen, it was found that Pd(OAc)2 alone as the catalyst is very sluggish at ambient temperature which provided a low yield of the olefination product, whereas adding non-redox metal ions to Pd(OAc)2 substantially improves its catalytic efficiency. In particular, it provided bis(indolyl)methane derivatives as the dominant product, a category of pharmacological molecules which could not be synthesized by Pd(II)-catalyzed oxidative coupling previously. Detailed investigations revealed that the reaction proceeds by heterobimetallic Pd(II)/Sc(III)-catalyzed oxidative coupling of an indole with an olefin followed by Sc(III)-catalyzed addition with a second indole molecule. DFT calculations disclosed that the formation of heterobimetallic Pd(II)/Sc(III) species substantially decreases the C–H bond activation energy barrier, and shifts the rate determining step from C–H bond activation of indole to the olefination step. This non-redox metal ion promoted Pd(II)-catalyzed C–H bond activation may offer a new opportunity for catalyst design in organic synthesis, which has not been fully recognized yet.
Co-reporter:Yujuan Wang, Song Shi, Huajun Wang, Dajian Zhu and Guochuan Yin
Chemical Communications 2012 - vol. 48(Issue 63) pp:NaN7834-7834
Publication Date(Web):2012/07/02
DOI:10.1039/C2CC33615D
The kinetics of hydrogen abstraction by manganese(IV) species having hydroxo or oxo group reveals that they have very similar reactive characters in the transition state of hydrogen abstraction.
Co-reporter:Jisheng Zhang, Yujuan Wang, Nengchao Luo, Zhuqi Chen, Kangbing Wu and Guochuan Yin
Dalton Transactions 2015 - vol. 44(Issue 21) pp:NaN9859-9859
Publication Date(Web):2015/04/20
DOI:10.1039/C5DT00804B
Utilization of dioxygen as the terminal oxidant at ambient temperature is always a challenge in redox chemistry, because it is hard to oxidize a stable redox metal ion like iron(III) to its high oxidation state to initialize the catalytic cycle. Inspired by the dioxygenation and co-oxidase activity of lipoxygenases, herein, we introduce an alternative protocol to activate the sluggish iron(III) species with non-redox metal ions, which can promote its oxidizing power to facilitate substrate oxidation with dioxygen, thus initializing the catalytic cycle. In oxidations of N,N-dimethylaniline and its analogues, adding Zn(OTf)2 to the [Fe(TPA)Cl2]Cl catalyst can trigger the amine oxidation with dioxygen, whereas [Fe(TPA)Cl2]Cl alone is very sluggish. In stoichiometric oxidations, it has also been confirmed that the presence of Zn(OTf)2 can apparently improve the electron transfer capability of the [Fe(TPA)Cl2]Cl complex. Experiments using different types of substrates as trapping reagents disclosed that the iron(IV) species does not occur in the catalytic cycle, suggesting that oxidation of amines is initialized by electron transfer rather than hydrogen abstraction. Combined experiments from UV-Vis, high resolution mass spectrometry, electrochemistry, EPR and oxidation kinetics support that the improved electron transfer ability of iron(III) species originates from its interaction with added Lewis acids like Zn2+ through a plausible chloride or OTf− bridge, which has promoted the redox potential of iron(III) species. The amine oxidation mechanism was also discussed based on the available data, which resembles the co-oxidase activity of lipoxygenases in oxidative dealkylation of xenobiotic metabolisms where an external electron donor is not essential for dioxygen activation.

![Lithium, (10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5-yl)-](http://img.cochemist.com/ccimg/540600/540512-99-0.png)
![Lithium, (10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5-yl)-](http://img.cochemist.com/ccimg/540600/540512-99-0_b.png)












![2-Propenoic acid, 3-[1-(phenylmethyl)-1H-indol-3-yl]-, ethyl ester, (E)-](http://img.cochemist.com/ccimg/92700/92637-35-9.png)
![2-Propenoic acid, 3-[1-(phenylmethyl)-1H-indol-3-yl]-, ethyl ester, (E)-](http://img.cochemist.com/ccimg/92700/92637-35-9_b.png)










![2-cyano-2-{5-[(4-ethoxy-3-methoxyphenyl)methylidene]-4-oxo-3-phenyl-1,3-thiazolidin-2-ylidene}-N-(2-phenylethyl)acetamide](http://img.cochemist.com/ccimg/5300/5279-62-9.png)
![2-cyano-2-{5-[(4-ethoxy-3-methoxyphenyl)methylidene]-4-oxo-3-phenyl-1,3-thiazolidin-2-ylidene}-N-(2-phenylethyl)acetamide](http://img.cochemist.com/ccimg/5300/5279-62-9_b.png)











![Benzenesulfonic acid,4-[2-[4-(dimethylamino)phenyl]diazenyl]-](http://img.cochemist.com/ccimg/600/502-02-3.png)
![Benzenesulfonic acid,4-[2-[4-(dimethylamino)phenyl]diazenyl]-](http://img.cochemist.com/ccimg/600/502-02-3_b.png)


















![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8.png)
![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8_b.png)





![Tungstate(3-),tetracosa-m-oxododecaoxo[m12-[phosphato(3-)-kO:kO:kO:kO':kO':kO':kO'':kO'':kO'':kO''':kO''':kO''']]dodeca-,hydrogen (1:3)](http://img.cochemist.com/ccimg/1400/1343-93-7.png)
![Tungstate(3-),tetracosa-m-oxododecaoxo[m12-[phosphato(3-)-kO:kO:kO:kO':kO':kO':kO'':kO'':kO'':kO''':kO''':kO''']]dodeca-,hydrogen (1:3)](http://img.cochemist.com/ccimg/1400/1343-93-7_b.png)
![Benzene, [(phenylmethyl)sulfinyl]-](/data/chemimg/1595300/833-82-9.png)
![Benzene, [(phenylmethyl)sulfinyl]-](/data/chemimg/1595300/833-82-9_b.png)