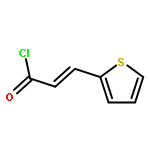
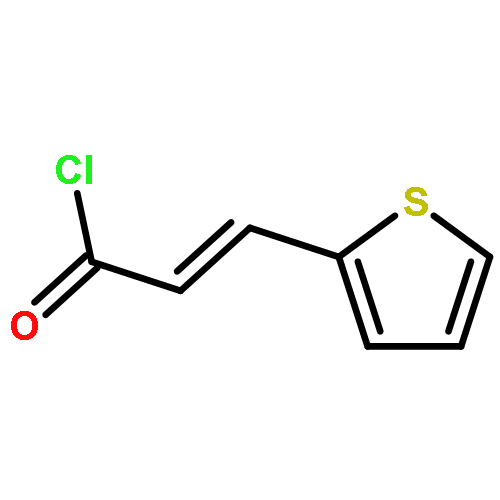
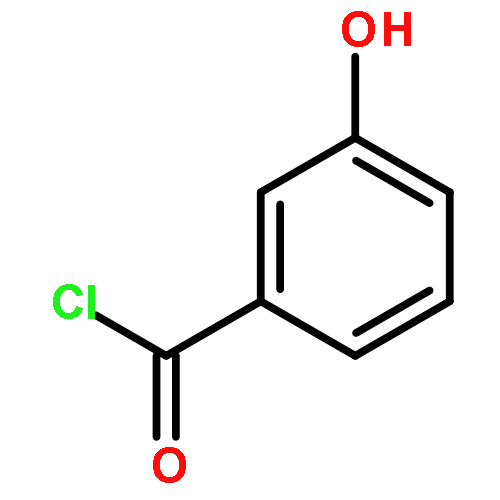
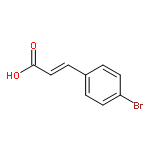
Co-reporter:Mi Yan, Ruixin Ma, Li Jia, Henrietta Venter, Shutao Ma
European Journal of Medicinal Chemistry 2017 Volume 127(Volume 127) pp:
Publication Date(Web):15 February 2017
DOI:10.1016/j.ejmech.2016.10.065
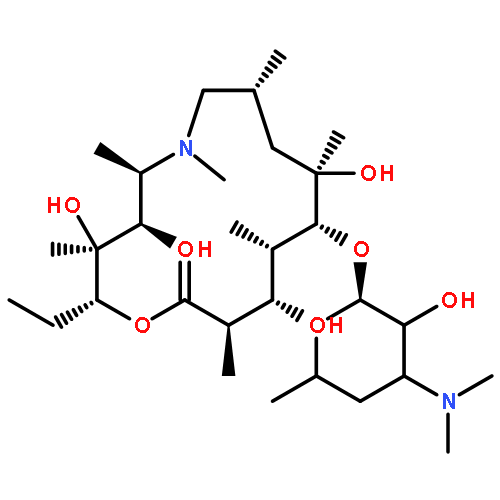
•3-O-Descladinosylazithromycin derivative were synthesized and evaluated.•They had moderate to favorable activity against susceptible bacterial strains.•They also showed greatly improved activity against resistant bacterial strains.•The inspirations and the future direction of ideal candidates are highlighted.Novel series of novel 3-O-arylalkylcarbamoyl descladinosylazithromycin derivatives with the 2′-O-acetyl and 11,12-cyclic carbonate groups, the 11,12-cyclic carbonate group and the 11-O-arylalkylcarbamoyl side chain, and 2′-O-arylalkylcarbamoyl descladinosylazithromycin with the 11,12-cyclic carbonate group were designed, synthesized and evaluated for their antibacterial activity using broth microdilution method. The results showed that the majority of the target compounds showed moderate to favorable activity against six kinds of susceptible strains and almost all of them displayed significantly improved activity compared with references against three erythromycin-resistant strains of S. pneumoniae B1 expressing the ermB gene, S. pneumoniae AB11 expressing the ermB and mefA genes, and S. pyogenes R1. In particular, compound 6h exhibited the most potent activity against susceptible B. subtilis ATCC9372 (0.5 μg/mL), penicillin-resistant S. epidermidis (0.125 μg/mL), and erythromycin-resistant S. pneumoniae B1 (1 μg/mL) and S. pneumoniae AB11 (1 μg/mL), which were 2-, 2-, 256-, 256-fold better activity than azithromycin, respectively. Additionally, compounds 6f (0.5 μg/mL) and 6g (0.25 μg/mL) were the most active against S. pneumoniae A22072, which were 8- and 16-fold better activity than azithromycin (4 μg/mL). As for erythromycin-resistant S. pyogenes R1, compound 5a presented the most excellent activity (8 μg/mL), showing 32- and 32-fold higher activity than azithromycin (256 μg/mL) and clarithromycin (256 μg/mL).11,3-Di-O-arylalkylcarbamoyl descladinosylazithromycin derivatives exhibited excellent in vitro antibacterial activity against Gram-positive and -negative bacteria, especially drug-resistant bacteria.Download high-res image (106KB)Download full-size image
Co-reporter:Yinhu Wang, Chao Cong, Wern Chern Chai, Ruiqian Dong, Li Jia, Di Song, Ziteng Zhou, Shutao Ma
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 16(Issue 16) pp:
Publication Date(Web):15 August 2017
DOI:10.1016/j.bmcl.2017.06.044
•1,2,3-Triazol analogs of azithromycin were designed and synthesized.•These analogs were evaluated for their in vitro antibacterial activity.•They exhibited excellent activity against susceptible and -resistant strains.•Compounds 9g, 9h, 9j and 9k exerted potent and broad spectrum antibacterial activity.Three novel structural series of 4″-O-(1-aralkyl-1,2,3-triazol-4-methyl-carbamoyl) azithromycin analogs were designed, synthesized and evaluated for their in vitro antibacterial activity. All the target compounds exhibited excellent activity against erythromycin-susceptible Streptococcus pyogenes, and significantly improved activity against three phenotypes of erythromycin-resistant Streptococcus pneumoniae compared with clarithromycin and azithromycin. Among the three series of azithromycin analogs, the novel series of 11,4″-disubstituted azithromycin analogs 9a–k exhibited the most effective and balanced activity against susceptible and resistant bacteria. Among them, compound 9j showed the most potent activity against Staphylococcus aureus ATCC25923 (0.008 µg/mL) and Streptococcus pyogenes R2 (1 µg/mL). Besides, all the 11,4″-disubstituted azithromycin analogs 9a–k except 9f shared the identical activity with the MIC value <0.002 µg/mL against Streptococcus pyogenes S2. Furthermore, compounds 9g, 9h, 9j and 9k displayed significantly improved activity compared with the references against all the three phenotypes of resistant S. pneumoniae. Particularly, compound 9k was the most effective (0.06, 0.03 and 0.125 µg/mL) against all the erythromycin-resistant S. pneumoniae expressing the erm gene, the mef gene and the erm and mef genes, exhibiting 2133, 133 and 2048-fold more potent activity than azithromycin, respectively.The novel 4″-O-(1-aralkyl-1,2,3-triazol-4-methyl-carbamoyl) azithromycin analogs exhibited excellent in vitro antibacterial activity against various susceptible and resistant pathogens.Download high-res image (50KB)Download full-size image
Co-reporter:Li Jia, Mi Yan, Yan Shen, Yinhui Qin, Shengsheng Qiang, Shutao Ma
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 16(Issue 16) pp:
Publication Date(Web):15 August 2017
DOI:10.1016/j.bmcl.2017.07.017
•11-O-carbamoyl clarithromycin ketolides were designed and synthesized.•These derivatives were evaluated for their in vitro antibacterial activity.•They exhibited excellent activity against susceptible and resistant strains.•Some compounds exerted potent and broad-spectrum antibacterial activity.A series of novel 11-O-carbamoyl clarithromycin ketolides were designed, synthesized and evaluated for their in vitro antibacterial activity. The results showed that the majority of the target compounds displayed improved activity compared with references against erythromycin-resistant S. pneumoniae A22072 expressing the mef gene, S. pneumoniae B1 expressing the erm gene and S. pneumoniae AB11 expressing the mef and erm genes. In particular, compounds 9, 18, 19 and 22 showed the most potent activity against erythromycin-resistant S. pneumoniae A22072 with the MIC values of 0.5 μg/mL. Furthermore, compounds 11, 18, 19, 24 and 29 were also found to exhibit favorable antibacterial activity against erythromycin-susceptible S. pyogenes with the MIC values of 0.125–1 μg/mL, and moderate activity against erythromycin-susceptible S. aureus ATCC25923 and B. subtilis ATCC9372.A series of novel 11-O-aralkylcarbamoyl-3-O-descladinosyl-3-oxo-clarithromycin derivatives were designed, synthesized and evaluated for their in vitro antibacterial activity. Some compounds exerted potent and broad-spectrum antibacterial activity.Download high-res image (173KB)Download full-size image
Co-reporter:Chaobin Jin
MedChemComm (2010-Present) 2017 vol. 8(Issue 3) pp:501-515
Publication Date(Web):2017/03/23
DOI:10.1039/C6MD00612D
Metabotropic glutamate receptors (mGlu receptors) belong to the G-protein-coupled receptors superfamily. They are divided into three groups, in which group II and group III belong to presynaptic receptors that negatively modulate glutamate and γ-aminobutyric acid (GABA) release when activated. In this review, we introduce not only the functions of mGlu receptors, but also the group II and group III allosteric modulators and agonists/antagonists reported over the past five years according to a classification of their structures, with a specific focus on their biological activity and selectivity. In particular, the structure of these compounds and the future directions of ideal candidates are highlighted.
Co-reporter:Fang Liu, Henrietta Venter, Fangchao Bi, Susan J. Semple, Jingru Liu, Chaobin Jin, Shutao Ma
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 15(Issue 15) pp:
Publication Date(Web):1 August 2017
DOI:10.1016/j.bmcl.2017.06.005
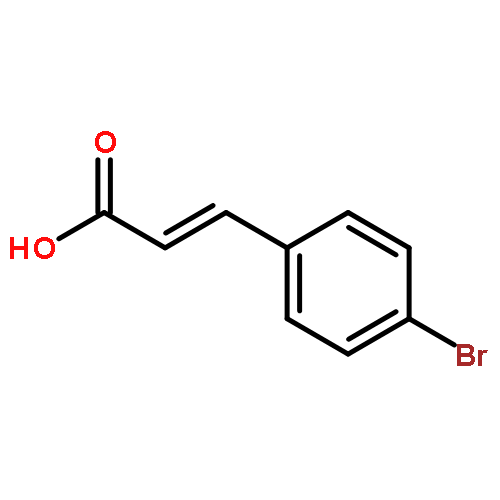
•5-Methylphenanthridium derivatives were designed and synthesized.•They were tested for their antibacterial and cell division inhibitory activities.•Compounds 5A2, 5B1, 5B2, 5B3, 5C1 and 5C2 exerted the best on-target activity.5-Methylphenanthridium derivatives were designed, synthesized and evaluated for their in vitro antibacterial activity and cell division inhibitory activity against various Gram-positive and -negative bacteria. Among them, compounds 5A2, 5B1, 5B2, 5B3, 5C1 and 5C2 displayed the best on-target antibacterial activity with an MIC value of 4 µg/mL against B. subtilis ATCC9372 and S. pyogenes PS, showing over 2-fold better activity than sanguinarine. The SARs showed that the 5-methylphenanthridium derivatives with the alkyl side chains at the 2-postion, especially the straight alkyl side chains exerted better on-target antibacterial activity.Novel 5-methylphenanthridium derivatives were designed, synthesized and evaluated for their in vitro antibacterial activity and cell division inhibitory activity against various Gram-positive and -negative bacteria. Among them, compounds 5A2, 5B1, 5B2, 5B3, 5C1 and 5C2 displayed the best on-target antibacterial activity with an MIC value of 4 µg/mL against B. subtilis ATCC9372 and S. pyogenes PS, showing over 2-fold better activity than sanguinarine.Download high-res image (102KB)Download full-size image
Co-reporter:Li Jia, Shutao Ma
European Journal of Medicinal Chemistry 2016 Volume 121() pp:209-220
Publication Date(Web):4 October 2016
DOI:10.1016/j.ejmech.2016.05.052
•We introduced the structure and function of heparanase.•We summarized recent advances of heparanase inhibitors as novel and potent anti-cancer agents.•We focused on the existing heparanase inhibitors, as well as their inhibitory activities and mechanisms of action.Heparanase, an only endo-β-d-glucuronidase capable of cleaving heparan sulfate (HS) side chains at specific sites, contributes to remodeling of the extracellular matrix (ECM) and releasing of HS-linked growth factors, cytokines and signaling proteins. In addition, heparanase also plays an indispensable role in tumor angiogenesis, invasion and metastasis, indicating that it is a promising target for the development of antitumor drugs. Recent progress leads to three classes of heparanase inhibitors, including active analogs of endogenous substance, synthetic small molecule compounds and natural products. In this review, following an outline on the heparanase structure and function, an overview of the advancement of heparanase inhibitors as novel and potent anti-cancer agents will be given, especially introducing various existing heparanase inhibitors, as well as their inhibitory activities and mechanisms of action.The heparanase has emerged as an attractive target for the development of anti-cancer therapy. This review highlights advances in the discovery of heparanase inhibitors.
Co-reporter:Shengsheng Qiang;Changde Wang;Henrietta Venter;Xin Li;Yi Wang;Liwei Guo;Ruixin Ma
Chemical Biology & Drug Design 2016 Volume 87( Issue 2) pp:257-264
Publication Date(Web):
DOI:10.1111/cbdd.12658
Novel series of 3-O-arylalkylbenzamide and 3-O-arylalkyl-2,6-difluorobenzamide derivatives were synthesized and evaluated for their on-target activity and antibacterial activity. The results indicated that the 3-O-arylalkyl-2,6-difluorobenzamide derivatives possessed much better on-target activity and antibacterial activity than the 3-O-arylalkylbenzamide derivatives. Among them, 3-O-chlorobenzyl derivative 36 was the most effective in antibacterial activity (0.5, 4, and 8 μg/mL) against Bacillus subtilis ATCC9372, methicillin-resistant Staphylococcus aureus ATCC29213, and penicillin-resistant Staphylococcus aureus PR, while 3-O-methylbenzyl derivative 41 only exhibited the most potent activity (2 μg/mL) against Staphylococcus aureus ATCC25923.
Co-reporter:Di Song ; Shutao Ma
ChemMedChem 2016 Volume 11( Issue 7) pp:646-659
Publication Date(Web):
DOI:10.1002/cmdc.201600041
Abstract
Clinically significant antibiotic resistance is one of the greatest challenges of the twenty-first century. However, new antibacterial agents are currently being developed at a much slower pace than our growing need for such drugs. Given their diverse biological activities and clinical applications, many bioactive heterocyclic compounds containing a benzimidazole nucleus have been the focus of interest for many researchers. The benzimidazole nucleus is a structural isostere of naturally occurring nucleotides. This advantage allows benzimidazoles to readily interact with the various biopolymers found in living systems. In view of this situation, much attention has been given to the exploration of benzimidazole-based antibacterial agents, leading to the discovery of many new chemical entities with intriguing profiles. In this minireview we summarize novel benzimidazole derivatives active against various bacterial strains. In particular, we outline the relationship between the structures of variously modified benzimidazoles and their antibacterial activity.
Co-reporter:Xin Li, Shutao Ma
European Journal of Medicinal Chemistry 2015 Volume 95() pp:1-15
Publication Date(Web):5 May 2015
DOI:10.1016/j.ejmech.2015.03.026
•We introduced the function and dynamic behaviors of FtsZ protein.•We reviewed action mechanism and biological activity of the known FtsZ inhibitors.•We described availability and structure-activity relationship of FtsZ inhibitors.•The inspirations and the future direction of ideal candidates are highlighted.Currently, wide-spread antimicrobials resistance among bacterial pathogens continues being a dramatically increasing and serious threat to public health, and thus there is a pressing need to develop new antimicrobials to keep pace with the bacterial resistance. Filamentous temperature-sensitive protein Z (FtsZ), a prokaryotic cytoskeleton protein, plays an important role in bacterial cell division. It as a very new and promising target, garners special attention in the antibacterial research in the recent years. This review describes not only the function and dynamic behaviors of FtsZ, but also the known natural and synthetic inhibitors of FtsZ. In particular, the small molecules recently developed and the future directions of ideal candidates are highlighted.FtsZ as a very new and promising target, garners special attention in the antibacterial research in the recent years. This review describes not only the function and dynamic behaviors of FtsZ, but also its known natural and synthetic inhibitors.
Co-reporter:Xin Li, Juzheng Sheng, Guihua Huang, Ruixin Ma, Fengxin Yin, Di Song, Can Zhao, Shutao Ma
European Journal of Medicinal Chemistry 2015 Volume 97() pp:32-41
Publication Date(Web):5 June 2015
DOI:10.1016/j.ejmech.2015.04.048
•Novel cinnamaldehyde derivatives as FtsZ inhibitors were designed, synthesized and evaluated.•These derivatives had excellent activity against S. aureus strains, especially S. aureus ATCC25923.•They also showed greatly improved activity against all the tested strains compared with their precursor.•The best compounds bearing 2-methylbenzimidazolyl moiety had have chlorine atom(s) at the 4- or/and 2-position of the benzene ring.In an attempt to discover potential antibacterial agents against the increasing bacterial resistance, novel cinnamaldehyde derivatives as FtsZ inhibitors were designed, synthesized and evaluated for their antibacterial activity against nine significant pathogens using broth microdilution method, and their cell division inhibitory activity against four representative strains. In the in vitro antibacterial activity, the newly synthesized compounds generally displayed better efficacy against Staphylococcus aureus ATCC25923 than the others. In particular, compounds 3, 8 and 10 exerted superior or comparable activity to all the reference drugs. In the cell division inhibitory activity, all the compounds showed the same trend as their in vitro antibacterial activity, exhibiting better activity against S. aureus ATCC25923 than the other strains. Additionally, compounds 3, 6, 7 and 8 displayed potent cell division inhibitory activity with an MIC value of below 1 μg/mL, over 256-fold better than all the reference drugs.The cinnamaldehyde derivatives, especially compound 10, exhibited potent FtsZ-targeted antibacterial activity with an MIC value of 4 μg/mL against Staphylococcus aureus and Staphylococcus epidermidis.
Co-reporter:Mi Yan, Xiaodong Ma, Ruiqian Dong, Xin Li, Can Zhao, Zhenzhen Guo, Yan Shen, Fang Liu, Ruixin Ma, Shutao Ma
European Journal of Medicinal Chemistry 2015 Volume 103() pp:506-515
Publication Date(Web):20 October 2015
DOI:10.1016/j.ejmech.2015.09.020
•4″-O-(trans-β-arylacrylamido)carbamoyl analogs were synthesized and evaluated.•They had potent activity against susceptible bacterial strains.•They also showed greatly improved activity against resistant bacterial strains.•The inspirations and the future direction of ideal candidates are highlighted.Novel 4″-O-(trans-β-arylacrylamido)carbamoyl azithromycin analogs were designed, synthesized and evaluated for their antibacterial activity against nine significant pathogens using broth microdilution method. A majority of these derivatives maintained the activity of azithromycin against susceptible Streptococcus pyogenes and all the compounds demonstrated remarkably improved activity compared with the references against all the three phenotypes of resistant Streptococcus pneumoniae. In particular, compound 24 exhibited the most potent activity against susceptible Staphylococcus aureus (MIC = 0.5 μg/mL), S. pneumoniae (MIC = 0.06 μg/mL) and S. pyogenes (MIC = 0.25 μg/mL). The most active compound 7 (MIC = 0.015 μg/mL) against resistant S. pneumoniae expressing the mefA gene, exhibited 512 and 256-fold more potent activity than erythromycin and azithromycin, respectively. Compounds 28 (MIC = 0.5 μg/mL), 29 (MIC = 0.25 μg/mL) and 30 (MIC = 0.5 μg/mL) demonstrated potent activity against resistant S. pneumoniae expressing the ermB gene, which were 256, 512 and 256-fold better than the references, respectively.The azithromycin derivatives bearing C-4″ carbamoyl moieties exhibited excellent in vitro antibacterial activity against various pathogens including Gram-positive and -negative bacteria.
Co-reporter:Yi Wang;Mi Yan;Ruixin Ma
Archiv der Pharmazie 2015 Volume 348( Issue 4) pp:266-274
Publication Date(Web):
DOI:10.1002/ardp.201400412
A series of novel 4-bromo-1H-indazole derivatives as filamentous temperature-sensitive protein Z (FtsZ) inhibitors were designed, synthesized, and assayed for their in vitro antibacterial activity against various phenotypes of Gram-positive and Gram-negative bacteria and their cell division inhibitory activity. The results indicated that this series showed better antibacterial activity against Staphylococcus epidermidis and penicillin-susceptible Streptococcus pyogenes than the other tested strains. Among them, compounds 12 and 18 exhibited 256-fold and 256-fold more potent activity than 3-methoxybenzamide (3-MBA) against penicillin-resistant Staphylococcus aureus, and compound 18 showed 64-fold better activity than 3-MBA but 4-fold weaker activity than ciprofloxacin in the inhibition of S. aureus ATCC29213. Particularly, compound 9 presented the best activity (4 µg/mL) against S. pyogenes PS, being 32-fold, 32-fold, and 2-fold more active than 3-MBA, curcumin, and ciprofloxacin, respectively, but it was four times less active than oxacillin sodium. In addition, some synthesized compounds displayed moderate inhibition of cell division against S. aureus ATCC25923, Escherichia coli ATCC25922, and Pseudomonas aeruginosa ATCC27853, sharing a minimum cell division concentration of 128 µg/mL.
Co-reporter:Can Zhao ; Shutao Ma
ChemMedChem 2014 Volume 9( Issue 11) pp:2425-2437
Publication Date(Web):
DOI:10.1002/cmdc.201402174
Abstract
N-Myristoyltransferase (NMT) is a cytosolic monomeric enzyme present in eukaryotes such as fungi and protozoa, but is not found in prokaryotes. The attachment of a 14-carbon saturated fatty acid, myristate, from myristoyl-CoA (14:0 CoA) to the N-terminal glycine residue in a specific set of cellular proteins is commonly called protein N-myristoylation. The myristoylation reaction catalyzed by the enzyme myristoyl CoA:NMT is both necessary for the growth of various organisms and conclusive for cellular proliferation. Therefore, NMT has been identified as a novel and promising target for antifungal, antiparasitic, and anticancer agents, and a large number of potent NMT inhibitors with antifungal, antiparasitic, and anticancer activities have been reported. Herein we describe recent advances in the discovery of NMT inhibitors. We introduce not only the functions of NMT, but also some representative natural and synthetic inhibitors, with a focus on their biological activity, selectivity, and structure–activity relationship (SAR) information. In particular, inspiration from NMT inhibitor structures and the future direction of these compounds are highlighted.
Co-reporter:Siti Ma, Chao Cong, Xiaohui Meng, Shasha Cao, Hongkun Yang, Yuanyuan Guo, Xueyi Lu, Shutao Ma
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 14) pp:4076-4079
Publication Date(Web):15 July 2013
DOI:10.1016/j.bmcl.2013.05.056
Novel 3-elongated arylalkoxybenzamide derivatives were designed, synthesized and evaluated for their cell division inhibitory activity and antibacterial activity. Among them, the subseries of 3-alkyloxybenzamide derivatives exhibited greatly improved on-target activity against Bacillus subtilis and Staphylococcus aureus, and remarkably increased antibacterial activity against B. subtilis ATCC9372, penicillin-susceptible S. aureus ATCC25923, methicillin-resistant S. aureus ATCC29213 (MRSA) and penicillin-resistant S. aureus PR compared with 3-methoxybenzamide. In contrast, the subseries of 3-phenoxyaklyloxybenzamide, 3-heteroarylalkyloxybenzamide and 3-heteroarylthioalkyloxybenzamide derivatives only showed a significant improvement in on-target activity and antibacterial activity against B. subtilis ATCC9372.
Co-reporter:Yi Wang ; Shutao Ma
ChemMedChem 2013 Volume 8( Issue 10) pp:1589-1608
Publication Date(Web):
DOI:10.1002/cmdc.201300209
Abstract
Bacterial infections are a constant and serious threat to human health. With the increase of multidrug resistance of clinically pathogenic bacteria, common antibiotic therapies have been less effective. Fatty acid synthesis type II (FASII) system enzymes are essential for bacterial membrane lipid biosynthesis and represent increasingly promising targets for the discovery of antibacterial agents with new mechanisms of action. This review highlights recent advances in inhibitors of bacterial FASII as potential antibacterial agents, paying special attention to the activities, mechanisms, and structure–activity relationships of those inhibitors that mainly target β-ketoacyl-ACP synthase, β-ketoacyl-ACP reductase, β-hydroxyacyl-ACP dehydratase, and enoyl-ACP reductase. Although inhibitors with low nanomolar and selective activity against various bacterial FASII have entered clinical trials, further research is needed to expand upon both available and yet unknown scaffolds to identify new FASII inhibitors that may have antibacterial potential, particularly against resistant bacterial strains.
Co-reporter:Siti Ma ; Shutao Ma
ChemMedChem 2012 Volume 7( Issue 7) pp:1161-1172
Publication Date(Web):
DOI:10.1002/cmdc.201200156
Abstract
The emergence and prevalence of bacterial resistance has resulted in a clear demand for novel antibacterial drugs. As a tubulin homologue, FtsZ is an essential cell-division protein in prokaryotic organisms and is showing increasing promise as a target for antibacterial drug discovery. This review describes the role of FtsZ in bacterial cytokinesis and various FtsZ inhibitors, with particular focus on their discovery, antibacterial activities, mechanisms of action, synthetic methods, and representative analogues.
Co-reporter:Mi Yan ; Shutao Ma
ChemMedChem 2012 Volume 7( Issue 12) pp:2063-2075
Publication Date(Web):
DOI:10.1002/cmdc.201200339
Abstract
Tuberculosis (TB) is a major health problem, with approximately one-third of the world′s population infected with Mycobacterium tuberculosis, eight million people in the active disease state, and two million dying annually. Furthermore, the prevalence of TB/HIV co-infection, and the emergence of multidrug-resistant tuberculosis (MDR-TB) and extensively drug-resistant tuberculosis (XDR-TB) have further aggravated the spread of this disease and thus mortality by it. There is an urgent need for novel antitubercular agents with improved properties, such as lower toxicity, shortened duration of therapy, rapid bactericidal action, and enhanced activity against MDR strains. Fortunately, a number of new potential antitubercular candidate drugs with heterocyclic rings, which are most likely to be effective against resistant strains, have entered clinical trials in recent years. This review highlights recent advances in the research of novel heterocyclic compounds, with particular focus on their antimycobacterial activity, mechanisms of action, toxicity, and structure–activity relationships (SARs).
Co-reporter:Chao Cong, Haiyang Wang, Yue Hu, Chen Liu, Siti Ma, Xin Li, Jichao Cao, Shutao Ma
European Journal of Medicinal Chemistry 2011 Volume 46(Issue 7) pp:3105-3111
Publication Date(Web):July 2011
DOI:10.1016/j.ejmech.2011.04.004
Novel 4″-O-benzimidazolyl clarithromycin derivatives were designed, synthesized and evaluated for their in vitro antibacterial activities. These benzimidazolyl derivatives exhibited excellent activity against erythromycin-susceptible strains better than the references, and some of them showed greatly improved activity against erythromycin-resistant strains. Compounds 16 and 17, which have the terminal 2-(4-methylphenyl)benzimidazolyl and 2-(2-methoxyphenyl)benzimidazolyl groups on the C-4″ bishydrazide side chains, were the most active against erythromycin-resistant Staphylococcus pneumoniae expressing the erm gene and the mef gene. In addition, compound 17 exhibited the highest activity against erythromycin-susceptible S. pneumoniae ATCC49619 and Staphylococcus aureus ATCC25923 as well. It is worth noting that the 4″-O-(2-aryl)benzimidazolyl derivatives show higher activity against erythromycin-susceptible and erythromycin-resistant strains than the 4″-O-(2-alkyl)benzimidazolyl derivatives.Novel 4″-O-benzimidazolyl clarithromycin derivatives were designed, synthesized and evaluated for their in vitro antibacterial activities. Compound 17 showed the highest activity against the erythromycin-susceptible and the erythromycin-resistant strains, much better than the references.Highlights►Novel 4″-O-benzimidazolyl clarithromycin derivatives were synthesized and evaluated. ►The derivatives showed excellent activity against erythromycin-susceptible strains. ►Some of them showed greatly improved activity against erythromycin-resistant strains. ►4″-O-(2-aryl)benzimidazolyl derivatives had higher activity than other derivatives. ►The other derivatives are the 4″-O-(2-alkyl)benzimidazolyl derivatives of CAM.
Co-reporter:Shutao Ma, Bo Jiao, Yongjing Ju, Manjie Zheng, Ruixin Ma, Lin Liu, Ling Zhang, Xuecui Shen, Chenchen Ma, Ya Meng, Hui Wang, Yunkun Qi, Xiaodong Ma, Wenping Cui
European Journal of Medicinal Chemistry 2011 Volume 46(Issue 2) pp:556-566
Publication Date(Web):February 2011
DOI:10.1016/j.ejmech.2010.11.035
Novel clarithromycin derivatives with C-4″ elongated arylalkyl groups were designed, synthesized and evaluated to probe the effect of different lengths of their C-4″ side chains on the activity against resistant bacterial strains. These derivatives had excellent activity against erythromycin-susceptible Streptococcus pneumoniae, Streptococcus aureus or Streptococcus pyogenes and some of them exhibited greatly improved activity against erythromycin-resistant strains. Compounds 18 and 16, which had the C-4″ elongated arylalkyl groups with eight atoms from the 4″-oxygen atom to the terminal benzene ring, were the most effective against S. pneumoniae expressing the erm gene and the erm and mef genes. In contrast, the most potent compounds 3, 5, 9, 17 and 18 against S. pneumoniae expressing the mef gene had C-4″ elongated arylalkyl groups with three to eight atoms between the 4″-oxygen atom and the terminal aromatic ring.33 novel clarithromycin derivatives with elongated C-4″ side chains were synthesized and evaluated to probe the effect of different lengths of their C-4″ side chains on activity against resistant bacterial strains.Research highlights► Novel clarithromycin derivatives with elongated C-42 side chains were synthesized. ► Their antibacterial activity was evaluated.► The derivatives had excellent activity against erythromycin-susceptible strains. ► Some of them showed greatly improved activity against erythromycin-resistant strains. ► The lengths of their C-42 side chains affected activity against resistant bacteria.
Co-reporter:Xiaodong Ma, Ling Zhang, Rongmei Wang, Jichao Cao, Chen Liu, Yi Fang, Jida Wang, Shutao Ma
European Journal of Medicinal Chemistry 2011 Volume 46(Issue 10) pp:5196-5205
Publication Date(Web):October 2011
DOI:10.1016/j.ejmech.2011.08.001
Three novel structural series of C-4″ modified azithromycin analogs with two amide groups, which were connected by different alkyl linkage, were designed, prepared and evaluated for their in vitro antibacterial activity against seven phenotypes of respiratory pathogens. Among them, 7d, 8j and 9j, as representatives of corresponding series, exhibited remarkably improved activity against erythromycin-resistant Streptococcus pneumoniae expressing the erm gene, the mef gene, and the erm and mef genes. In addition, 7a–c, 7f–h, 7j, 8d, 8g, 8i, 9a–b and 9i displayed favorable efficacy against erythromycin-resistant S. pneumoniaeA22072 expressing the mef gene.Novel structural series of C-4″ bisamide azithromycin analogs were designed, prepared and evaluated in vitro. Compound 9j, as the representative derivative, exhibited remarkably improved antibacterial activity against erythromycin-resistant Streptococcus pneumoniae than the references.Highlights► Three series of C-4″ bisamide azithromycin analogs were synthesized and evaluated. ► They have two amide groups connected by different linkage in the C-4″ side chain. ► Some of them showed greatly improved activity against erythromycin-resistant strains. ► Their resistance is encoded by the erm gene, the mef gene, and the erm and mef genes.
Co-reporter:Ling Zhang, Bo Jiao, Xiangrui Yang, Lin Liu and Shutao Ma
The Journal of Antibiotics 2011 64(3) pp:243-247
Publication Date(Web):January 19, 2011
DOI:10.1038/ja.2010.166
A series of new 4″-O-carbamates of 11,12-cyclic carbonate erythromycin A 6,9-imino ether were synthesized and evaluated for their in vitro antibacterial activity. All the desired compounds demonstrated favorable activity (0.03 μg ml–1) against erythromycin-susceptible Streptococcus pneumoniae comparable to the references, exhibiting 133-fold higher activity than precursor 2 or 3. Similarly, all of the analogs exhibited improved activity against the erythromycin-resistant S. pneumoniae encoded by the erm gene and the erm and mef genes, showing 4–32-fold more effectiveness than erythromycin A.
Co-reporter:Yunkun Qi ; Shutao Ma
ChemMedChem 2011 Volume 6( Issue 3) pp:399-409
Publication Date(Web):
DOI:10.1002/cmdc.201000534
Abstract
Marine natural products have become a major source of new chemical entities in the discovery of potential anticancer agents that potently suppress various molecular targets. In particular, the marine macrolides, which include an array of novel biomolecules endowed with outstanding cytotoxic and/or antiproliferative activities, are a prominent class of marine natural products that offer continued promise for breakthroughs in anticancer research. Herein we highlight some recent studies of promising marine macrolides, paying particular attention to their discovery, anticancer activities, mechanisms of action, chemical synthesis, and representative analogues.
Co-reporter:Ling Zhang, Linchen Song, Zhaopeng Liu, Hui Li, Yingdong Lu, Zerong Li, Shutao Ma
European Journal of Medicinal Chemistry 2010 Volume 45(Issue 3) pp:915-922
Publication Date(Web):March 2010
DOI:10.1016/j.ejmech.2009.11.032
Two series of novel 3-O-carbamoyl derivatives of clarithromycin and 11,12-cyclic carbonate azithromycin were designed, synthesized and evaluated for their in vitro antibacterial activities. Compounds 4j and 4k were the most potent activity against erythromycin-susceptible Staphylococcus aureus, Streptococcus pyogenes and Streptococcus pneumoniae, which were comparable to those of clarithromycin and azithromycin. Compounds 4d, 4h and 4i showed potent activity against erythromycin-resistant S. pneumoniae encoded by the mef gene and compounds 4h and 4i displayed greatly improved activity against erythromycin-resistant S. pneumoniae encoded by the erm gene. Compound 7c exhibited improved activity against erythromycin-resistant S. pneumoniae encoded by the erm and mef genes.Novel 3-O-carbamoyl derivatives of clarithromycin and 11,12-cyclic carbonate azithromycin were synthesized and evaluated. Some derivatives showed excellent activity against erythromycin-susceptible bacteria and greatly improved activity against erythromycin-resistant Streptococcus pneumoniae.
Co-reporter:Yunkun Qi;Bo Jiao;Xiaodong Ma;Wenping Cui
Archiv der Pharmazie 2010 Volume 343( Issue 8) pp:458-464
Publication Date(Web):
DOI:10.1002/ardp.200900288
Abstract
Novel 4″-O-carbamoyl erythromycin-A derivatives were designed, synthesized, and evaluated for their in-vitro antibacterial activities. All of the 4″-O-carbamoyl derivatives showed excellent activity against erythromycin-susceptible Staphylococcus aureus ATCC25923, Streptococcus pyogenes, and Streptococcus pneumoniae ATCC49619. Most of the 4″-O-arylalkylcarbamoyl derivatives displayed potent activity against erythromycin-resistant S. pneumoniae encoded by the mef gene and greatly improved activity against erythromycin-resistant S. pneumoniae encoded by the erm gene or the erm and mef genes. In particular, the 4″-O-arylalkyl derivatives 4c–4e and 4g were found to possess the most potent activity against all the tested erythromycin-susceptible strains, which were comparable to those of erythromycin, clarithromycin, or azithromycin. 4″-O-Arylalkyl derivatives 4e and 4g were the most effective against erythromycin-resistant S. pneumoniae encoded by the mef gene (0.25 and 0.25 µg/mL). 4″-O-Arylalkyl derivatives 4a and 4b exhibited significantly improved activity against erythromycin-resistant S. pneumoniae encoded by the erm gene. In contrast, the 4″-O-alkylcarbamoyl derivatives hardly showed improved activity against all the tested erythromycin-resistant strains.
Co-reporter:Xue Cui Shen, Bo Jiao, Shu Tao Ma
Chinese Chemical Letters 2010 Volume 21(Issue 3) pp:257-260
Publication Date(Web):March 2010
DOI:10.1016/j.cclet.2009.11.023
A series of novel 4″-O-carbamoyl analogs of clarithromycin were synthesized and evaluated for their in vitro antibacterial activity. All of the desired compounds showed excellent activity against erythromycin-susceptible S. pneumoniae. Particularly, 4-fluorobenzyl carbamate 7a demonstrated potent activity against erythromycin-resistant S. pneumoniae encoded by the mef gene, and remarkably improved activity against erythromycin-resistant S. pneumoniae encoded by the erm gene, and the erm and mef genes.
Co-reporter:Yongjing Ju, Ruiqing Xian, Ling Zhang, Ruixin Ma, Jichao Cao, Shutao Ma
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 11) pp:3272-3274
Publication Date(Web):1 June 2010
DOI:10.1016/j.bmcl.2010.04.051
Novel series of novel 4″-O-arylalkylcarbamoyl and 4″-O-((arylalkylamino)-4-oxo-butyl)carbamoyl clarithromycin derivatives were designed, synthesized and evaluated for their in vitro antibacterial activities. These derivatives retained excellent activity against the erythromycin-susceptible strains and showed significantly improved activity against all of the tested erythromycin-resistant strains. Among them, compound 4c was the most effective (0.06 μg/mL) against Streptococcus pneumonia encoded by the erm gene and compound 4a was had the most potent activity (0.25 μg/mL) against S. pneumonia encoded by the erm and mef genes.Novel 4″-O-arylalkylcarbamoyl and 4″-O-((arylalkylamino)-4-oxo-butyl)carbamoyl clarithromycin derivatives were designed, synthesized and evaluated for their in vitro antibacterial activities. Among them, compound 4c was the most effective against Streptococcus pneumoniae encoded by the erm gene.
Co-reporter:Chenchen Ma, Zhaopeng Liu, Hualong Song, Rentao Jiang, Fawen He and Shutao Ma
The Journal of Antibiotics 2010 63(1) pp:3-8
Publication Date(Web):November 13, 2009
DOI:10.1038/ja.2009.108
A series of novel 11,12-cyclic carbonate azithromycin 4″-O-carbamate derivatives were designed, synthesized and evaluated for their in vitro antibacterial activities. Compounds 7b and 7d were the most effective (0.5 and 0.5 μg ml−1) against two strains of erythromycin-resistant Streptococcus pneumoniae whose resistance was encoded by the erm gene and the erm and mef genes, respectively. Compounds 7a, 7e and 7g showed significantly potent activity against erythromycin-susceptible strains such as Staphylococcus aureus and S. pyogenes. These results suggest that the introduction of the prolonged arylalkylcarbamoyl group to the C-4″ position can dramatically enhance the activity against erythromycin-resistant bacteria encoded by the erm gene or the erm and mef genes.
Co-reporter:Ling Zhang
ChemMedChem 2010 Volume 5( Issue 6) pp:811-822
Publication Date(Web):
DOI:10.1002/cmdc.201000006
Abstract
Multidrug resistance (MDR) is the cause of an ever-increasing number of problems in the treatment of cancers and bacterial infections. The active efflux of drugs contributes significantly to this phenomenon. This minireview summarizes recent advances in combating MDR, with particular emphasis on natural and synthetic efflux pump inhibitors of P-glycoprotein in resistant tumor cells and of the NorA MDR pump in Staphylococcus aureus.
Co-reporter:Shutao Ma, Ruixin Ma, Zhaopeng Liu, Chenchen Ma, Xuecui Shen
European Journal of Medicinal Chemistry 2009 Volume 44(Issue 10) pp:4010-4020
Publication Date(Web):October 2009
DOI:10.1016/j.ejmech.2009.04.030
4″-Carbamate, 11,12-cyclic carbonate-4″-carbamate and 11,4″-di-O-arylcarbamoyl analogs of azithromycin were designed, synthesized and evaluated. The 4″-carbamate analogs retained excellent activity against erythromycin-susceptible Staphylococcus pneumoniae and showed improved activity against erythromycin-resistant Staphylococcus pneumoniae. Compared with 4″-carbamate analogs, 11,12-cyclic carbonate-4″-carbamate analogs exhibited improved activity against erythromycin-resistant Staphylococcus pneumoniae encoded by the mef gene or the erm and mef genes, and 11,4″-di-O-arylalkylcarbamoyl analogs showed greatly improved activity (0.25–0.5 μg/mL) against erythromycin-resistant Staphylococcus pneumoniae encoded by the erm gene. Among them, the novel series of 11,4″-di-O-arylalkylcarbamoyl analogs 7a–k exhibited potent and balanced activity against susceptible and resistant bacteria. In particular, compounds 7f and 7k were the most effective against susceptible bacteria and resistant bacteria encoded by the erm gene or the mef gene.Three series of 15-membered macrolide analogs were designed, synthesized and evaluated for their antibacterial activities. Among them, 11,4″-di-O-arylalkylcarbamoyl analogs exhibited potent and balanced activity against susceptible and resistant bacteria.
Co-reporter:Shu Tao Ma, Rui Xin Ma, Rui Qing Xian, Bo Jiao
Chinese Chemical Letters 2009 Volume 20(Issue 8) pp:931-934
Publication Date(Web):August 2009
DOI:10.1016/j.cclet.2009.03.015
A series of novel dimers of 15-membered macrolides was synthesized and evaluated. The dimers exhibited excellent activity against erythromycin-susceptible S. pneumonia, but did not show any improved activity against erythromycin-resistant S. pneumoniae encoded by erm gene.
Co-reporter:Shutao Ma, Bo Jiao, Zhaopeng Liu, Hui Wang, Ruiqing Xian, Manjie Zheng, Hongxiang Lou
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 6) pp:1698-1701
Publication Date(Web):15 March 2009
DOI:10.1016/j.bmcl.2009.01.092
A series of new 4″,11-di-O-arylalkylcarbamoyl azithromycin derivatives were designed, synthesized and evaluated for their in vitro antibacterial activities. Some derivatives exhibited greatly improved activity against erythromycin-resistant bacteria. Among them, compounds 5f and 5k were found to have potent activity against erythromycin-resistant Streptococcus pneumoniae whose resistance was encoded by the erm or mef gene.A series of new 4″,11-di-O-arylalkylcarbamoyl azithromycin derivatives were designed, synthesized and evaluated for their in vitro antibacterial activities. Some derivatives exhibited greatly improved activity against erythromycin-resistant Streptococcus pneumoniae encoded by the erm or mef gene.
Co-reporter:Yinhu Wang, Rumana Mowla, Liwei Guo, Abiodun D. Ogunniyi, Taufiq Rahman, Miguel A. De Barros Lopes, Shutao Ma, Henrietta Venter
Bioorganic & Medicinal Chemistry Letters (15 February 2017) Volume 27(Issue 4) pp:
Publication Date(Web):15 February 2017
DOI:10.1016/j.bmcl.2017.01.042
Drug efflux pumps confer multidrug resistance to dangerous pathogens which makes these pumps important drug targets. We have synthesised a novel series of compounds based on a 2-naphthamide pharmacore aimed at inhibiting the efflux pumps from Gram-negative bacteria. The archeatypical transporter AcrB from Escherichia coli was used as model efflux pump as AcrB is widely conserved throughout Gram-negative organisms. The compounds were tested for their antibacterial action, ability to potentiate the action of antibiotics and for their ability to inhibit Nile Red efflux by AcrB. None of the compounds were antimicrobial against E. coli wild type cells. Most of the compounds were able to inhibit Nile Red efflux indicating that they are substrates of the AcrB efflux pump. Three compounds were able to synergise with antibiotics and reverse resistance in the resistant phenotype. Compound A3, 4-(isopentyloxy)-2-naphthamide, reduced the MICs of erythromycin and chloramphenicol to the MIC levels of the drug sensitive strain that lacks an efflux pump. A3 had no effect on the MIC of the non-substrate rifampicin indicating that this compound acts specifically through the AcrB efflux pump. A3 also does not act through non-specific mechanisms such as outer membrane or inner membrane permeabilisation and is not cytotoxic against mammalian cell lines. Therefore, we have designed and synthesised a novel chemical compound with great potential to further optimisation as inhibitor of drug efflux pumps.
Co-reporter:Fangchao Bi, Liwei Guo, Yinhu Wang, Henrietta Venter, Susan J. Semple, Fang Liu, Shutao Ma
Bioorganic & Medicinal Chemistry Letters (15 February 2017) Volume 27(Issue 4) pp:
Publication Date(Web):15 February 2017
DOI:10.1016/j.bmcl.2016.12.081
•Novel 2,6-difluorobenzamide derivatives were synthesized and evaluated.•Some compounds showed potent activity against both susceptible and resistant strains.•They exerted their effects by inhibition of bacterial cell division protein FtsZ.•Replacing the benzothiazole with benzimidazole lost its antibacterial activity.Novel series of 3-substituted 2,6-difluorobenzamide derivatives as FtsZ inhibitors were designed, synthesized and evaluated for their in vitro antibacterial activity against various phenotype of Gram-positive and Gram-negative bacteria, and their cell division inhibitory activity against three representative strains. As a result, 3-chloroalkoxy derivative 7, 3-bromoalkoxy derivative 12 and 3-alkyloxy derivative 17 were found to exhibit the best antibacterial activity against Bacillus subtilis with MICs of 0.25–1 μg/mL, and good activity (MIC < 10 μg/mL) against both susceptible and resistant Staphylococcus aureus. Additionally, all the three compounds displayed potent cell division inhibitory activity with MIC values of below 1 μg/mL against Bacillus subtilis and Staphylococcus aureus.The 2,6-difluorobenzamide derivative (17) bearing 3-O-flexible alkyl side chains exhibited modest in vitro antibacterial and cell division inhibitory activities, while the 3-O-rigid heteroaryl substituted derivative (25) showed weak antibacterial activity although it had very similar structure to previously discovered compound 6g.


















![[5-(HYDROXYMETHYL)-4-METHOXY-3-METHYL-2-PYRIDINYL]METHYL ACETATE](http://img.cochemist.com/ccimg/76300/76228-15-4.png)
![[5-(HYDROXYMETHYL)-4-METHOXY-3-METHYL-2-PYRIDINYL]METHYL ACETATE](http://img.cochemist.com/ccimg/76300/76228-15-4_b.png)