Co-reporter:Dingding Gao, Yingxia Li
Bioorganic & Medicinal Chemistry 2017 Volume 25, Issue 14(Issue 14) pp:
Publication Date(Web):15 July 2017
DOI:10.1016/j.bmc.2017.05.017
Indoleamine 2,3-dioxygenase 1 (IDO1) plays a vital role in the catabolism of tryptophan along with the kynurenine pathway which is involved in many human diseases including cancer, Alzheimer’s disease, etc. In this study, compound 1 bearing a 1-Indanone scaffold was identified as a novel IDO1 inhibitor by structure-based virtual screening, with moderate to good enzymatic and cellular inhibitory activities. Also, surface plasmon resonance analysis validated the direct interaction between compound 1 and IDO1 protein. The preliminary SAR was further explored and the binding mode with IDO1 protein was predicted by experiment along with molecular docking. Subsequent ADME properties of these active compounds were analyzed in silico, and the results showed good pharmacokinetic efficiencies. We believe this study contributes a lot to the structural diversity for the future development of highly potent IDO1 inhibitors.Novel IDO1 inhibitor 1 bearing a 1-Indanone scaffold was discovered via structure-based virtual screening, with enzymatic and cellular IC50 values of 2.780 μM and 9.171 μM respectively. The preliminary SAR was discussed and the binding mode with IDO1 protein was predicted by experiment along with molecular docking.Download high-res image (130KB)Download full-size image
Co-reporter:Jing Cui, Xia Peng, Dingding Gao, Yang Dai, Jing Ai, Yingxia Li
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 16(Issue 16) pp:
Publication Date(Web):15 August 2017
DOI:10.1016/j.bmcl.2017.06.068
Fibroblast growth factor receptor (FGFR) is a potential target for cancer therapy because of its critical role in promoting cancer formation and progression. In a continuing effort to improve the cellular activity of hit compound 7r bearing an indazole scaffold, which was previously discovered by our group, several compounds harnessing fluorine substituents were designed, synthesized and biological evaluated. Besides, the region extended out to the ATP binding pocket toward solvent was also explored. Among them, compound 2a containing 2,6-difluoro-3-methoxyphenyl residue exhibited the most potent activities (FGFR1: less than 4.1 nM, FGFR2: 2.0 ± 0.8 nM). More importantly, compound 2a showed an improved antiproliferative effect against KG1 cell lines and SNU16 cell lines with IC50 values of 25.3 ± 4.6 nM and 77.4 ± 6.2 nM respectively.In a continuing effort to develop more potent cellular activity of hit compound 7r which was previously discovered by our group, several compounds harnessing fluorine substituents were designed, synthesized and biological evaluated. Additionally, the region extended out to the ATP binding pocket toward solvent was also explored.Download high-res image (104KB)Download full-size image
Co-reporter:Hongliang Zhang, Hengtao Wang, Qiulong Xu, Rui Lu, Yunhua Cao, Zhefeng Wang, Pingping Tang, Feng Lin, Yingxia Li
Carbohydrate Research 2017 Volume 448(Volume 448) pp:
Publication Date(Web):7 August 2017
DOI:10.1016/j.carres.2017.05.012
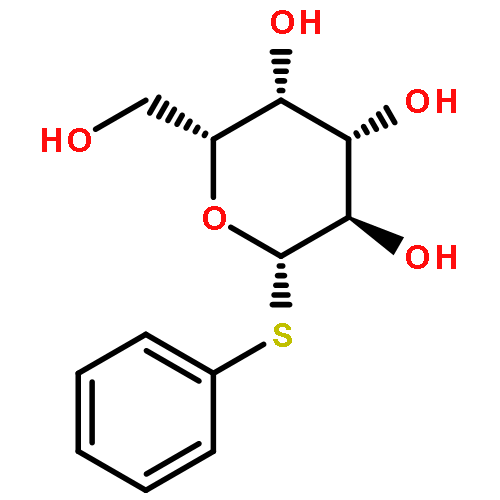
•The first synthesis of β-D-fructopyranosyl hexamer.•N-phenyltrifluoroacetimidate glycosylation was vital for oligomer synthesis.•Provided an interesting method for selective glycosylation of ketose.•Fructopyranosyl oligomer provide new backbone, with promising applications.The hexa-fructopyranoside was synthesized with N-phenyltrifluoroacetimidate glycosylation. The synthesis was achieved by regioselective glycosylation on the 1-OH of fructopyranosyl acceptor. Fructosyl oligosaccharides were elongated with β-(2 → 1)-difructopyranosyl unit in every two steps, without any further protection/deprotection step. This work proved N-phenyltrifluoroacetimidate glycosylation a practical method for oligo-fructopyranoside synthesis.Download high-res image (132KB)Download full-size image
Co-reporter:Dingding Gao, Qiang Xiao, Mingming Zhang, Yingxia Li
Bioorganic & Medicinal Chemistry 2016 Volume 24(Issue 11) pp:2549-2558
Publication Date(Web):1 June 2016
DOI:10.1016/j.bmc.2016.04.022
STAT3 signaling pathway has been validated as a vital therapeutic target for cancer therapy. Based on the novel STAT3 inhibitor of a benzyloxyphenyl-methylaminophenol scaffold hit (1) discovered through virtual screening, a series of analogues had been designed and synthesized for more potent inhibitors. The preliminary SAR had been discussed and the unique binding site in SH2 domain was predicted by molecular docking. Among them, compounds 4a and 4b exhibited superior activities than hit compound (1) against IL-6/STAT3 signaling pathway with IC50 values as low as 7.71 μM and 1.38 μM, respectively. Compound 4a also displayed potent antiproliferative activity against MDA-MB-468 cell line with an IC50 value of 9.61 μM. We believe that these benzyloxyphenyl-methylaminophenol derivatives represent a unique mechanism for interrogating STAT3 as well as a potential structure type for further exploration.A series of novel benzyloxyphenyl-methylaminophenol derivatives were synthesized and evaluated as inhibitors of STAT3 signaling pathway.
Co-reporter:Qiang Xiao, Rong Qu, Dingding Gao, Qi Yan, Linjiang Tong, Wei Zhang, Jian Ding, Hua Xie, Yingxia Li
Bioorganic & Medicinal Chemistry 2016 Volume 24(Issue 12) pp:2673-2680
Publication Date(Web):15 June 2016
DOI:10.1016/j.bmc.2016.04.032
To overcome the drug-resistance of first generation EGFR inhibitors and the nonselective toxicities of second generation inhibitors among NSCLC patients, a series of 5-(methylthio)pyrimidine derivatives were discovered as novel EGFR inhibitors, which harbored not only potent enzymatic and antiproliferative activities against EGFRL858R/T790M mutants, but good selectivity over wide-type form of the receptor. This goal was achieved by employing structure-based drug design and traditional optimization strategies, based on WZ4002 and CO1686. These derivatives inhibited the enzymatic activity of EGFRL858R/T790M mutants with IC50 values in subnanomolar ranges, while exhibiting hundreds of fold less potency on EGFRWT. These compounds also strongly inhibited the proliferation of H1975 non-small cell lung cancer cells bearing EGFRL858R/T790M, while being significantly less toxic to A431 human epithelial carcinoma cells with overexpressed EGFRWT. The EGFR kinase inhibitory and antiproliferative activities were further validated by Western blot analysis for activation of EGFR and the downstream signaling in cancer cells.
Co-reporter:Qi Yan, Ning Ding, Yingxia Li
Carbohydrate Research 2016 Volume 421() pp:1-8
Publication Date(Web):8 February 2016
DOI:10.1016/j.carres.2015.10.011
•Novel dioxa-bicycle C-aryl glucosides were synthesized.•SGLT2 inhibitory activities were evaluated.•The most potent compound showed an IC50 value of 714 nM.A series of novel C-aryl glucosides containing dioxa-bicycle were synthesized and evaluated for inhibition activity against hSGLT2. Among the compounds tested, compound 6a showed moderate SGLT2 inhibition activities at 700 nM. The results could benefit the discovery of new SGLT2 inhibitors.
Co-reporter:Hong Cui, Xia Peng, Jian Liu, Chunhua Ma, Yinchun Ji, Wei Zhang, Meiyu Geng, Yingxia Li
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 18) pp:4483-4486
Publication Date(Web):15 September 2016
DOI:10.1016/j.bmcl.2016.07.077
A series of 2-oxo-1,2-dihydroquinoline-containing c-Met inhibitors were designed, synthesized and evaluated for their in vitro activities targeting c-Met. Most compounds showed high potency against c-Met with IC50 values in the single-digit nM range. Among these compounds, two target compounds, namely 1h and 1n, stood out as the most potent c-Met inhibitors with IC50s of 0.6 and 0.7 nM, respectively. And 1a exhibited higher potency than BMS-777607 did with respect to the inhibition of cell proliferation. The introduction of electron-donating substituent was favorable for the activities of the compounds to some extent. Furthermore, molecular docking studies also gave encouraging results that supported this work.
Co-reporter:Ruwen Yin, Wei Zhang, Guoyun Liu, Ping Wu, Choiwan Lau, Yingxia Li
Tetrahedron 2016 Volume 72(27–28) pp:3823-3831
Publication Date(Web):7 July 2016
DOI:10.1016/j.tet.2016.04.050
Apratoxin A, a cyclodepsipeptide isolated from a marine cyanobacterium, demonstrates potent cytotoxicity against cancer cell lines by a unique mechanism. A lactam analogue of apratoxin A, named as amidapratoxin A, was efficiently synthesized over 22 linear steps in 2.1% overall yield for the first time. The further conformation analysis was conducted by NMR techniques and computer-based molecular modeling. The results showed that the orientation of the benzene ring in tyrosine moiety, the iso-butyl in isoleucine moiety and hydroxyl group in Ahtmna moiety are quite different from that of apratoxin A, which might result in a significant decrease of the cytotoxicity. While further investigation is on the way, these results provide increased understanding of conformation-activity relationship for apratoxin family members which is an important complement to structure-activity relationship.Apratoxin A is a cyclodepsipeptide with potent cytotoxicity against several human cancer cell lines. To further explore the conformation-activity relationship of apratoxin family members, we designed a lactam analogue of apratoxin A named as amidapratoxin A and completed its first synthesis with a concise approach. Amidapratoxin A showed three orders of magnitude less inhibitory potency compared to apratoxin A. Based on the molecular modeling with constraints from the NMR data, we presented the structural model of amidapratoxin A in solvent and illustrated that the orientation variation of the benzene ring in tyrosine moiety, the iso-butyl in isoleucine moiety and hydroxyl group in Ahtmna moiety may play critical role in cytotoxicity decrease. While further investigation is underway, the results above provide an important reference for structural optimization and efficient synthesis of apratoxin A.
Co-reporter:Jian Liu, Xia Peng, Yang Dai, Wei Zhang, Sumei Ren, Jing Ai, Meiyu Geng and Yingxia Li
Organic & Biomolecular Chemistry 2015 vol. 13(Issue 28) pp:7643-7654
Publication Date(Web):01 Jun 2015
DOI:10.1039/C5OB00778J
Fibroblast growth factor receptor (FGFR) is a potential target for cancer therapy. Based on the structure of AZD4547 and NVPBGJ-398, we designed novel 1H-indazol-3-amine scaffold derivatives by utilizing scaffold hopping and molecular hybridization strategies. Consequently, twenty-eight new compounds were synthesized and evaluated for their inhibitory activity against FGFR1. Compound 7n bearing a 6-(3-methoxyphenyl)-1H-indazol-3-amine scaffold was first identified as a potent FGFR1 inhibitor, with good enzymatic inhibition (IC50 = 15.0 nM) and modest cellular inhibition (IC50 = 642.1 nM). The crystal structure of 7n bound to FGFR1 was obtained, which might provide a new basis for potent inhibitor design. Further structural optimization revealed that compound 7r stood out as the most potent FGFR1 inhibitor with the best enzyme inhibitory (IC50 = 2.9 nM) and cellular activity (IC50 = 40.5 nM).
Co-reporter:Jian Liu, Wuyan Chen, Yechun Xu, Sumei Ren, Wei Zhang, Yingxia Li
Bioorganic & Medicinal Chemistry 2015 Volume 23(Issue 9) pp:1963-1974
Publication Date(Web):1 May 2015
DOI:10.1016/j.bmc.2015.03.034
Nineteen new derivatives based on the structure of marine natural product tasiamide B were designed, synthesized, and evaluated for their inhibitory activity against BACE1, a potential therapeutic target for Alzheimer’s disease. The hydrophobic substituents Val at P3 position, Leu at P1′ position, Ala at P2′ position, and Phe at P3′ position were found to significantly affect the inhibition. Free carboxylic acid at C-terminus was also found to be important to the activity. In addition, the structure–activity relationships (SARs) were supported by molecular docking simulation.
Co-reporter:Wei Zhang, Jing Ai, Dakuo Shi, Xia Peng, Yinchun Ji, Jian Liu, Meiyu Geng, Yingxia Li
European Journal of Medicinal Chemistry 2014 80() pp: 254-266
Publication Date(Web):
DOI:10.1016/j.ejmech.2014.04.056
Co-reporter:Mingming Zhang, Weiliang Zhu, Yingxia Li
European Journal of Medicinal Chemistry 2013 Volume 62() pp:301-310
Publication Date(Web):April 2013
DOI:10.1016/j.ejmech.2013.01.009
Inhibition of the signal transducer and activator of transcription 3 (STAT3) signaling pathway has been considered a novel therapeutic strategy to treat human cancers that harbor aberrantly-active STAT3. In this study, nearly 204,000 compounds in Specs database were screened by virtual screening, and samples of top 100 compounds identified as candidate small-molecule inhibitors of STAT3 were evaluated by STAT3-dependent luciferase reporter assay as well as other cell-based assays. A benzothiazole core scaffold containing compound, 9, was identified as an inhibitor of IL-6/STAT3 signaling with an IC50 of 3.567 μM. It is the first time to discover benzothiazole scaffold as a potent STAT3 signaling inhibitor. We further investigated the (structure–activity relationship) SAR of the benzothiazole analogues, and discovered compound 16w as a better small-molecule inhibitor. Both compounds inhibited the phosphorylation of STAT3 and had no obvious effect on upstream JAK2 kinase.Graphical abstractDocking mode of compound 9 with STAT3 generated by GLIDE. Compound 9 formed three hydrogen bonds with Gln644, Glu638, and Gln635.Highlights► Novel enzothiazole scaffold against STAT3 signaling by virtual screening. ► Showed potent activity in STAT3-dependent luciferase reporter assay. ► Showed potent activity in cell line harboring constitutively active STAT3. ► Inhibited (Tyr705) phosphorylation of STAT3. ► Had no obvious effect on upstream tyrosine kinases.
Co-reporter:Qing-Chao Liu, Tian-Tian Guo, Lei Zhang, Yue Yu, Peng Wang, Ju-Fang Yang, Ying-Xia Li
European Journal of Medicinal Chemistry 2013 Volume 63() pp:511-522
Publication Date(Web):May 2013
DOI:10.1016/j.ejmech.2013.03.001
Twenty-four sugar-substituted oleanolic acid derivatives (1a–1f, 2a–2j, and 3a–3h) were synthesized in a concise and efficient strategy and their effects on the inhibition of protein tyrosine phosphatase 1B (PTP1B) and insulin-sensitizing response were evaluated in vitro. Several derivatives showed moderate to good inhibitory activities against PTP1B, and especially compounds 2f, 2h, 3d and 3e exhibited the most potent inhibitory activities with the IC50 values of 1.91, 12.2, 9.21 and 0.56 μM against PTP1B, respectively. Furthermore, compounds 2g–2h and 3b–3e displayed good insulin-sensitizing activities with the response values ranging from 21.52% to 59.58%. Structure–activity relationship study of these sugar-substituted oleanolic acid derivatives demonstrated that PTP1B inhibitory activity and insulin-sensitizing response were strongly influenced by both the carbohydrate moiety at the C-3 position and the long acidic chain at C-28 position of oleanolic acid.Graphical abstractHighlights► A series of sugar-substituted oleanolic acid derivatives were synthesized. ► PTP1B activities and insulin-sensitizing response of all compounds were evaluated. ► Compounds 2f, 2h, 3d and 3e showed the most potent PTP1B inhibitory activities.
Co-reporter:Qingchao Liu, Hongchun Liu, Lei Zhang, Tiantian Guo, Peng Wang, Meiyu Geng, Yingxia Li
European Journal of Medicinal Chemistry 2013 Volume 64() pp:1-15
Publication Date(Web):June 2013
DOI:10.1016/j.ejmech.2013.04.016
•Sixteen oleanolic acid triterpenoid saponins were synthesized.•The cytotoxic activity against cancer cells of all compounds was evaluated.•The structure–activity relationships of compounds have been discussed.Twenty-six naturally occurring oleanolic acid saponins and their derivatives, 16 of which were synthesized in this study, were preliminarily evaluated against human cancer cells. From SAR studies, the presence of α-l-rhamnosyl residue at the terminal of both C-3 and C-28 position for oleanolic acid bidesmosides was important to enhance cytotoxicity, and introducing more sugar residues at C3–OH of compound 12 with C-28 carboxylic acid is a favorable modification to ameliorate the anticancer activity. Furthermore, α-l-rhamnosyl moiety linked to C2–OH of the first monosaccharide (α-l-alabinose, β-d-xylose, β-d-galactose or β-d-glucose) in C3–OH of oleanolic acid was helpful to improve the cytotoxicity. According to the predicted log P values, lipophilicity of the synthesized saponins was not an important factor for cytotoxicity.
Co-reporter:Junlong Xiong;Zhichao Lu;Ning Ding;Sumei Ren
European Journal of Organic Chemistry 2013 Volume 2013( Issue 27) pp:6158-6166
Publication Date(Web):
DOI:10.1002/ejoc.201300575
Abstract
The synthesis of the pentasaccharide moiety of thornasteroside A, the first asterosaponin isolated from starfish in 1978 has been achieved for the first time. Initially, a [3+2] convergent strategy was attempted, but the β(14) glycosidic linkage between galactopyranose (sugar IV) and xylopyranose (sugar II) was formed with a low stereoselectivity and in low yield. Subsequently, a [3+1+1] strategy was adopted. A galactopyranosyl donor (18) equipped with a neighboring participating Lev (levulinoyl) group at the 2-position was first coupled with a trisaccharide acceptor to construct the β(14) glycosidic bond. Then the Lev group was selectively removed, and subsequent glycosylation with a perbenzoylated D-fucopyranosyl Schmidt donor efficiently gave the desired pentasaccharide.
Co-reporter:Yongfeng Tao, Ning Ding, Sumei Ren, Yingxia Li
Tetrahedron Letters 2013 Volume 54(Issue 45) pp:6101-6104
Publication Date(Web):6 November 2013
DOI:10.1016/j.tetlet.2013.08.118
An effective Heck-type cross-coupling reaction between halo-exo-glycals and endo-glycals to achieve C-glycosidic disaccharides has been developed. Using Pd(OAc)2 as the catalyst, dppp as ligand and K2CO3 as base, the reactions gave C-glycosidic products in good to excellent yields with exclusive stereochemistry.
Co-reporter:Mingming Zhang, Weiliang Zhu, Ning Ding, Wei Zhang, Yingxia Li
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 7) pp:2225-2229
Publication Date(Web):1 April 2013
DOI:10.1016/j.bmcl.2013.01.056
Inhibition of the signal transducer and activator of transcription 3 (STAT3) signaling pathway has been considered a novel therapeutic strategy to treat human cancers that harbor aberrantly-active STAT3. In this study, a series of small molecules were identified as novel inhibitors of STAT3 signaling pathway through virtual screening. A tricyclic scaffold containing compound, 6, was identified as an inhibitor of IL-6/STAT3 signaling with an IC50 of 26.68 μM. In addition, this compound inhibited Tyr705 phosphorylation of STAT3 and had no obvious effect on upstream tyrosine kinases. Thus, compound 6 is a potential lead structure and valuable for further drug development.
Co-reporter:Yanxia Liu ; Wei Zhang ; Li Li ; Lilibeth A. Salvador ; Tiantian Chen ; Wuyan Chen ; Kevin M. Felsenstein ; Thomas B. Ladd ; Ashleigh R. Price ; Todd E. Golde ; Jianhua He ; Yechun Xu ; Yingxia Li ;Hendrik Luesch
Journal of Medicinal Chemistry 2012 Volume 55(Issue 23) pp:10749-10765
Publication Date(Web):November 26, 2012
DOI:10.1021/jm301630s
Inspired by marine cyanobacterial natural products, we synthesized modified peptides with a central statine-core unit, characteristic for aspartic protease inhibition. A series of tasiamide B analogues inhibited BACE1, a therapeutic target in Alzheimer’s disease. We probed the stereospecificity of target engagement and determined additional structure–activity relationships with respect to BACE1 and related aspartic proteases, cathepsins D and E. We cocrystallized selected inhibitors with BACE1 to reveal the structural basis for the activity. Hybrid molecules that combine features of tasiamide B and an isophthalic acid moiety-containing sulfonamide showed nanomolar cellular activity. Compounds were screened in a series of rigorous complementary cell-based assays. We measured secreted APP ectodomain (sAPPβ), membrane bound carboxyl terminal fragment (CTF), levels of β-amyloid (Aβ) peptides and selectivity for β-secretase (BACE1) over γ-secretase. Prioritized compounds showed reasonable stability in vitro and in vivo, and our most potent inhibitor showed efficacy in reducing Aβ levels in the rodent brain.
Co-reporter:Ning Ding, Qing Chen, Wei Zhang, Sumei Ren, Ying Guo, Yingxia Li
European Journal of Medicinal Chemistry 2012 Volume 53() pp:316-326
Publication Date(Web):July 2012
DOI:10.1016/j.ejmech.2012.04.022
The occurrence of highly pathogenic avian influenza virus H5N1 highlights the urgent need for new classes of antiviral drugs. Theoretically, each of steps in influenza viral life cycle can be a target of antiviral therapeutics. However, up to date, no licenced entry inhibitor drug is available for H5N1 or any other influenza viruses. Our strategy for developing new anti-influenza therapeutics is to target the interaction between HA and sialic acid which is influenza viral receptor presented on host cell surface. Here, based on our previously discovered small molecule inhibitor saponin 1, intensive SAR studies around the sugar chain and aglycone were conducted. The results showed that both the chacotriosyl residue and the chlorogenin moiety of active compound 1 are important for the antiviral activity, although several subtle modifications can be made on particular positions.Graphical abstractIntensive SAR studies around the sugar chain and aglycone were conducted on highly pathogenic H5N1 influenza viral entry inhibitory activity.Highlights► Saponin derivatives were discovered as small molecule H5N1 viral entry inhibitors. ► Intensive SAR studies around the sugar chain and aglycone of the saponin derivatives were conducted. ► Both the chacotriosyl and chlorogenin moiety of active compound 1 are important for the antiviral activity.
Co-reporter:Da-Kuo Shi, Wei Zhang, Ning Ding, Ming Li, Ying-Xia Li
European Journal of Medicinal Chemistry 2012 Volume 47() pp:424-431
Publication Date(Web):January 2012
DOI:10.1016/j.ejmech.2011.11.011
Recently, a novel glycosylated diphyllin derivative 11 which exhibiting potent anticancer activity by targeting topoisomerase IIα was reported by our group. In order to provide more molecules for structure-activity relationship (SAR) studies, 12 new glycosylated diphyllin analogs have been designed, synthesized, and evaluated for their biological activities. The SAR analysis revealed that (i) the sugar moiety on the diphyllin is essential for the anticancer activity; (ii) equatorial C4′–OH on the sugar is superior to the axial one, and (iii) a proper cyclic lipophilic group at the C4′ and C6′ of sugar might enhance the anticancer activity.A series of glycosylated diphyllin derivatives were designed for anti-tumor activity and SAR studies were revealed.Highlights► 12 new glycosylated diphyllin derivatives were designed for anti-tumor activity. ► The sugar moiety on the C4 is a key element for its bioactivity. ► Glycosides with equatorial C4′–OH are more active than those with axial one. ► Acetal glucosides showed outstanding cytotoxicity and Topo II inhibitory activity.
Co-reporter:Siming Yang, Wei Zhang, Ning Ding, Jeannette Lo, Yanxia Liu, Michael J. Clare-Salzler, Hendrik Luesch, Yingxia Li
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 15) pp:4774-4780
Publication Date(Web):1 August 2012
DOI:10.1016/j.bmc.2012.05.077
The linear depsipeptide grassystatin A, a valuable probe for the study of cathepsin E function, has been synthesized by a [4+6] strategy. It exhibited specific inhibitory activity against cathepsin E with an IC50 value of 0.8 nM. Our studies indicated that inhibition of cathepsin E did not have an impact on ovalbumin antigen processing and peptide presentation, unique from studies of other aspartic protease inhibitors.The linear depsipeptide grassystatin A, a valuable probe for the study of cathepsin E function, has been synthesized by a [4+6] strategy. It exhibited specific inhibitory activity against cathepsin E with an IC50 value of 0.8 nM. And our studies indicated that inhibition of cathepsin E did not have an impact on ovalbumin antigen processing and peptide presentation, unique from studies of other aspartic protease inhibitors.
Co-reporter:Shiqiang Yan, Ning Ding, Wei Zhang, Peng Wang, Yingxia Li, Ming Li
Carbohydrate Research 2012 Volume 354() pp:6-20
Publication Date(Web):1 June 2012
DOI:10.1016/j.carres.2012.02.021
An efficient, chemoselective, and environment-friendly method for the deprotection of tert-butyldiphenylsilyl ethers mediated by triflic acid supported on silica gel is reported. A wide range of tert-butyldiphenylsilyl ethers derived from carbohydrate and saponin residues can be smoothly cleaved in the presence of various types of other protecting groups in good to excellent yields in acetonitrile. This heterogeneous reaction does not require aqueous workup, and the supported catalyst can be readily recycled.
Co-reporter:Ning Ding, Wei Zhang, Hua Ling Xiao, Peng Wang, Ying Xia Li
Chinese Chemical Letters 2012 Volume 23(Issue 5) pp:529-532
Publication Date(Web):May 2012
DOI:10.1016/j.cclet.2012.03.016
The synthesis and biological evaluation of two series of salicylanilide derivatives on the EGFR and ErbB-2 tyrosine kinases inhibitory activities were conducted. Of the tested compounds those having an additional aryl group substituted on the anilino ring were active on the EGFR tyrosine kinase inhibition (7a–c and 13a, 13c, 13d, 13f). The inhibitory activities were all in the low micromolar or submicromolar range. In addition, compound 13a was found to have dual inhibitory activities both on EGFR and ErbB-2 tyrosine kinases (1.654 ± 1.280 and 7.134 ± 1.265 μmol/L).
Co-reporter:Ning Ding;Yuexing Chun;Wei Zhang
Chinese Journal of Chemistry 2012 Volume 30( Issue 2) pp:409-412
Publication Date(Web):
DOI:10.1002/cjoc.201180487
Abstract
4,6-O-Benzylidenation of D-galactal with PhCH(OCH3)2 catalyzed by bromodimethylsulfonium bromide leads to methyl 2-dexoy-4,6-O-benzylidene galactopyranoside efficiently, which serves as a key intermediate to the ready preparation of 2,3- and 2,6-dideoxy galactopyranosides.
Co-reporter:Zhichao Lu, Ning Ding, Wei Zhang, Peng Wang, Yingxia Li
Tetrahedron Letters 2011 Volume 52(Issue 26) pp:3320-3323
Publication Date(Web):29 June 2011
DOI:10.1016/j.tetlet.2011.04.060
A convenient synthesis of the core trisaccharide of the N-glycans was described. Orthogonal one-pot glycosylation of three monosaccharide building blocks was performed to furnish β-glucosyl chitobiose, which was then transformed to β-mannosyl chitobiose by intramolecular epimerization of the C-2 position of the β-glucoside. The key glucosyl donor 7c with differentiated 2,3-OH was prepared following the 4,6-O-benzylidene-protected 1,2-orthoester strategy.
Co-reporter:Ning Ding;Wei Zhang;Guokai Lv
Archiv der Pharmazie 2011 Volume 344( Issue 12) pp:786-793
Publication Date(Web):
DOI:10.1002/ardp.201000335
Abstract
The synthesis and in-vitro biological evaluation of antifungal activities of a series of 1,2-trans glycosphingolipids (GSL) against Candida albicans, Candida parapsilosis, and Candida tropicalis were described. The preliminary study indicated that the sort of sugar moiety of GSLs affected their antifungal activities and selectivities towards the three Candida species, and the permeability might not be the sole parameter affecting their antifungal properties as presumed before, which would bring new clues to understand the antifungal profile for these types of compounds.
Co-reporter:Guangfa Wang, Zhichao Lu, Ning Ding, Wei Zhang, Peng Wang, Yingxia Li
Carbohydrate Research 2011 Volume 346(Issue 15) pp:2368-2373
Publication Date(Web):8 November 2011
DOI:10.1016/j.carres.2011.08.008
A facile and efficient method to differentiate the 2,3-diols of glucopyranosides based on 1,2-orthoesters strategy was developed. Stable thioglucosides were employed as the starting materials to prepare the corresponding 1,2-orthoesters. When treated with HCl aqueous solution and followed with Et3N, differentiation of the 2,3-diols was efficiently achieved along with the generation of a convertible anomeric hydroxyl group. In addition, an easy and practical method based on NOE was proposed to determine whether the 1,2-orthoesters were endo-type or exo-type.
Co-reporter:Ning Ding, Xiaoguang Du, Wei Zhang, Zhichao Lu, Yingxia Li
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 12) pp:3531-3535
Publication Date(Web):15 June 2011
DOI:10.1016/j.bmcl.2011.04.145
Naphtho[2,1-α]pyrrolo[3,4-c]carbazole-5,7(6H,12H)-dione (NPCD) is known to be a very potent and selective cyclin D1-CDK4 inhibitors and could induce strong G1 phase arrest in breast tumor cell lines. In this work, the synthesis of five NPCD glycosides and their cytotoxic activities against eight tumor cell lines are presented, as well as the investigation of their cell cycle arrest profiles. The results showed that the introduction of a sugar moiety onto NPCD did not affect much of their cytotoxic activities, while the subtle structure of the sugar moiety affected the underlying mechanism strongly. In addition, NPCD showed distinct cell-cycle arrest profiles in BxPC3 prostate cells and MCF-7 breast cells, while NPCD glycosides shared similar cell cycle arrest profiles in MCF-7 and BxPC3 cells, which also indicated that not only the indolocarbazole framework as well known before but the sugar moiety can have a profound impact on the mechanism of action for these types of compounds.
Co-reporter:Yuexing Chun, Shiqiang Yan, Xiangpeng Li, Ning Ding, Wei Zhang, Peng Wang, Ming Li, Yingxia Li
Tetrahedron Letters 2011 Volume 52(Issue 46) pp:6196-6198
Publication Date(Web):16 November 2011
DOI:10.1016/j.tetlet.2011.09.055
A new and efficient method for the preparation of 1,6-anhydro sugars using silica supported perchloric acid as a catalyst is described. The catalyst is heterogeneous and 1,6-anhydro sugars could be formed within a few minutes with good yields.
Co-reporter:Ning Ding, Zaihong Zhang, Wei Zhang, Yuexing Chun, Peng Wang, Huimin Qi, Shan Wang, Yingxia Li
Carbohydrate Research 2011 Volume 346(Issue 14) pp:2126-2135
Publication Date(Web):18 October 2011
DOI:10.1016/j.carres.2011.07.028
A series of novel oligorhamnoside derivatives (1–10) and naturally occurring cleistrioside-5 were synthesized and evaluated for their in vitro antibacterial activities. Among them, dirhamnoside derivative 7 and cleistrioside-5 displayed similar antibacterial profiles and exhibited moderate to good inhibitory activities on bacterial growth against a panel of Gram-positive bacteria (MICs ⩽ 4–32 μg/mL). The results revealed that these two compounds showed selectivity towards bacterial species strictly, without being affected by the antibiotic-resistant/susceptible properties of one species, which suggested that they might have the potential to avoid antibiotic cross-resistance. In addition, the preliminary SARs of this type of oligorhamnoside derivatives on the antibacterial activities were determined.Partially acetylated oligorhamnoside derivatives as antibacterial agents (MICs ⩽ 4–32 μg/mL).
Co-reporter:Jun-Fei Wang;Ning Ding;Wei Zhang;Peng Wang;Ying-Xia Li
Helvetica Chimica Acta 2011 Volume 94( Issue 12) pp:2221-2230
Publication Date(Web):
DOI:10.1002/hlca.201100162
Abstract
Baicalein, an important active constituent of the traditional Chinese herb Scutellaria baicalensis, exhibited antitumor activity and inhibitory activity against P-gp 170. The syntheses of 25 baicalein derivatives, 2–26 (Table), are described here (Scheme 1). These compounds were systematically modified with O-alkylation and O-acylation at HOC(5), HOC(6), and HOC(7), singly or in combination, on the ring A of baicalein in order to evaluate the effects of such modifications on their inhibitory activities against multidrug-resistant tumor cell lines and P-gp 170. Highly selective and efficient alkylations at HOC(7) of peracetylated baicalein were the key to the distinction between HOC(6) and HOC(7) of baicalein.
Co-reporter:Gaopeng Song, Hongchun Liu, Wei Zhang, Meiyu Geng, Yingxia Li
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 14) pp:5183-5193
Publication Date(Web):15 July 2010
DOI:10.1016/j.bmc.2010.05.064
A series of anthracene l-rhamnopyranosides were designed and synthesized in a practical way and their cytotoxic activity was examined in vitro. Most compounds exhibited both potent cytotoxicity against several tumor cell lines and high DNA binding capacity. The preliminary results showed that subtle modifications of rhamnosyl moiety in anthracene rhamnosides with acetyl group had a selective toxicity for different tumor cells and the displacement of C-10 carbonyl group in emodin by acetylmethylene group was helpful to improve the inhibitory activity. Lipophilicity of the anthracene glycosides was not a crucial factor for cytotoxicity and most molecules with good cytotoxicity could inhibit the catalytic activity of Top2α.A series of anthracene l-rhamnopyranosides were designed and synthesized in a practical way. Most compounds exhibited both potent cytotoxicity against several tumor cell lines and high DNA binding capacity.
Co-reporter:Zaihong Zhang, Chengli Zong, Gaopeng Song, Guokai Lv, Yuexing Chun, Peng Wang, Ning Ding, Yingxia Li
Carbohydrate Research 2010 Volume 345(Issue 6) pp:750-760
Publication Date(Web):19 April 2010
DOI:10.1016/j.carres.2010.01.015
The first total synthesis of caminoside B, a novel marine antimicrobial glycolipid isolated from the marine sponge Caminus sphaeroconia, was developed. This marine small molecule inhibitor (IC50 = 20 μM) targeting type III secretory pathway of bacterial pathogenesis was assembled in good yield via a ‘2+2+1’ strategy based on stereocontrolled construction of the four glycosidic linkages.The first total synthesis of caminoside B was developed via a ‘2+2+1’ strategy based on stereocontrolled construction of the four glycosidic linkages.
Co-reporter:Chengli Zong, Zhongzhen Li, Tiantian Sun, Peng Wang, Ning Ding, Yingxia Li
Carbohydrate Research 2010 Volume 345(Issue 11) pp:1522-1532
Publication Date(Web):19 July 2010
DOI:10.1016/j.carres.2010.04.006
Two types of sulfated octyl tetra- to octaoligofucosides with different sulfation patterns were synthesized employing a combination of stepwise elongation and convergent strategies in which trichloroacetimidates and thioglycosides were selected as the glycosyl donors.
Co-reporter:Peng Wang, Jun Wang, TianTian Guo, Yingxia Li
Carbohydrate Research 2010 Volume 345(Issue 5) pp:607-620
Publication Date(Web):30 March 2010
DOI:10.1016/j.carres.2010.01.002
Fourteen ursolic acid and oleanolic acid saponins with N-acetyl-β-d-glucosamine, and (1→4)-linked and (1→6)-linked N-acetyl-β-d-glucosamine oligosaccharide residues were synthesized in a convergent manner. The structures of all compounds were confirmed by 1H NMR and 13C NMR spectroscopy and by mass spectrometry, and their cytotoxic activities were assayed in three cancer cell lines. Only oleanolic acid-3-yl β-d-GluNAc showed significant cytotoxicity against HL-60 and BGC-823.Fourteen ursolic acid and oleanolic acid saponins, where R = GlcNAc or (1→4)-linked and (1→6)-linked GlcNAc oligosaccharide residues were synthesized in a convergent manner and evaluated for their cytotoxic activity in three cancer cell lines. R1 and R2 = H or CH3.
Co-reporter:Shan Jiang;Ning Ding;Wei Zhang;Yichun Zhang;Meiyu Geng
Archiv der Pharmazie 2010 Volume 343( Issue 4) pp:215-221
Publication Date(Web):
DOI:10.1002/ardp.200900217
Abstract
A series of novel conjugates of aspirin with natural phenolic acid antioxidants connected through a diol linker were designed and synthesized as potential bifunctional agents combining antioxidant and anti-inflammatory activity for reducing gastrointestinal toxicity. In general, the conjugates were found to be efficient antioxidants and many of them demonstrated much more potent anti-inflammatory activity than aspirin. Among them, 5a and 5b which bear the best anti-inflammatory activity exhibited significantly reduced ulcerogenic potency and toxicity compared to aspirin. However, it is evident that the anti-inflammatory activity of these dual-acting molecules in vivo, was not simply consistent with their antioxidant ability in vitro.
Co-reporter:Hualing Xiao;Guangfa Wang;Peng Wang
Chinese Journal of Chemistry 2010 Volume 28( Issue 7) pp:1229-1232
Publication Date(Web):
DOI:10.1002/cjoc.201090213
Abstract
The rare and expensive D-talose was conveniently synthesized from readily available D-galactose in four steps with an overall yield of 58%. The key step was the inversion of equatorial 2-OH of galactose to the axial one by SN2 reaction under the modified Lattrell-Dax reaction conditions.
Co-reporter:Jian Liu, Xia Peng, Yang Dai, Wei Zhang, Sumei Ren, Jing Ai, Meiyu Geng and Yingxia Li
Organic & Biomolecular Chemistry 2015 - vol. 13(Issue 28) pp:NaN7654-7654
Publication Date(Web):2015/06/01
DOI:10.1039/C5OB00778J
Fibroblast growth factor receptor (FGFR) is a potential target for cancer therapy. Based on the structure of AZD4547 and NVPBGJ-398, we designed novel 1H-indazol-3-amine scaffold derivatives by utilizing scaffold hopping and molecular hybridization strategies. Consequently, twenty-eight new compounds were synthesized and evaluated for their inhibitory activity against FGFR1. Compound 7n bearing a 6-(3-methoxyphenyl)-1H-indazol-3-amine scaffold was first identified as a potent FGFR1 inhibitor, with good enzymatic inhibition (IC50 = 15.0 nM) and modest cellular inhibition (IC50 = 642.1 nM). The crystal structure of 7n bound to FGFR1 was obtained, which might provide a new basis for potent inhibitor design. Further structural optimization revealed that compound 7r stood out as the most potent FGFR1 inhibitor with the best enzyme inhibitory (IC50 = 2.9 nM) and cellular activity (IC50 = 40.5 nM).

![L-Valinamide,N,N-dimethyl-L-valyl-N-[(1S,2R)-2-methoxy-4-[(2S)-2-[(1R,2R)-1-methoxy-2-methyl-3-oxo-3-[(2-phenylethyl)amino]propyl]-1-pyrrolidinyl]-1-[(1S)-1-methylpropyl]-4-oxobutyl]-N-methyl-](http://img.cochemist.com/ccimg/149700/149606-27-9.png)
![L-Valinamide,N,N-dimethyl-L-valyl-N-[(1S,2R)-2-methoxy-4-[(2S)-2-[(1R,2R)-1-methoxy-2-methyl-3-oxo-3-[(2-phenylethyl)amino]propyl]-1-pyrrolidinyl]-1-[(1S)-1-methylpropyl]-4-oxobutyl]-N-methyl-](http://img.cochemist.com/ccimg/149700/149606-27-9_b.png)
![L-Valinamide,N,N-dimethyl-L-valyl-N-[(1S,2R)-2-methoxy-4-[(2S)-2-[(1R,2R)-1-methoxy-2-methyl-3-oxo-3-[[(1S)-2-phenyl-1-(2-thiazolyl)ethyl]amino]propyl]-1-pyrrolidinyl]-1-[(1S)-1-methylpropyl]-4-oxobutyl]-N-methyl-](http://img.cochemist.com/ccimg/110500/110417-88-4.png)
![L-Valinamide,N,N-dimethyl-L-valyl-N-[(1S,2R)-2-methoxy-4-[(2S)-2-[(1R,2R)-1-methoxy-2-methyl-3-oxo-3-[[(1S)-2-phenyl-1-(2-thiazolyl)ethyl]amino]propyl]-1-pyrrolidinyl]-1-[(1S)-1-methylpropyl]-4-oxobutyl]-N-methyl-](http://img.cochemist.com/ccimg/110500/110417-88-4_b.png)
![N-[3-[5-CHLORO-2-[2-METHOXY-4-(4-METHYLPIPERAZIN-1-YL)ANILINO]PYRIMIDIN-4-YL]OXYPHENYL]PROP-2-ENAMIDE](http://img.cochemist.com/ccimg/1213300/1213269-23-8.png)
![N-[3-[5-CHLORO-2-[2-METHOXY-4-(4-METHYLPIPERAZIN-1-YL)ANILINO]PYRIMIDIN-4-YL]OXYPHENYL]PROP-2-ENAMIDE](http://img.cochemist.com/ccimg/1213300/1213269-23-8_b.png)














![3-Pyridinecarboxamide,N-[4-[(6,7-dimethoxy-4-quinolinyl)oxy]-3-fluorophenyl]-1-(4-fluorophenyl)-1,2-dihydro-2-oxo-](http://img.cochemist.com/ccimg/929300/929252-41-5.png)
![3-Pyridinecarboxamide,N-[4-[(6,7-dimethoxy-4-quinolinyl)oxy]-3-fluorophenyl]-1-(4-fluorophenyl)-1,2-dihydro-2-oxo-](http://img.cochemist.com/ccimg/929300/929252-41-5_b.png)
![2-HEPTEN-1-OL, 5-[(4-METHOXYPHENYL)METHOXY]-3,6,6-TRIMETHYL-, (2E,5S)-](http://img.cochemist.com/ccimg/878700/878626-77-8.png)
![2-HEPTEN-1-OL, 5-[(4-METHOXYPHENYL)METHOXY]-3,6,6-TRIMETHYL-, (2E,5S)-](http://img.cochemist.com/ccimg/878700/878626-77-8_b.png)
![1H-PYRROLO[2,3-D]PYRIMIDINE, 4-(2-FLUORO-4-NITROPHENOXY)-](http://img.cochemist.com/ccimg/875800/875764-99-1.png)
![1H-PYRROLO[2,3-D]PYRIMIDINE, 4-(2-FLUORO-4-NITROPHENOXY)-](http://img.cochemist.com/ccimg/875800/875764-99-1_b.png)



![Heptanal, 3,6,6-trimethyl-5-[(triethylsilyl)oxy]-, (3R,5S)-](http://img.cochemist.com/ccimg/780800/780770-95-8.png)
![Heptanal, 3,6,6-trimethyl-5-[(triethylsilyl)oxy]-, (3R,5S)-](http://img.cochemist.com/ccimg/780800/780770-95-8_b.png)










![Heptanal, 5-[(4-methoxyphenyl)methoxy]-3,6,6-trimethyl-, (3R,5S)-](http://img.cochemist.com/ccimg/676400/676324-77-9.png)
![Heptanal, 5-[(4-methoxyphenyl)methoxy]-3,6,6-trimethyl-, (3R,5S)-](http://img.cochemist.com/ccimg/676400/676324-77-9_b.png)
![1-HEPTANOL, 5-[(4-METHOXYPHENYL)METHOXY]-3,6,6-TRIMETHYL-, (3S,5S)-](http://img.cochemist.com/ccimg/676400/676324-76-8.png)
![1-HEPTANOL, 5-[(4-METHOXYPHENYL)METHOXY]-3,6,6-TRIMETHYL-, (3S,5S)-](http://img.cochemist.com/ccimg/676400/676324-76-8_b.png)
![Pentanal, 3-[(4-methoxyphenyl)methoxy]-4,4-dimethyl-, (3S)-](http://img.cochemist.com/ccimg/676400/676324-73-5.png)
![Pentanal, 3-[(4-methoxyphenyl)methoxy]-4,4-dimethyl-, (3S)-](http://img.cochemist.com/ccimg/676400/676324-73-5_b.png)
![2-HEXANONE, 4-[[(1,1-DIMETHYLETHYL)DIMETHYLSILYL]OXY]-5,5-DIMETHYL-, (4S)-](http://img.cochemist.com/ccimg/612500/612492-55-4.png)
![2-HEXANONE, 4-[[(1,1-DIMETHYLETHYL)DIMETHYLSILYL]OXY]-5,5-DIMETHYL-, (4S)-](http://img.cochemist.com/ccimg/612500/612492-55-4_b.png)


















![Ruthenium, bis(acetato-kO,kO')[1,1'-(1S)-[1,1'-binaphthalene]-2,2'-diylbis[1,1-diphenylphosphine-kP]]-, (OC-6-22)-](http://img.cochemist.com/ccimg/262000/261948-85-0.png)
![Ruthenium, bis(acetato-kO,kO')[1,1'-(1S)-[1,1'-binaphthalene]-2,2'-diylbis[1,1-diphenylphosphine-kP]]-, (OC-6-22)-](http://img.cochemist.com/ccimg/262000/261948-85-0_b.png)
























![ACETIC ACID, [[(4-METHYLPHENYL)AMINO]SULFONYL]-, ETHYL ESTER](http://img.cochemist.com/ccimg/78400/78374-64-8.png)
![ACETIC ACID, [[(4-METHYLPHENYL)AMINO]SULFONYL]-, ETHYL ESTER](http://img.cochemist.com/ccimg/78400/78374-64-8_b.png)
![ACETIC ACID, [[(4-METHYLPHENYL)AMINO]SULFONYL]-](http://img.cochemist.com/ccimg/78400/78374-09-1.png)
![ACETIC ACID, [[(4-METHYLPHENYL)AMINO]SULFONYL]-](http://img.cochemist.com/ccimg/78400/78374-09-1_b.png)




![Propanedioic acid,2-[(2-nitrophenyl)methylene]-, 1,3-dimethyl ester](http://img.cochemist.com/ccimg/66000/65974-52-9.png)
![Propanedioic acid,2-[(2-nitrophenyl)methylene]-, 1,3-dimethyl ester](http://img.cochemist.com/ccimg/66000/65974-52-9_b.png)

![Acetic acid, [(phenylamino)sulfonyl]-, ethyl ester](http://img.cochemist.com/ccimg/56200/56146-84-0.png)
![Acetic acid, [(phenylamino)sulfonyl]-, ethyl ester](http://img.cochemist.com/ccimg/56200/56146-84-0_b.png)
![Benzoic acid, 5-chloro-2-[(4-chlorophenyl)methoxy]-](http://img.cochemist.com/ccimg/52900/52803-76-6.png)
![Benzoic acid, 5-chloro-2-[(4-chlorophenyl)methoxy]-](http://img.cochemist.com/ccimg/52900/52803-76-6_b.png)
![ALANINE, N-[(1,1-DIMETHYLETHOXY)CARBONYL]LEUCYL-, METHYL ESTER](http://img.cochemist.com/ccimg/43200/43189-51-1.png)
![ALANINE, N-[(1,1-DIMETHYLETHOXY)CARBONYL]LEUCYL-, METHYL ESTER](http://img.cochemist.com/ccimg/43200/43189-51-1_b.png)
![L-Isoleucine, N-[(1,1-dimethylethoxy)carbonyl]-N-methyl-, methyl ester](http://img.cochemist.com/ccimg/24200/24164-05-4.png)
![L-Isoleucine, N-[(1,1-dimethylethoxy)carbonyl]-N-methyl-, methyl ester](http://img.cochemist.com/ccimg/24200/24164-05-4_b.png)

![ACETIC ACID, [(PHENYLAMINO)SULFONYL]-](http://img.cochemist.com/ccimg/7200/7117-21-7.png)
![ACETIC ACID, [(PHENYLAMINO)SULFONYL]-](http://img.cochemist.com/ccimg/7200/7117-21-7_b.png)


![3-Pyridinecarboxamide, N-[4-[(2-amino-3-chloro-4-pyridinyl)oxy]-3-fluorophenyl]-4-ethoxy-1-(4-fluorophenyl)-1,2-dihydro-2-oxo-](/data/chemimg/315000/1025720-94-8.png)
![3-Pyridinecarboxamide, N-[4-[(2-amino-3-chloro-4-pyridinyl)oxy]-3-fluorophenyl]-4-ethoxy-1-(4-fluorophenyl)-1,2-dihydro-2-oxo-](/data/chemimg/315000/1025720-94-8_b.png)
![Benzenamine, 3-fluoro-4-(1H-pyrrolo[2,3-d]pyrimidin-4-yloxy)-](http://img.cochemist.com/ccimg/875800/875765-00-7.png)
![Benzenamine, 3-fluoro-4-(1H-pyrrolo[2,3-d]pyrimidin-4-yloxy)-](http://img.cochemist.com/ccimg/875800/875765-00-7_b.png)
![Benzenamine, 4-[(6,7-dimethoxy-4-quinolinyl)oxy]-3-fluoro-](http://img.cochemist.com/ccimg/347200/347161-74-4.png)
![Benzenamine, 4-[(6,7-dimethoxy-4-quinolinyl)oxy]-3-fluoro-](http://img.cochemist.com/ccimg/347200/347161-74-4_b.png)



