Co-reporter:Timothy M. Acker, Jonathan E. Gable, Markus-Frederik Bohn, Priyadarshini Jaishankar, Michael C. Thompson, James S. Fraser, Adam R. Renslo, and Charles S. Craik
Journal of the American Chemical Society August 30, 2017 Volume 139(Issue 34) pp:11650-11650
Publication Date(Web):July 31, 2017
DOI:10.1021/jacs.7b04030
Targeting of cryptic binding sites represents an attractive but underexplored approach to modulating protein function with small molecules. Using the dimeric protease (Pr) from Kaposi’s sarcoma-associated herpesvirus (KSHV) as a model system, we sought to dissect a putative allosteric network linking a cryptic site at the dimerization interface to enzyme function. Five cryogenic X-ray structures were solved of the monomeric protease with allosteric inhibitors bound to the dimer interface site. Distinct coordinated movements captured by the allosteric inhibitors were also revealed as alternative states in room-temperature X-ray data and comparative analyses of other dimeric herpesvirus proteases. A two-step mechanism was elucidated through detailed kinetic analyses and suggests an enzyme isomerization model of inhibition. Finally, a representative allosteric inhibitor from this class was shown to be efficacious in a cellular model of viral infectivity. These studies reveal a coordinated dynamic network of atomic communication linking cryptic binding site occupancy and allosteric inactivation of KHSV Pr that can be exploited to target other members of this clinically relevant family of enzymes.
Co-reporter:Nicole O. Meyer, Anthony J. O’Donoghue, Ursula Schulze-Gahmen, Matthew Ravalin, Steven M. Moss, Michael B. Winter, Giselle M. Knudsen, and Charles S. Craik
Analytical Chemistry April 18, 2017 Volume 89(Issue 8) pp:4550-4550
Publication Date(Web):March 21, 2017
DOI:10.1021/acs.analchem.6b05002
The more than 500 protein kinases comprising the human kinome catalyze hundreds of thousands of phosphorylation events to regulate a diversity of cellular functions; however, the extended substrate specificity is still unknown for many of these kinases. We report here a method for quantitatively describing kinase substrate specificity using an unbiased peptide library-based approach with direct measurement of phosphorylation by tandem liquid chromatography–tandem mass spectrometry (LC–MS/MS) peptide sequencing (multiplex substrate profiling by mass spectrometry, MSP-MS). This method can be deployed with as low as 10 nM enzyme to determine activity against S/T/Y-containing peptides; additionally, label-free quantitation is used to ascertain catalytic efficiency values for individual peptide substrates in the multiplex assay. Using this approach we developed quantitative motifs for a selection of kinases from each branch of the kinome, with and without known substrates, highlighting the applicability of the method. The sensitivity of this approach is evidenced by its ability to detect phosphorylation events from nanogram quantities of immunoprecipitated material, which allows for wider applicability of this method. To increase the information content of the quantitative kinase motifs, a sublibrary approach was used to expand the testable sequence space within a peptide library of approximately 100 members for CDK1, CDK7, and CDK9. Kinetic analysis of the HIV-1 Tat (transactivator of transcription)-positive transcription elongation factor b (P-TEFb) interaction allowed for localization of the P-TEFb phosphorylation site as well as characterization of the stimulatory effect of Tat on P-TEFb catalytic efficiency.
Co-reporter:Dr. Jonathan E. Gable;Dr. Gregory M. Lee;Dr. Timothy M. Acker;Kaitlin R. Hulce;Eric R. Gonzalez;Dr. Patrick Schweigler;Dr. Samu Melkko;Dr. Christopher J. Farady; Charles S. Craik
ChemMedChem 2016 Volume 11( Issue 8) pp:862-869
Publication Date(Web):
DOI:10.1002/cmdc.201500526
Abstract
Fragment-based drug discovery has shown promise as an approach for challenging targets such as protein–protein interfaces. We developed and applied an activity-based fragment screen against dimeric Kaposi's sarcoma-associated herpesvirus protease (KSHV Pr) using an optimized fluorogenic substrate. Dose–response determination was performed as a confirmation screen, and NMR spectroscopy was used to map fragment inhibitor binding to KSHV Pr. Kinetic assays demonstrated that several initial hits also inhibit human cytomegalovirus protease (HCMV Pr). Binding of these hits to HCMV Pr was also confirmed by NMR spectroscopy. Despite the use of a target-agnostic fragment library, more than 80 % of confirmed hits disrupted dimerization and bound to a previously reported pocket at the dimer interface of KSHV Pr, not to the active site. One class of fragments, an aminothiazole scaffold, was further explored using commercially available analogues. These compounds demonstrated greater than 100-fold improvement of inhibition. This study illustrates the power of fragment-based screening for these challenging enzymatic targets and provides an example of the potential druggability of pockets at protein–protein interfaces.
Co-reporter:Jonathan E. Gable, Timothy M. Acker, and Charles S. Craik
Chemical Reviews 2014 Volume 114(Issue 22) pp:11382
Publication Date(Web):October 2, 2014
DOI:10.1021/cr500255e
Co-reporter:Jonathan E. Gable, Gregory M. Lee, Priyadarshini Jaishankar, Brian R. Hearn, Christopher A. Waddling, Adam R. Renslo, and Charles S. Craik
Biochemistry 2014 Volume 53(Issue 28) pp:4648-4660
Publication Date(Web):June 30, 2014
DOI:10.1021/bi5003234
Herpesviruses rely on a homodimeric protease for viral capsid maturation. A small molecule, DD2, previously shown to disrupt dimerization of Kaposi’s sarcoma-associated herpesvirus protease (KSHV Pr) by trapping an inactive monomeric conformation and two analogues generated through carboxylate bioisosteric replacement (compounds 2 and 3) were shown to inhibit the associated proteases of all three human herpesvirus (HHV) subfamilies (α, β, and γ). Inhibition data reveal that compound 2 has potency comparable to or better than that of DD2 against the tested proteases. Nuclear magnetic resonance spectroscopy and a new application of the kinetic analysis developed by Zhang and Poorman [Zhang, Z. Y., Poorman, R. A., et al. (1991) J. Biol. Chem. 266, 15591–15594] show DD2, compound 2, and compound 3 inhibit HHV proteases by dimer disruption. All three compounds bind the dimer interface of other HHV proteases in a manner analogous to binding of DD2 to KSHV protease. The determination and analysis of cocrystal structures of both analogues with the KSHV Pr monomer verify and elaborate on the mode of binding for this chemical scaffold, explaining a newly observed critical structure–activity relationship. These results reveal a prototypical chemical scaffold for broad-spectrum allosteric inhibition of human herpesvirus proteases and an approach for the identification of small molecules that allosterically regulate protein activity by targeting protein–protein interactions.
Co-reporter:Cheryl A. Tajon, Daeha Seo, Jennifer Asmussen, Neil Shah, Young-wook Jun, and Charles S. Craik
ACS Nano 2014 Volume 8(Issue 9) pp:9199
Publication Date(Web):August 28, 2014
DOI:10.1021/nn502959q
Caspases are proteases involved in cell death, where caspase-3 is the chief executioner that produces an irreversible cutting event in downstream protein substrates and whose activity is desired in the management of cancer. To determine such activity in clinically relevant samples with high signal-to-noise, plasmon rulers are ideal because they are sensitively affected by their interparticle separation without ambiguity from photobleaching or blinking effects. A plasmon ruler is a noble metal nanoparticle pair, tethered in close proximity to one another via a biomolecule, that acts through dipole–dipole interactions and results in the light scattering to increase exponentially. In contrast, a sharp decrease in intensity is observed when the pair is confronted by a large interparticle distance. To align the mechanism of protease activity with building a sensor that can report a binary signal in the presence or absence of caspase-3, we present a caspase-3 selective plasmon ruler (C3SPR) composed of a pair of Zn0.4Fe2.6O4@SiO2@Au core–shell nanoparticles connected by a caspase-3 cleavage sequence. The dielectric core (Zn0.4Fe2.6O4@SiO2)-shell (Au) geometry provided a brighter scattering intensity versus solid Au nanoparticles, and the magnetic core additionally acted as a purification handle during the plasmon ruler assembly. By monitoring the decrease in light scattering intensity per plasmon ruler, we detected caspase-3 activity at single molecule resolution across a broad dynamic range. This was observed to be as low as 100 fM of recombinant material or 10 ng of total protein from cellular lysate. By thorough analyses of single molecule trajectories, we show caspase-3 activation in a drug-treated chronic myeloid leukemia (K562) cancer system as early as 4 and 8 h with greater sensitivity (2- and 4-fold, respectively) than conventional reagents. This study provides future implications for monitoring caspase-3 as a biomarker and efficacy of drugs.Keywords: caspase; gold nanoparticles; leukemia; plasmon coupling; single molecule;
Co-reporter:Aaron M. LeBeau;Minhee Lee;Stephanie T. Murphy;Byron C. Hann;Robert S. Warren;Romelyn Delos Santos;John Kurhanewicz;Samir M. Hanash;Henry F. VanBrocklin;
Proceedings of the National Academy of Sciences 2013 110(1) pp:93-98
Publication Date(Web):December 17, 2012
DOI:10.1073/pnas.1218694110
Proteases responsible for the increased peritumoral proteolysis associated with cancer represent functional biomarkers for
monitoring tumorigenesis. One attractive extracellular biomarker is the transmembrane serine protease matriptase. Found on
the surface of epithelial cells, the activity of matriptase is regulated by its cognate inhibitor hepatocyte growth factor
activator inhibitor-1 (HAI-1). Quantitative mass spectrometry allowed us to show that, in selected cancers, HAI-1 expression
decreases, leading to active matriptase. A preclinical probe specific for the measurement of emergent active matriptase was
developed. Using an active-site–specific, recombinant human antibody for matriptase, we found that the selective targeting
of active matriptase can be used to visualize the tumorigenic epithelium. Live-cell fluorescence imaging validated the selectivity
of the antibody in vitro by showing that the probe localized only to cancer cell lines with active matriptase on the surface.
Immunofluorescence with the antibody documented significant levels of active matriptase in 68% of primary and metastatic colon
cancer sections from tissue microarrays. Labeling of the active form of matriptase in vivo was measured in human colon cancer
xenografts and in a patient-derived xenograft model using near-infrared and single-photon emission computed tomography imaging.
Tumor uptake of the radiolabeled antibody, 111In-A11, by active matriptase was high in xenografts (28% injected dose per gram) and was blocked in vivo by the addition of
a matriptase-specific variant of ecotin. These findings suggest, through a HAI-1–dependent mechanism, that emergent active
matriptase is a functional biomarker of the transformed epithelium and that its proteolytic activity can be exploited to noninvasively
evaluate tumorigenesis in vivo.
Co-reporter:Gregory M. Lee, Eaman Balouch, David H. Goetz, Ana Lazic, James H. McKerrow, and Charles S. Craik
Biochemistry 2012 Volume 51(Issue 50) pp:
Publication Date(Web):November 26, 2012
DOI:10.1021/bi301305k
Cruzain is a member of the papain/cathepsin L family of cysteine proteases, and the major cysteine protease of the protozoan Trypanosoma cruzi, the causative agent of Chagas disease. We report an autoinduction methodology that provides soluble cruzain in high yields (>30 mg/L in minimal medium). These increased yields provide sufficient quantities of active enzyme for use in nuclear magnetic resonance (NMR)-based ligand mapping. Using circular dichroism and NMR spectroscopy, we also examined the solution-state structural dynamics of the enzyme in complex with a covalently bound vinyl sulfone inhibitor (K777). We report the backbone amide and side chain carbon chemical shift assignments of cruzain in complex with K777. These resonance assignments were used to identify and map residues located in the substrate binding pocket, including the catalytic Cys25 and His162. Selective [15N]Cys, [15N]His, and [13C]Met labeling was performed to quickly assess cruzain–ligand interactions for a set of eight low-molecular weight compounds exhibiting micromolar binding or inhibition. Chemical shift perturbation mapping verified that six of the eight compounds bind to cruzain at the active site. Three different binding modes were delineated for the compounds, namely, covalent, noncovalent, and noninteracting. These results provide examples of how NMR spectroscopy can be used to screen compounds for fast evaluation of enzyme–inhibitor interactions to facilitate lead compound identification and subsequent structural studies.
.jpg)
![Carbamic acid,N-[(1S,2R)-3-[(1,3-benzodioxol-5-ylsulfonyl)(2-methylpropyl)amino]-2-hydroxy-1-[[4-[(2-methyl-4-thiazolyl)methoxy]phenyl]methyl]propyl]-,(3R,3aS,6aR)-hexahydrofuro[2,3-b]furan-3-yl ester](http://img.cochemist.com/ccimg/313700/313682-08-5.png)
![Carbamic acid,N-[(1S,2R)-3-[(1,3-benzodioxol-5-ylsulfonyl)(2-methylpropyl)amino]-2-hydroxy-1-[[4-[(2-methyl-4-thiazolyl)methoxy]phenyl]methyl]propyl]-,(3R,3aS,6aR)-hexahydrofuro[2,3-b]furan-3-yl ester](http://img.cochemist.com/ccimg/313700/313682-08-5_b.png)
![Morpholine,4-[2-bromo-4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/892600/892502-13-5.png)
![Morpholine,4-[2-bromo-4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/892600/892502-13-5_b.png)
![Propanamide, N-[2-chloro-4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/839700/839689-22-4.png)
![Propanamide, N-[2-chloro-4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/839700/839689-22-4_b.png)






















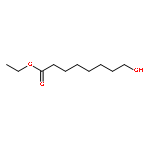
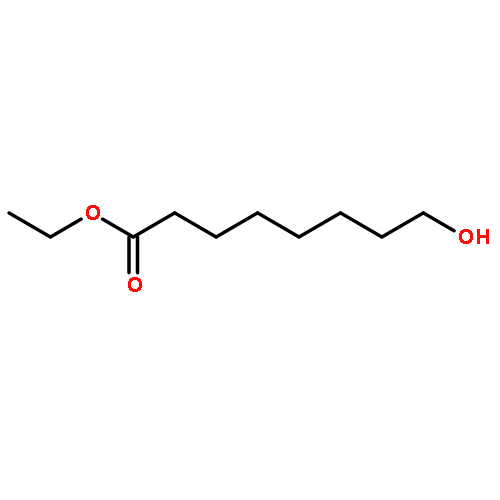
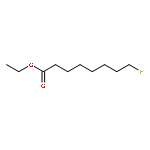
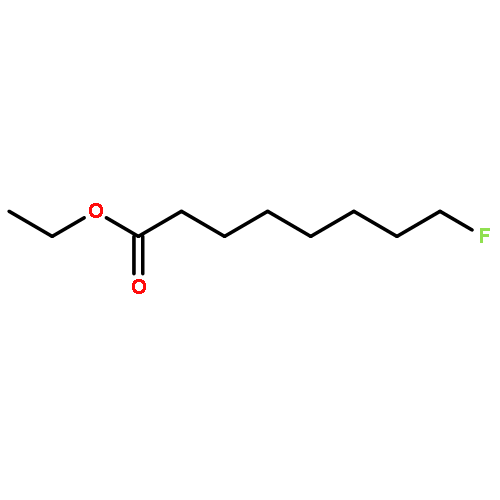
![Octanoic acid, 8-[[(4-methylphenyl)sulfonyl]oxy]-, ethyl ester](http://img.cochemist.com/ccimg/144700/144682-03-1.png)
![Octanoic acid, 8-[[(4-methylphenyl)sulfonyl]oxy]-, ethyl ester](http://img.cochemist.com/ccimg/144700/144682-03-1_b.png)


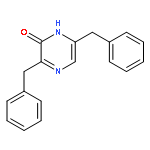
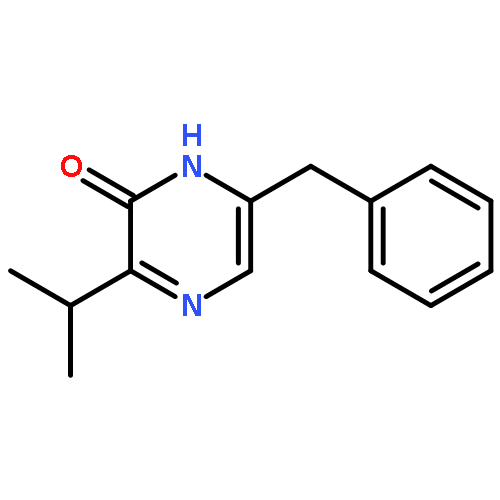
![Benzamide, N-[2-(aminocarbonyl)phenyl]-4-methyl-](http://img.cochemist.com/ccimg/53000/52910-88-0.png)
![Benzamide, N-[2-(aminocarbonyl)phenyl]-4-methyl-](http://img.cochemist.com/ccimg/53000/52910-88-0_b.png)
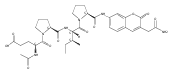
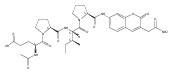
![N-(3-METHYLBUTANOYL)-L-VALYL-N-[(3S,4S)-1-{[(2S)-1-{[(2S,3S)-1-CA<WBR />RBOXY-2-HYDROXY-5-METHYL-3-HEXANYL]AMINO}-1-OXO-2-PROPANYL]AMINO}<WBR />-3-HYDROXY-6-METHYL-1-OXO-4-HEPTANYL]-L-VALINAMIDE](http://img.cochemist.com/ccimg/39400/39324-30-6.png)
![N-(3-METHYLBUTANOYL)-L-VALYL-N-[(3S,4S)-1-{[(2S)-1-{[(2S,3S)-1-CA<WBR />RBOXY-2-HYDROXY-5-METHYL-3-HEXANYL]AMINO}-1-OXO-2-PROPANYL]AMINO}<WBR />-3-HYDROXY-6-METHYL-1-OXO-4-HEPTANYL]-L-VALINAMIDE](http://img.cochemist.com/ccimg/39400/39324-30-6_b.png)
![2-AMINO-6,7,8,9-TETRAHYDRODIBENZO[B,D]FURAN 97%](http://img.cochemist.com/ccimg/38100/38084-44-5.png)
![2-AMINO-6,7,8,9-TETRAHYDRODIBENZO[B,D]FURAN 97%](http://img.cochemist.com/ccimg/38100/38084-44-5_b.png)












































![L-Proline,N-[[(2S,3S)-3-[(propylamino)carbonyl]-2-oxiranyl]carbonyl]-L-isoleucyl-](http://img.cochemist.com/ccimg/134500/134448-10-5.png)
![L-Proline,N-[[(2S,3S)-3-[(propylamino)carbonyl]-2-oxiranyl]carbonyl]-L-isoleucyl-](http://img.cochemist.com/ccimg/134500/134448-10-5_b.png)






![L-Leucinamide,N-acetyl-L-leucyl-N-[(1S)-4-[(aminoiminomethyl)amino]-1-formylbutyl]-](http://img.cochemist.com/ccimg/55200/55123-66-5.png)
![L-Leucinamide,N-acetyl-L-leucyl-N-[(1S)-4-[(aminoiminomethyl)amino]-1-formylbutyl]-](http://img.cochemist.com/ccimg/55200/55123-66-5_b.png)