Co-reporter:Tongliang Zhou;Lingfei Yang;Lei Liang;Hui Liu;Yuanjun Zhu;Mengyang Shui;Lan Yuan;Fengrong Xu;Yan Niu;Chao Wang
European Journal of Organic Chemistry 2017 Volume 2017(Issue 22) pp:3274-3281
Publication Date(Web):2017/06/16
DOI:10.1002/ejoc.201700355
A class of pyridazinone derivatives as near-infrared optical probes in fluorescence microscopy images was designed. The design strategy consisted of the stepwise extension and modification of pyridazinone by expansion of the electron-donating moiety to a larger π-conjugated system and anchoring a subcellular directing group such as triphenylphosphine or morpholine. All the desired products were successfully applied in cell imaging with high subcellular colocalization. Furthermore, these fluorescent probes showed excellent performance in mouse-brain imaging.
Co-reporter:Gang Yan, Lina Hao, Yan Niu, Wenjie Huang, Wei Wang, Fengrong Xu, Lei Liang, Chao Wang, Hongwei Jin, Ping Xu
European Journal of Medicinal Chemistry 2017 Volume 137(Volume 137) pp:
Publication Date(Web):8 September 2017
DOI:10.1016/j.ejmech.2017.06.020
•Guided by docking studies, a series of compounds were designed and synthesized.•Enzyme activity, affinity, permeability and cytotoxicity have been evaluated.•Sixteen compounds were found to have μM to sub-μM level activities towards BACE1.•Compound 41 exhibited high activities and the most optimized drug-like profiles.In this work, a series of 2-substituted-thio-N-(4-substituted-thiazol/1H-imidazol-2-yl)acetamide derivatives were developed as β-secretase (BACE-1) inhibitors. Supported by docking study, a small library of derivatives were designed, synthesized and biologically evaluated in vitro. In addition, the selected compounds were tested with affinity (KD) towards BACE-1, blood brain barrier (BBB) permeability and cytotoxicity. The studies revealed that the most potent analog 41 (IC50 = 4.6 μM) with high predicted BBB permeability and low cellular cytotoxicity, could serve as a good lead structure for further optimization.Download high-res image (306KB)Download full-size image
Co-reporter:Hongyue Li, Tongliang Zhou, Hui Liu, Fengrong Xu, Yan Niu, Chao Wang, Lei Liang, Ping Xu
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 10(Issue 10) pp:
Publication Date(Web):15 May 2017
DOI:10.1016/j.bmcl.2017.03.022
A series of Schiff base ligands (L1–L5) and their cobalt(II) complexes (1–5) were designed and synthesized for MEK1 binding experiment. The biological evaluation results showed that Bis(N,N′-disalicylidene)-3,4-phenylenediamine-cobalt(II) 1 and Bis(N,N′-disalicylidene)-1,2-cyclohexanediamine-cobalt(II) 2 are much more effective than the parent Schiff bases (L1 and L2). Importantly, 2 exhibited MEK1 binding affinity with IC50 71 nM, which is so far the best result for metal complexes and more potent than U0126 (7.02 μM) and AZD6244 (2.20 μM). Docking study was used to elucidate the binding modes of complex 2 with MEK1. Thus cobalt(II) complex 2 may be further developed as a novel MEK1 inhibitor.Download high-res image (87KB)Download full-size image
Co-reporter:Jing Sun, Yan Niu, Chao Wang, Hao Zhang, Bingyu Xie, Fengrong Xu, Hongwei Jin, Yihong Peng, Lei Liang, Ping Xu
Bioorganic & Medicinal Chemistry 2016 Volume 24(Issue 16) pp:3472-3482
Publication Date(Web):15 August 2016
DOI:10.1016/j.bmc.2016.05.055
Enterovirus 71 (EV71) is a kind of RNA virus and one of the two causes of Hand, foot and mouth disease (HFMD). Inhibitors that target key components of Ras/Raf/MEK/ERK pathway in host cells could impair replication of EV71. A series of 3-benzyl-1,3-benzoxazine-2,4-diones were designed from a specific MEK inhibitor G8935, by replacing the double bond between C3 and C4 within the coumarin scaffold with amide bond. One compound (9f) showed submicromolar inhibitory activity among the 12 derivatives. Further optimization on 9f led to two active compounds (9k and 9m) with nanomolar bioactivities (55 nM and 60 nM). The results of enzymatic assays also demonstrated that this series of compounds were allosteric inhibitors of unphosphorylated MEK1. The binding mode of compound 9k was predicted by molecular dynamic simulation and the key interactions were same as published MEK1/2 allosteric inhibitors. In the cell-based assays, compounds 9k and 9m could effectively suppress the ERK1/2 pathway, expression of EV71 VP1, and EV71 induced cytopathic effect (CPE) in rhabdomyosarcoma (RD) cells.
Co-reporter:Lingfei Yang;Dr. Yuanjun Zhu;Mengyang Shui;Tongliang Zhou;Yuanbo Cai;Wei Wang;Fengrong Xu;Yan Niu;Chao Wang;Dr. Jun-Long Zhang;Dr. Ping Xu;Dr. Lan Yuan;Dr. Lei Liang
Chemistry - A European Journal 2016 Volume 22( Issue 35) pp:12363-12370
Publication Date(Web):
DOI:10.1002/chem.201601499
Abstract
Phthalazinone derivatives were designed as optical probes for one- and two-photon fluorescence microscopy imaging. The design strategy involves stepwise extension and modification of pyridazinone by 1) expansion of pyridazinone to phthalazinone, a larger conjugated system, as the electron acceptor, 2) coupling of electron-donating aromatic groups such as N,N-diethylaminophenyl, thienyl, naphthyl, and quinolyl to the phthalazinone, and 3) anchoring of an alkyl chain to the phthalazinone with various terminal substituents such as triphenylphosphonio, morpholino, triethylammonio, N-methylimidazolio, pyrrolidino, and piperidino. Theoretical calculations were utilized to verify the initial design. The desired fluorescent probes were synthesized by two different routes in considerable yields. Twenty-two phthalazinone derivatives were synthesized and their photophysical properties were measured. Selected compounds were applied in cell imaging, and valuable information was obtained. Furthermore, the designed compounds showed excellent performance in two-photon microscopic imaging of mouse brain slices.
Co-reporter:Dr. Qi Sun;Dr. Bo Xu;Dr. Yan Niu;Fengrong Xu;Dr. Lei Liang;Dr. Chao Wang;Jiapei Yu;Gang Yan;Wei Wang;Dr. Hongwei Jin; Ping Xu
ChemMedChem 2015 Volume 10( Issue 3) pp:498-510
Publication Date(Web):
DOI:10.1002/cmdc.201402484
Abstract
Proteasome inhibitors are promising compounds for a number of therapies, including cardiovascular and eye diseases, diabetes, and cancers. We previously reported a series of furan-based peptidic inhibitors with moderate potencies against the proteasome β5 subunit, hypothesizing that the C-terminal furyl ketone motif could form a covalent bond with the catalytic residue, threonine 1. In this context, we describe further optimizations of the furan-based peptides, and a series of dipeptidic and tripeptidic inhibitors were designed and synthesized, aiming at improved potency and better solubility. Most of the tripeptidic inhibitors demonstrated improved potency and selectivity as β5 subunit inhibitors in both enzymatic and cellular assays, and good antineoplastic activities in various tumor cell lines were also observed. However, no inhibitory effects were observed for the dipeptidic compounds, which led us to presume that a noncovalent binding mode is adopted. Docking studies and molecular dynamics simulations were carried out to verify this presumption, with results showing that the distance between the furyl ketone motif and Thr1 is slightly too long to form covalent bond.
Co-reporter:Peng Liu, Yan Niu, Chao Wang, Qi Sun, Yaya Zhai, Jiapei Yu, Jing Sun, Fengrong Xu, Gang Yan, Wenjie Huang, Lei Liang, Ping Xu
European Journal of Medicinal Chemistry 2014 Volume 79() pp:413-421
Publication Date(Web):22 May 2014
DOI:10.1016/j.ejmech.2014.04.025
•Novel 4-oxo-1,4-dihydro-quinoline-3-carboxamide motif with BACE-1 were reported.•A series of 6-substituted derivatives as novel BACE-1 inhibitors were synthesized.•Compounds 14d and 14e were good drug-like profiles for further modification.In this work, we report a series of new 4-oxo-1,4-dihydro-quinoline-3-carboxamide derivatives as β-secretase (BACE-1) inhibitors. Supported by docking study, a small library of derivatives were designed, synthesized and biologically evaluated in vitro. The studies revealed that the most potent analog 14e (IC50 = 1.89 μM) with low cellular cytotoxicity and high predicted blood brain barrier permeability, could serve as a good structure for further modification.A series of 4-oxo-1,4-dihydro-quinoline-3-carboxamids were designed from 1 as BACE-1 inhibitors, among which 14e exhibited better drugabilities including improved inhibitory potency, low cytotoxicity and possibility to penetrate BBB.
Co-reporter:Wei Wang;Lei Liang;Fengrong Xu;Wenjie Huang;Yan Niu;Qi Sun
European Journal of Organic Chemistry 2014 Volume 2014( Issue 31) pp:6863-6867
Publication Date(Web):
DOI:10.1002/ejoc.201402986
Abstract
A ruthenium-catalyzed switchable N–H/C–H alkenylation reaction of 6-phenyl-(dihydro)pyridazin-3(2H)-ones triggered by a nitrogen/oxygen atmosphere was developed. To achieve switchable modification of the two important sites of the widely used pharmacophore, a simple and efficient procedure containing two optimized ruthenium catalytic systems was utilized, which afforded excellent activity, high selectivity, and good tolerance of a wide range of functional groups.
Co-reporter:Lei Liang, Wei Wang, Fengrong Xu, Yan Niu, Qi Sun, and Ping Xu
Organic Letters 2013 Volume 15(Issue 11) pp:2770-2773
Publication Date(Web):May 14, 2013
DOI:10.1021/ol401105x
A Cu-catalyzed tandem dehydrogenation/dehalogenation sequential reaction along with N-arylation has been developed for the synthesis of pyridazinone derivatives in an aerobic and aqueous environment. To achieve the transformation of three chemical bonds in a one-pot reaction, a multifunctional copper catalyst was used which afforded excellent activity, high selectivity, and recyclability. The catalytic system consists of a water-soluble Cusalen complex and Na2CO3 in neat water and an air atmosphere.
Co-reporter:Lei Liang;Guanyu Yang;Wei Wang;Fengrong Xu;Yan Niu;Qi Sun
Advanced Synthesis & Catalysis 2013 Volume 355( Issue 7) pp:1284-1290
Publication Date(Web):
DOI:10.1002/adsc.201300026
Abstract
A copper-catalyzed cascade dehydrogenative and dehalogenative reaction of halogenated 6-phenyl-4,5-dihydropyridazin-3(2H)-ones to 6-phenylpyridazin-3(2H)-ones has been developed. Moreover, the catalytic system consisting of copper(II) acetate/sodium carbonate/pyridine exhibits high reactivity and selectivity with oxygen as the terminal oxidant.
Co-reporter:Lei Liang;Guanyu Yang;Fengrong Xu;Yan Niu;Qi Sun
European Journal of Organic Chemistry 2013 Volume 2013( Issue 27) pp:6130-6136
Publication Date(Web):
DOI:10.1002/ejoc.201300640
Abstract
A simple and efficient procedure for the synthesis of pyridazin-3(2H)-ones through copper-catalyzed dehydrogenation of a single C–C bond of 4,5-dihydropyridazin-3(2H)-ones to a C=C bond with oxygen as the terminal oxidant is described. Functional groups including hydroxy, carboxylic, bromo, chloro, cyano, nitro and alkoxy were all tolerated under the reaction conditions. Moreover, this methodology was applied to the preparation of a series of structurally similar N-substituted 6-phenylpyridazinone compounds containing fluorine. The dehydrogenation reactions exhibit good yields and selectivity.
Co-reporter:Dr. Lei Liang;Wei Wang;Jun Wu;Fengrong Xu;Dr. Yan Niu;Dr. Bo Xu;Dr. Ping Xu
Chemistry - A European Journal 2013 Volume 19( Issue 41) pp:13774-13782
Publication Date(Web):
DOI:10.1002/chem.201302495
Abstract
There is widespread interest in the application, optimization, and evolution of the transition-metal-catalyzed arylation of N-heteroarenes to discover full-color tunable fluorescent core frameworks. Inspired by the versatile roles of pyridazinone in organic synthesis and medicinal chemistry, herein, we report a simple and efficient copper-catalyzed cross-coupling reaction for the N-functionalization of pyridazinones in neat water. To achieve the efficient conversion of pyridazinones and organic halides in aqueous phase, a series of copper-salen complexes composed of different Schiff base ligands were investigated by rational design. A final choice of fine-tuned hydrophilicity balanced with lipophilicity among the candidates was confirmed, which affords excellent activity towards the reaction of a wide range of pyridazinones and organic halides. More importantly, the products as N-substituted pyridazinones were synthesized rationally by this methodology as full-color tunable fluorescent agents (426–612 nm). The N2 position of pyridazinones was modified by different aryl group such as benzothiazole, N,N-dimethylaniline, 3-quinoline, 4-isoquinoline and 2-thiophene, resulting in a series of full-color tunable fluorescent reagents. Meanwhile, the effects of electron-donating and electron-withdrawing groups of the 6-substituted phenyl ring have also been investigated to optimize the fluorescent properties. These fluorescent core frameworks were studied in several cell lines as fluorescent dyes. Different colors from blue to red were observed by using fluorescence microscopy and confocal microscopy.
Co-reporter:Yan Niu;Chao Ma;Hongwei Jin;Fengrong Xu;Haifei Gao;Peng Liu;Yongjian Li;Chao Wang;Guanyu Yang
Chemical Biology & Drug Design 2012 Volume 79( Issue 6) pp:972-980
Publication Date(Web):
DOI:10.1111/j.1747-0285.2012.01367.x
This article describes the identification of two small molecular inhibitors for β-secretase by integrating virtual screening with fluorescence resonance energy transfer bioassay. A ligand-based pharmacophore model was developed, and the sequential virtual screening of ZINC database was performed using the acquired pharmacophore model and molecular docking. Biological evaluation of 10 virtual hits led to the identification of two novel inhibitors with IC50 values of 4.76 and 0.31 μm, respectively. These two moderate inhibitors could represent new potentials for the development of anti-Alzheimer’s disease agents.
Co-reporter:Yan Niu;Haifei Gao;Fengrong Xu;Chao Wang;Peng Liu;Guanyu Yang;Qi Sun
Chemical Biology & Drug Design 2012 Volume 80( Issue 5) pp:775-780
Publication Date(Web):
DOI:10.1111/cbdd.12016
A series of 3, 5-disubstituted benzimidamides were synthesized and biologically evaluated as potential BACE1 inhibitors. Both the targeted compounds (benzimidamides) and the synthetic intermediates (benzonitriles) were tested for their BACE1 inhibitory activities in a cell-free FRET assay. All the synthesized benzimidamides were active as BACE1 inhibitors and compound 6d showed the lowest IC50 value of 3.35 μm. Molecular docking study proposed a binding mode, which would help to the further optimization on 6d to achieve more potent, BBB penetrant BACE1 inhibitors.
Co-reporter:Yan Niu, Yuehua Wang, Xiaomin Zou, Xiaoming Yang, Chao Ma, Yang Lü, Bo Zhou, Yue Yuan, Guanhua Du, Ping Xu
European Journal of Medicinal Chemistry 2010 Volume 45(Issue 5) pp:2089-2094
Publication Date(Web):May 2010
DOI:10.1016/j.ejmech.2010.01.044
Based on the structure of OM99-2 and the X-ray crystal structure of its complex with β-secretase, a series of compounds containing the Leu*Ala hydroxyethylene isostere as a scissile bond substitution were designed. 31 compounds were synthesized and their β-secretase inhibition activities were measured. It was found that isobutyl group was a better R3 substitution as C-terminus in our target compounds, and 4-nitrobenzyl group was the best R2 side chain. With the aid of molecular modeling, the binding modes of compounds 9 and 22 with β-secretase were compared. The result revealed a stronger bonding mode of 22 than 9. This explored that the optimal length of this series of peptidomimetic inhibitors was P3–P2′. The molecular weights of compounds with this length are around 600.31 Compounds containing the Leu*Ala hydroxyethylene isostere as a scissile bond substitution were designed, synthesized and evaluated with their β-secretase inhibition activities. It was found that isobutyl group was a better R3 substitution as C-terminus, and 4-nitrobenzyl group was the best R2 side chain. With the aid of molecular modeling, the binding modes of compounds 9 and 22 with β-secretase were compared.
Co-reporter:Wenjing Chen, Ke Mou, Bo Xu, Xiaomei Ling, Jingrong Cui, Ping Xu
Analytical Biochemistry 2009 Volume 394(Issue 1) pp:62-67
Publication Date(Web):1 November 2009
DOI:10.1016/j.ab.2009.07.020
A method for studying 20S proteasome inhibitors by capillary electrophoresis (CE) has been developed. Proteasome plays a fundamental role in degrading key regulatory proteins. The 20S proteasome can degrade intrinsically disordered proteins in an ATP-independent manner without additional “helper” molecules. The discovery of new proteasome inhibitors with little or no toxicity is highly desirable in anticancer therapy. In this study, the inhibitory effects of MG132 and MG115 on the 20S proteasome were evaluated by CE for the first time. The optimized CE conditions were as follows: fused-silica capillary of 30 cm effective length and 75 μm internal diameter, pressure injection of 0.5 psi for 5 s, 50 mM Hepes buffer (pH 7.6) with 2% dimethyl sulfoxide, constant voltage of 20 kV, and detection wavelength at 340 nm. Also, the new method was used to study the inhibitory effects of 30 novel peptidyl vinyl ester derivatives of MG132. The 50% inhibition concentrations (IC50 values) of MG132 and MG115 were 40.0 and 84.7 nM, respectively. Two new compounds, XP32 and XP35, showed considerable inhibitory effects on the 20S proteasome. When the concentrations of them were fixed at 172 nM, their inhibition rates were 36.2% and 29.1%, respectively. The results showed that the CE method was powerful, sensitive, and fast and required little sample. It could be employed as one of the reliable drug screening methods for 20S proteasome inhibitors.
Co-reporter:Yuanjun Zhu, Tongliang Zhou, Lingfei Yang, Lan Yuan, Lei Liang, Ping Xu
Biochemical and Biophysical Research Communications (13 May 2017) Volume 486(Issue 4) pp:904-908
Publication Date(Web):13 May 2017
DOI:10.1016/j.bbrc.2017.03.121
Co-reporter:Tongliang Zhou, Yuanbo Cai, Lei Liang, Lingfei Yang, Fengrong Xu, Yan Niu, Chao Wang, Jun-Long Zhang, Ping Xu
Bioorganic & Medicinal Chemistry Letters (1 December 2016) Volume 26(Issue 23) pp:5780-5784
Publication Date(Web):1 December 2016
DOI:10.1016/j.bmcl.2016.10.043
We reported the synthesis, characterization and biological activity of several copper(II) Schiff base complexes, which exhibit high proteasome inhibitory activities with particular selectivity of β2 subunit. Structure–activity relationships information obtained from complex Na2[Cu(a4s1)] demonstrated that distinct bonding modes in β2 and β5 subunits determines its selectivity and potent inhibition for β2 subunit.
Co-reporter:Tongliang Zhou, Yuanbo Cai, Lei Liang, Lingfei Yang, Fengrong Xu, Yan Niu, Chao Wang, Jun-Long Zhang, Ping Xu
Bioorganic & Medicinal Chemistry Letters (1 December 2016) Volume 26(Issue 23) pp:
Publication Date(Web):1 December 2016
DOI:10.1016/j.bmcl.2016.10.043
We reported the synthesis, characterization and biological activity of several copper(II) Schiff base complexes, which exhibit high proteasome inhibitory activities with particular selectivity of β2 subunit. Structure–activity relationships information obtained from complex Na2[Cu(a4s1)] demonstrated that distinct bonding modes in β2 and β5 subunits determines its selectivity and potent inhibition for β2 subunit.

![Copper,[[2,2'-[1,2-ethanediylbis[(nitrilo-kN)methylidyne]]bis[phenolato-kO]](2-)]-, (SP-4-2)-](http://img.cochemist.com/ccimg/14200/14167-15-8.png)
![Copper,[[2,2'-[1,2-ethanediylbis[(nitrilo-kN)methylidyne]]bis[phenolato-kO]](2-)]-, (SP-4-2)-](http://img.cochemist.com/ccimg/14200/14167-15-8_b.png)


![Benzoic acid, 3,4-bis[[(2-hydroxyphenyl)methylene]amino]-](http://img.cochemist.com/ccimg/87600/87578-89-0.png)
![Benzoic acid, 3,4-bis[[(2-hydroxyphenyl)methylene]amino]-](http://img.cochemist.com/ccimg/87600/87578-89-0_b.png)




![L-TYROSINE, N-[(PHENYLMETHOXY)CARBONYL]-L-LEUCYL-O-METHYL-](http://img.cochemist.com/ccimg/886800/886758-89-0.png)
![L-TYROSINE, N-[(PHENYLMETHOXY)CARBONYL]-L-LEUCYL-O-METHYL-](http://img.cochemist.com/ccimg/886800/886758-89-0_b.png)




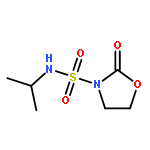
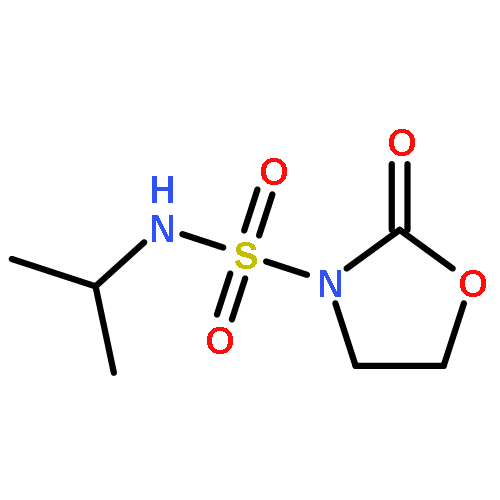














![L-Leucinamide,N-[(phenylmethoxy)carbonyl]-L-leucyl-N-[(1S)-1-formyl-3-methylbutyl]-](http://img.cochemist.com/ccimg/133500/133407-82-6.png)
![L-Leucinamide,N-[(phenylmethoxy)carbonyl]-L-leucyl-N-[(1S)-1-formyl-3-methylbutyl]-](http://img.cochemist.com/ccimg/133500/133407-82-6_b.png)
![L-Tyrosine,O-methyl-N-[(phenylmethoxy)carbonyl]-, methyl ester](http://img.cochemist.com/ccimg/121800/121778-71-0.png)
![L-Tyrosine,O-methyl-N-[(phenylmethoxy)carbonyl]-, methyl ester](http://img.cochemist.com/ccimg/121800/121778-71-0_b.png)
![L-Tyrosine, O-methyl-N-[N-[(phenylmethoxy)carbonyl]-L-phenylalanyl]-](http://img.cochemist.com/ccimg/118300/118253-10-4.png)
![L-Tyrosine, O-methyl-N-[N-[(phenylmethoxy)carbonyl]-L-phenylalanyl]-](http://img.cochemist.com/ccimg/118300/118253-10-4_b.png)


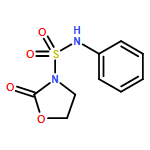
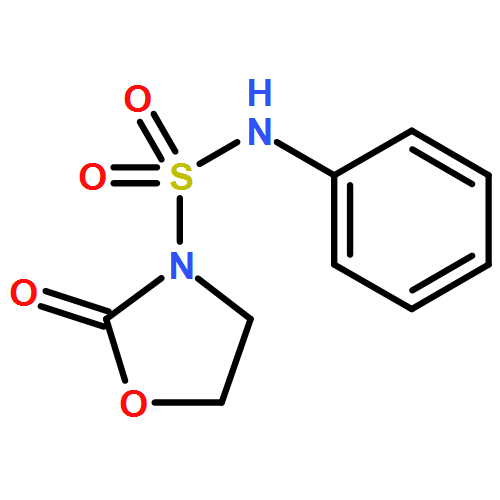





















![L-Tyrosine, N-[N-[(phenylmethoxy)carbonyl]-L-phenylalanyl]-, methylester](http://img.cochemist.com/ccimg/19400/19391-50-5.png)
![L-Tyrosine, N-[N-[(phenylmethoxy)carbonyl]-L-phenylalanyl]-, methylester](http://img.cochemist.com/ccimg/19400/19391-50-5_b.png)
![L-Tyrosine,O-methyl-N-[(phenylmethoxy)carbonyl]-](http://img.cochemist.com/ccimg/17600/17554-34-6.png)
![L-Tyrosine,O-methyl-N-[(phenylmethoxy)carbonyl]-](http://img.cochemist.com/ccimg/17600/17554-34-6_b.png)
![L-Phenylalanine,N-[(phenylmethoxy)carbonyl]-L-leucyl-](http://img.cochemist.com/ccimg/6500/6401-63-4.png)
![L-Phenylalanine,N-[(phenylmethoxy)carbonyl]-L-leucyl-](http://img.cochemist.com/ccimg/6500/6401-63-4_b.png)


![Methyl 3-phenyl-2-[[3-phenyl-2-(phenylmethoxycarbonylamino)propanoyl]amino]propanoate](http://img.cochemist.com/ccimg/4900/4892-10-8.png)
![Methyl 3-phenyl-2-[[3-phenyl-2-(phenylmethoxycarbonylamino)propanoyl]amino]propanoate](http://img.cochemist.com/ccimg/4900/4892-10-8_b.png)
![L-PHENYLALANINE, N-[(PHENYLMETHOXY)CARBONYL]-L-LEUCYL-, METHYL ESTER](http://img.cochemist.com/ccimg/4900/4818-00-2.png)
![L-PHENYLALANINE, N-[(PHENYLMETHOXY)CARBONYL]-L-LEUCYL-, METHYL ESTER](http://img.cochemist.com/ccimg/4900/4818-00-2_b.png)


![L-Tyrosine,N-[N-[(phenylmethoxy)carbonyl]-L-phenylalanyl]- (9CI)](http://img.cochemist.com/ccimg/2600/2537-91-9.png)
![L-Tyrosine,N-[N-[(phenylmethoxy)carbonyl]-L-phenylalanyl]- (9CI)](http://img.cochemist.com/ccimg/2600/2537-91-9_b.png)








![4-amino-N-[3-(trifluoromethyl)phenyl]benzenesulfonamide](http://img.cochemist.com/ccimg/400/339-40-2.png)
![4-amino-N-[3-(trifluoromethyl)phenyl]benzenesulfonamide](http://img.cochemist.com/ccimg/400/339-40-2_b.png)



