Co-reporter:Qiao Wang, Fuyi Chen, Yaxing Liu, Nan Zhang, Liang An, and Roy L. Johnston
ACS Applied Materials & Interfaces October 18, 2017 Volume 9(Issue 41) pp:35701-35701
Publication Date(Web):September 27, 2017
DOI:10.1021/acsami.7b05186
Incorporating an oxophilic metal into a noble metal to produce a cost-effective Ag3Sn nanointermetallic catalyst is an emerging approach to enhance the catalytic activity of monometallic Ag in fuel cells, which is different from previous notions that consider a transition metal to increase the catalytic activity of Pt. The Ag3Sn electrocatalyst is prepared by a facile electrodeposition method and exhibits high catalytic performance for the oxygen reduction reaction (ORR) and borohydride oxidation reaction (BOR). The Ag3Sn electrocatalyst has an ORR specific activity of 0.246 mA cm–2, 1.3 times greater than the value of commercial Pt/C (0.187 mA cm–2) and a long-term stability with an 11 mV decrement in the half-wave potential and 7.01% loss of the diffusion-limiting current density after 2000 cycles, superior to that of Pt/C. Moreover, the Ag3Sn electrocatalyst delivers a surprisingly higher BOR current density of 11.332 mA cm–2 than most bimetallic Ag alloys. The better ORR catalytic activities of Ag-based alloys may arise from the ensemble effect, in which Sn atoms may promote the oxygen adsorption and Ag atoms may contribute to the removal of reaction products.Keywords: borohydride oxidation reaction; ensemble effect; intermetallic Ag3Sn; oxophilic metal; oxygen reduction reaction;
Co-reporter:Tian-E Fan;Ilker Demiroglu;Heider A. Hussein;Tun-Dong Liu
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 39) pp:27090-27098
Publication Date(Web):2017/10/11
DOI:10.1039/C7CP04811D
The structures and surface adsorption sites of Pd–Ir nanoalloys are crucial to the understanding of their catalytic performance because they can affect the activity and selectivity of nanocatalysts. In this article, density functional theory (DFT) calculations are performed on bare Pd–Ir nanoalloys to systematically explore their stability and chemical ordering properties, before studying the adsorption of CO on the nanoalloys. First, the structural stability of 38-atom and 79-atom truncated octahedral (TO) Pd–Ir nanoalloys are investigated. Then the adsorption properties and preferred adsorption sites of CO on 38-atom Pd–Ir nanoalloys are considered. The PdshellIrcore structure, which has the lowest energy of all the considered isomers, exhibits the highest structural stability, while the PdcoreIrshell configuration is the least stable. In addition, the adsorption strength of CO on Ir atoms is found to be greater than on Pd for Pd–Ir nanoclusters. The preferred adsorption sites of CO on pure Pd and Ir clusters are in agreement with calculations and experiments on extended Pd and Ir surfaces. In addition, d-band center and charge effects on CO adsorption strength on Pd–Ir nanoalloys are analyzed by comparison with pure clusters. The study provides a valuable theoretical insight into catalytically active Pd–Ir nanoalloys.
Co-reporter:Jiali Wang;Fuyi Chen;Yachao Jin;Yimin Lei
Advanced Functional Materials 2017 Volume 27(Issue 23) pp:
Publication Date(Web):2017/06/01
DOI:10.1002/adfm.201700260
A novel one-pot approach for synthesizing the dealloyed nanomaterials at room temperature is introduced for the first time. In such a synthetic strategy, applying modulated potentials effectively simplifies the traditional dealloying route, which usually requires additional corrosion process to dissolve nonprecious metals. The dealloyed AuNi nanodendrites (AuNi NDs) with tunable composition and uniformly elemental distribution are well developed by the one-pot strategy. Impressively, the as-synthesized AuNi NDs exhibit a higher electrochemically active area and definite improvements in electrocatalytic activity for oxygen reduction reaction (ORR) and borohydride oxidation reaction (BOR) compared to the commercial Pt/C. In particular, the AuNi NDs are 81 mV more positive in half-wave potential and about 3.1 times higher in specific activity (at 0.85 V) for the ORR than Pt/C, together with excellent stability and methanol tolerance. The superior BOR activity is highly promising compared to the previously reported catalysts. The unique nanodendritic structure with Au-rich surface and bimetallic electronic effect is the main factor to greatly enhance the bifunctional catalytic performance for the AuNi NDs. Furthermore, such a newly developed facile method is of great significance because it is one of the first examples to effectively engineer dealloyed bimetallic nanostructures via the practical and low-cost route for electrocatalytic applications.
Co-reporter:Ilker Demiroglu, Z.Y. Li, Laurent Piccolo, Roy L. Johnston
Computational and Theoretical Chemistry 2017 Volume 1107(Volume 1107) pp:
Publication Date(Web):1 May 2017
DOI:10.1016/j.comptc.2017.02.012
•Metal-to-support charge transfer depends on which metal atoms lie at the interface with the support.•Molecular adsorption strengths on Janus segregated structures are lower than on pure Rh clusters.•Higher molecular adsorption energies are predicted for the less stable AucoreRhshell structure.•The d-band model works better for larger and supported alloy clusters due to reduced mechanical effects.AuRh/TiO2 nanocatalysts have proved their efficiency in several catalytic reactions. In this work, density functional theory calculations are performed to investigate the effect of the TiO2 support on the structures of fcc 38-atom and 79-atom AuRh nanoalloys and their adsorption properties towards the reactant molecules CO and O2. d-band centre analysis shows that the d-band model captures the trends better for both larger and supported alloy clusters due to reduced mechanical effects. Calculations reveal metal-to-support electron transfer, depending mainly on which metal atoms lie at the interface with the support. The adsorption strengths of CO and O2 molecules on experimentally-relevant Janus segregated structures are slightly lower than on pure Rh clusters, which may reduce poisoning effects, while maintaining the high reactivity of Rh. In addition, higher adsorption energies are predicted for the less stable AucoreRhshell structure, which may lead to adsorption-induced restructuring under reaction conditions.Download high-res image (276KB)Download full-size image
Co-reporter:Xiaoqiang Wu, Fuyi Chen, Nan Zhang, Adnan Qaseem and Roy L. Johnston
Journal of Materials Chemistry A 2016 vol. 4(Issue 9) pp:3527-3537
Publication Date(Web):29 Jan 2016
DOI:10.1039/C5TA09266C
Highly active electrocatalysts with good long term stability are vital for the commercialization of metal air batteries and alkaline fuel cells which involve the oxygen reduction reaction (ORR) at the cathode end. Herein, we developed a pulsed laser deposition (PLD) technique for the precise fabrication of silver–copper metallic glass (AgCu-MG) electrocatalysts. This PLD technique provides excellent control over the surface microtopography along with high flexibility for the deposition of different compositions of silver–copper metallic glass electrocatalysts onto nickel foam. Among all investigated Ag-based catalysts, AgCu-MG catalysts exhibit high electrocatalytic activity with a half-wave potential of 0.67 V (vs. RHE) which can be in situ enhanced by dealloying treatment in N2 saturated 0.1 M KOH solution. In situ dealloying of the AgCu-MG provides exceptional ORR catalytic activity with a half-wave potential of 0.78 V (vs. RHE) at 1600 rpm, which is comparable to 0.81 V (vs. RHE) of commercial Pt/C-20%. The AgCu-MG electrocatalyst showed excellent long-term stability in rechargeable zinc–air batteries. After 1000 charge–discharge cycles at 20 mA cm−2, the discharge voltage of batteries was stable at 1.0 V demonstrating the potential application of AgCu-MG as an alternative to Pt/C-20% in zinc–air batteries and alkaline fuel cells.
Co-reporter:Adnan Qaseem, Fuyi Chen, Xiaoqiang Wu and Roy L. Johnston
Catalysis Science & Technology 2016 vol. 6(Issue 10) pp:3317-3340
Publication Date(Web):04 Mar 2016
DOI:10.1039/C5CY02270C
The oxygen reduction reaction (ORR) plays a crucial role in electrochemical energy conversion and storage devices such as alkaline fuel cells and metal–air batteries. These systems, which could employ non-platinum catalysts for oxygen reduction, are cheaper and stable alternatives to their expensive counterparts like proton exchange membrane fuel cells (PEMFCs) working on platinum based catalysts. Various binary and ternary silver nanoalloys have been reported to act as efficient electrocatalysts for the ORR in alkaline fuel cells and batteries. Herein, we present a critical review on the recent advances made in silver nanoalloy electrocatalysts for the ORR in alkaline media. The mechanism of ORR on nanoalloys is described; the effect of structure and composition of various silver nanoalloys (including Ag–Cu, Ag–Pd, Ag–Au, Ag–Co etc.) on their ORR activity and stability is discussed. The rational design of electrocatalysts in order to maximize the number of catalytically active sites on the surface of the electrocatalysts for the ORR is also reviewed. Finally, we provide insights into the remaining challenges and directions for future perspectives and research.
Co-reporter:Mikail Aslan, Jack B. A. Davis and Roy L. Johnston
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 9) pp:6676-6682
Publication Date(Web):04 Feb 2016
DOI:10.1039/C6CP00342G
The global optimisation of small bimetallic PdCo binary nanoalloys are systematically investigated using the Birmingham Cluster Genetic Algorithm (BCGA). The effect of size and composition on the structures, stability, magnetic and electronic properties including the binding energies, second finite difference energies and mixing energies of Pd–Co binary nanoalloys are discussed. A detailed analysis of Pd–Co structural motifs and segregation effects is also presented. The maximal mixing energy corresponds to Pd atom compositions for which the number of mixed Pd–Co bonds is maximised. Global minimum clusters are distinguished from transition states by vibrational frequency analysis. HOMO–LUMO gap, electric dipole moment and vibrational frequency analyses are made to enable correlation with future experiments.
Co-reporter:Jack B. A. Davis
The Journal of Physical Chemistry C 2016 Volume 120(Issue 7) pp:3759-3765
Publication Date(Web):February 2, 2016
DOI:10.1021/acs.jpcc.5b10226
The direct density functional theory global optimization of MgO(100)-supported AuIr sub-nanoalloys is performed using the Birmingham parallel genetic algorithm (BPGA). The BPGA is a pool-based genetic algorithm for the structural characterization of nanoalloys. The parallel pool methodology utilized within the BPGA allows the code to characterize the structures of N = 4–6 AunIrN–n clusters in the presence of the MgO(100) surface. The use of density functional theory allows the code to capture quantum size effects in the systems, which determine their structures. The searches reveal significant differences in structure and chemical ordering between the surface-supported and gas-phase global minimum structures.
Co-reporter:John C. Hey, Lewis C. Smeeton, Mark T. Oakley, and Roy L. Johnston
The Journal of Physical Chemistry A 2016 Volume 120(Issue 23) pp:4008-4015
Publication Date(Web):May 25, 2016
DOI:10.1021/acs.jpca.6b01495
Hydrated ions are crucially important in a wide array of environments, from biology to the atmosphere, and the presence and concentration of ions in a system can drastically alter its behavior. One way in which ions can affect systems is in their interactions with proteins. The Hofmeister series ranks ions by their ability to salt-out proteins, with kosmotropes, such as sulfate, increasing their stability and chaotropes, such as perchlorate, decreasing their stability. We study hydrated perchlorate clusters as they are strongly chaotropic and thus exhibit different properties than sulfate. In this study we simulate small hydrated perchlorate clusters using a basin-hopping geometry optimization search with empirical potentials. We compare topological features of these clusters to data from both computational and experimental studies of hydrated sulfate ions and draw some conclusions about ion effects in the Hofmeister series. We observe a patterning conferred to the water molecules within the cluster by the presence of the perchlorate ion and compare the magnitude of this effect to that observed in previous studies involving sulfate. We also investigate the influence of the overall ionic charge on the low-energy structures adopted by these clusters.
Co-reporter:Jack B. A. Davis, Armin Shayeghi, Sarah L. Horswell and Roy L. Johnston
Nanoscale 2015 vol. 7(Issue 33) pp:14032-14038
Publication Date(Web):22 Jul 2015
DOI:10.1039/C5NR03774C
A new open-source parallel genetic algorithm, the Birmingham parallel genetic algorithm, is introduced for the direct density functional theory global optimisation of metallic nanoparticles. The program utilises a pool genetic algorithm methodology for the efficient use of massively parallel computational resources. The scaling capability of the Birmingham parallel genetic algorithm is demonstrated through its application to the global optimisation of iridium clusters with 10 to 20 atoms, a catalytically important system with interesting size-specific effects. This is the first study of its type on Iridium clusters of this size and the parallel algorithm is shown to be capable of scaling beyond previous size restrictions and accurately characterising the structures of these larger system sizes. By globally optimising the system directly at the density functional level of theory, the code captures the cubic structures commonly found in sub-nanometre sized Ir clusters.
Co-reporter:Xiaoqiang Wu, Fuyi Chen, Yachao Jin, Nan Zhang, and Roy L. Johnston
ACS Applied Materials & Interfaces 2015 Volume 7(Issue 32) pp:17782
Publication Date(Web):July 22, 2015
DOI:10.1021/acsami.5b04061
A carbon-free and binder-free catalyst layer composed of a Ag–Cu nanoalloy on Ni foam was used as the air cathode in a zinc–air battery for the first time. The Ag–Cu catalyst was prepared using pulsed laser deposition. The structures of the catalysts were found to consist of crystalline Ag–Cu nanoalloy particles with an average size of 2.58 nm embedded in amorphous Cu films. As observed in the X-ray photoelectron spectra, the Ag 3d core levels shifted to higher binding energies, whereas the Cu 2p core levels shifted to lower binding energies, indicating alloying of the silver and copper. Rotating disk electrode measurements indicated that the oxygen reduction reaction (ORR) proceeded through a four-electron pathway on the Ag50Cu50 and Ag90Cu10 nanoalloy catalysts in alkaline solution. Moreover, the catalytic activity of Ag50Cu50 in the ORR is more efficient than that of Ag90Cu10. By performing charge and discharge cycling measurements, the Ag50Cu50 catalyst layer was confirmed to have a maximum power density of approximately 86.3 mW cm–2 and an acceptable cell voltage at 0.863 V for current densities up to 100 mA cm–2 in primary zinc–air batteries. In addition, a round-trip efficiency of approximately 50% at a current density of 20 mA cm–2 was also obtained in the test.Keywords: nanoalloy; oxygen reduction reaction; primary zinc−air battery; pulsed laser deposition; rechargeable zinc−air battery
Co-reporter:Lewis C. Smeeton, James D. Farrell, Mark T. Oakley, David J. Wales, and Roy L. Johnston
Journal of Chemical Theory and Computation 2015 Volume 11(Issue 5) pp:2377-2384
Publication Date(Web):March 30, 2015
DOI:10.1021/acs.jctc.5b00151
The sulfate ion is the most kosmotropic member of the Hofmeister series, but the chemical origins of this effect are unclear. We present a global optimization and energy landscape mapping study of microhydrated sulfate ions, SO42–(H2O)n, in the size range 3 ≤ n ≤ 50. The clusters are modeled using a rigid-body empirical potential and optimized using basin-hopping Monte Carlo in conjunction with a move set including cycle inversions to explore hydrogen bond topologies. For clusters containing a few water molecules (n ≤ 6) we are able to reproduce ab initio global minima, either as global minima of the empirical potential, or as low-energy isomers. This result justifies applications to larger systems. Experimental studies have shown that dangling hydroxyl groups are present on the surfaces of pure water clusters, but absent in hydrated sulfate clusters up to n ≈ 43. Our global optimization results agree with this observation, with dangling hydroxyl groups absent from the low-lying minima of small clusters, but competitive in larger clusters.
Co-reporter:A. Shayeghi, D. Götz, J. B. A. Davis, R. Schäfer and R. L. Johnston
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 3) pp:2104-2112
Publication Date(Web):01 Dec 2014
DOI:10.1039/C4CP04323E
The Birmingham cluster genetic algorithm is a package that performs global optimisations for homo- and bimetallic clusters based on either first principles methods or empirical potentials. Here, we present a new parallel implementation of the code which employs a pool strategy in order to eliminate sequential steps and significantly improve performance. The new approach meets all requirements of an evolutionary algorithm and contains the main features of the previous implementation. The performance of the pool genetic algorithm is tested using the Gupta potential for the global optimisation of the Au10Pd10 cluster, which demonstrates the high efficiency of the method. The new implementation is also used for the global optimisation of the Au10 and Au20 clusters directly at the density functional theory level.
Co-reporter:Riccardo Ferrando, Roy L. Johnston and Catherine Louis
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 42) pp:27920-27921
Publication Date(Web):12 Aug 2015
DOI:10.1039/C5CP90142A
A graphical abstract is available for this content
Co-reporter:Jack B. A. Davis, Francesca Baletto, and Roy L. Johnston
The Journal of Physical Chemistry A 2015 Volume 119(Issue 37) pp:9703-9709
Publication Date(Web):August 31, 2015
DOI:10.1021/acs.jpca.5b05710
The effect of dispersion corrections at a range of theory levels on the chemisorption properties of metallic nanoparticles is presented. The site preference for CO on Pt, Au, Pd, and Ir nanoparticles is determined for two geometries, the 38-atom truncated octahedron and the 55-atom icosahedron using density functional theory (DFT). The effects of Grimme’s DFT-D2 and DFT-D3 corrections and the optPBE vdW-DF on the site preference of CO is then compared to the “standard” DFT results. Functional behavior is shown to depend not only on the metal but also on the geometry of the nanoparticle with significant effects seen for Pt and Au. There are both qualitative and quantitative differences between the functionals, with significant energetic differences in the chemical ordering of inequivalent sites and adsorption energies varying by up to 1.6 eV.
Co-reporter:Christopher J. Heard, Sven Heiles, Stefan Vajda and Roy L. Johnston
Nanoscale 2014 vol. 6(Issue 20) pp:11777-11788
Publication Date(Web):07 Aug 2014
DOI:10.1039/C4NR03363A
The novel surface mode of the Birmingham Cluster Genetic Algorithm (S-BCGA) is employed for the global optimisation of noble metal tetramers upon an MgO (100) substrate at the GGA-DFT level of theory. The effect of element identity and alloying in surface-bound neutral subnanometre clusters is determined by energetic comparison between all compositions of PdnAg(4−n) and PdnPt(4−n). While the binding strengths to the surface increase in the order Pt > Pd > Ag, the excess energy profiles suggest a preference for mixed clusters for both cases. The binding of CO is also modelled, showing that the adsorption site can be predicted solely by electrophilicity. Comparison to CO binding on a single metal atom shows a reversal of the 5σ–d activation process for clusters, weakening the cluster–surface interaction on CO adsorption. Charge localisation determines homotop, CO binding and surface site preferences. The electronic behaviour, which is intermediate between molecular and metallic particles allows for tunable features in the subnanometre size range.
Co-reporter:Paul C. Jennings, Hristiyan A. Aleksandrov, Konstantin M. Neyman and Roy L. Johnston
Nanoscale 2014 vol. 6(Issue 2) pp:1153-1165
Publication Date(Web):15 Nov 2013
DOI:10.1039/C3NR04750D
Density functional theory calculations are performed on 38 and 79 metal atom truncated octahedron clusters to study oxygen dissociation as a model for the initial stage of the oxygen reduction reaction. Pure platinum and alloyed platinum–titanium core–shell systems are investigated. It is found that barrierless oxygen dissociation occurs on the (111) facet of the pure platinum clusters. A barrier of ∼0.3 eV is observed on the (100) facet. For the alloyed cluster, dissociation barriers are found on both facets, typically ∼0.6 eV. The differences between the two systems are attributed to the ability of oxygen to distort the (111) surface of the pure platinum clusters. We show that flexibility of the platinum shell is crucial in promotion of fast oxygen dissociation. However, the titanium core stabilises the platinum shell upon alloying, resulting in a less easily distortable surface. Therefore, whilst an alloyed platinum-titanium electrocatalyst has certain advantages over the pure platinum electrocatalyst, we suggest alloying with a more weakly interacting metal will be beneficial for facilitating oxygen dissociation.
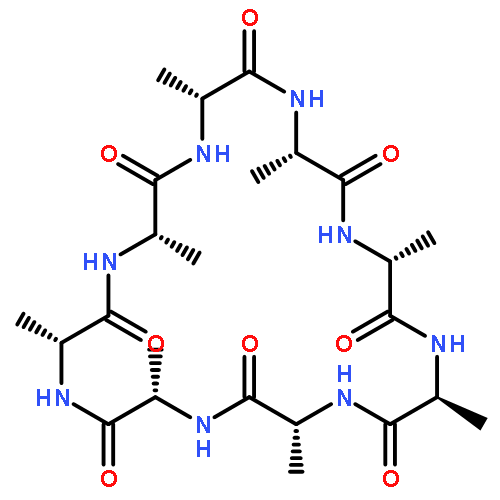
Co-reporter:Mark T. Oakley and Roy L. Johnston
Journal of Chemical Theory and Computation 2014 Volume 10(Issue 4) pp:1810-1816
Publication Date(Web):February 25, 2014
DOI:10.1021/ct500004k
Self-assembled cyclic peptide nanotubes have attracted much attention because of their antimicrobial properties. Here, we present calculations on the formation of cyclic peptide dimers using basin-hopping and discrete path sampling. We present an analysis of the basin-hopping move sets that most efficiently explore the conformations of cyclic peptides. Group rotation moves, in which sections of the ring are rotated as a rigid body, are the most effective for cyclic peptides containing up to 20 residues. For cyclic peptide dimers, we find that a combination of group rotation intramolecular moves and rigid body intermolecular moves performs well. Discrete path sampling calculations on the cyclic peptide dimers show significant differences in the dimerization of hexa- and octapeptides.
Co-reporter:Christopher J. Heard and Roy L. Johnston
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 39) pp:21039-21048
Publication Date(Web):03 Mar 2014
DOI:10.1039/C3CP55507K
The structures and optical response of helical clusters (“Bernal spirals”) with compositions Ag12Cu1+ and Ag1Cu12+ are calculated within Kohn–Sham density functional theory and the configuration interaction singles variant of time dependent density functional theory. The effects of dopant position within the cluster on the vertical excitation spectrum are investigated according to the underlying electronic structure of the major transitions. The roles of symmetry and geometry are investigated by calculating the optical response of helical, icosahedral and nanorod-like clusters of Ag13+, finding local structure to be significant in driving the resultant optical response at the subnanometre scale. Further, it is noted that helical clusters have optical properties which are quite distinct from those of nanorods of similar dimensions. The effect of multiple doping is studied by introducing copper atoms into the centre of the silver helix, over the composition range Ag13+ to Ag6Cu7+. There is a complex variation of the major plasmon-like peak over this range, attributed to subtle variations in the influence of the copper 3d band on the excitations and charge transfer for different sites within the cluster. This work suggests that coinage metal nanohelices have unusual, tunable electronic properties, which in addition to their inherent chirality makes them interesting systems to study for chiral catalysis and optoelectronics.
Co-reporter:Paul C. Jennings, Hristiyan A. Aleksandrov, Konstantin M. Neyman and Roy L. Johnston
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 48) pp:26539-26545
Publication Date(Web):15 Jul 2014
DOI:10.1039/C4CP02147A
Density functional theory calculations are performed to investigate oxygen dissociation on 116-atom truncated octahedron platinum particles. This work builds on results presented previously [Jennings et al., Nanoscale, 2014, 6, 1153], where it was shown that shell flexibility played an important role in facilitating fast oxygen dissociation. In this study, through investigation of the larger particle size, it is shown that oxygen dissociation on the (111) facet of pure platinum species is still aided by shell flexibility at larger sizes. Only the hollow sites close to the edges of the (111) facet mediate oxygen dissociation; oxygen is bound too weakly at other hollow sites for dissociation to occur. Further studies are performed on the (100) facet, which is larger for the Pt116 particle than for either the Pt38 or Pt79 ones. Much higher dissociation barriers are found on the (100) facet compared to the (111) facet, where the bridge sites are favourable for oxygen dissociation.
Co-reporter:Christopher J. Heard ; Stefan Vajda
The Journal of Physical Chemistry C 2014 Volume 118(Issue 7) pp:3581-3589
Publication Date(Web):January 30, 2014
DOI:10.1021/jp411019t
The effect of cluster size, oxidation state, and the support upon the structures and energetics of subnanometer palladium nanoparticles is investigated within a density functional framework. Gas phase global minima of Pd4 and Pd10 along with their suboxide counterparts are determined using a genetic algorithm and deposited upon MgO (001) and a high-index alumina surface. It is observed that there is an oxidation-dependent transition in the smaller clusters from three-dimensional to two-dimensional structures both in the gas phase and when supported by a surface. MgO strongly promotes a change from tetrahedral- and icosahedral-based structures toward cubic forms, while alumina induces significant distortion of the cluster and the breaking of Pd–Pd bonds. Increased oxygenation contributes cooperatively to these effects, causing disruption of the Pd–Pd bond network, favoring the incorporation of oxygen into the cluster structure, further complicating unambiguous structure prediction.
Co-reporter:Jack B. A. Davis, Sarah L. Horswell, and Roy L. Johnston
The Journal of Physical Chemistry A 2014 Volume 118(Issue 1) pp:208-214
Publication Date(Web):December 13, 2013
DOI:10.1021/jp408519z
The global optimization of PdnIr(N–n) N = 8–10 clusters has been performed using the Birmingham Cluster Genetic Algorithm (BCGA). Structures were evaluated directly using density functional theory (DFT), which has allowed the identification of Ir and Ir-rich PdIr cubic global minima, displaying a strong tendency to segregate. The ability of the searches to find the putative global minimum has been assessed using a homotop search method, which shows a high degree of success. The role of spin in the system has been considered through a series of spin-restricted reoptimizations of BCGA-DFT minima. The preferred spin of the clusters is found to vary widely with composition, showing no overall trend in lowest-energy multiplicities.
Co-reporter:Alina Bruma, Ramli Ismail, L. Oliver Paz-Borbón, Haydar Arslan, Giovanni Barcaro, Alessandro Fortunelli, Z. Y. Li and Roy L. Johnston
Nanoscale 2013 vol. 5(Issue 2) pp:646-652
Publication Date(Web):23 Nov 2012
DOI:10.1039/C2NR32517A
The energetics, structures and segregation of 98-atom AuPd nanoclusters are investigated using a genetic algorithm global optimization technique with the Gupta empirical potential (comparing three different potential parameterisations) followed by local minimizations using Density Functional Theory (DFT) calculations. A shell optimization program algorithm is employed in order to study the energetics of the highly symmetric Leary Tetrahedron (LT) structure and optimization of the chemical ordering of a number of structural motifs is carried out using the Basin Hopping Monte Carlo approach. Although one of the empirical potentials is found to favour the LT structure, it is shown that Marks Decahedral and mixed FCC-HCP motifs are lowest in energy at the DFT level.
Co-reporter:Andrew J. Logsdail, Z. Y. Li and Roy L. Johnston
Physical Chemistry Chemical Physics 2013 vol. 15(Issue 21) pp:8392-8400
Publication Date(Web):15 Apr 2013
DOI:10.1039/C3CP50978H
The structural preferences of nanoparticles are important for understanding their chemical properties and potential applications, and remain widely debated. Based on recent experimental observations, we present calculations on the stability of high-symmetry AuN and PdN clusters of various structural motifs, performing a systematic search of faceting preferences using mathematical constructs, a semi-empirical potential with two different parameter sets, and a quasi-Newtonian minimisation technique. We have studied the preferred ratios of (100) and (111) faces for two experimentally observed nanostructures: (a) FCC crystals, comparing octahedra with 8 (111) faces to cuboctahedra where the vertices have been systematically removed (for N < 1500); and (b) Marks-decahedra, with differing “stellation” depths (for N < 6000). For PdN and AuN we see preference towards minimisation of (100) surfaces using the parameter sets of both Cleri and Rosato [Cleri and Rosato, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 22] and Baletto et al. [Baletto et al., J. Chem. Phys., 2002, 116, 3856]. Fully stellated Marks-decahedra are found to be unfavourable at large sizes, with truncated facets identified which are similar to recent experimental observations. We find however that these stellations are deeper in PdN particles than AuN. Truncated-octahedra are found to prefer much reduced (100) surfaces and increased (111) surface areas.
Co-reporter:P.C. Jennings, R.L. Johnston
Computational and Theoretical Chemistry 2013 Volume 1021() pp:91-100
Publication Date(Web):1 October 2013
DOI:10.1016/j.comptc.2013.06.033
•A genetic algorithm-DFT approach is used to search for stable Ti- and V-doped Pt clusters.•Spin multiplicity and charge effects are studied for 3–6 atom clusters.•Partial spin quenching is observed when Pt clusters are doped with Ti or V.High level GA-DFT searches are performed on small platinum clusters doped with early transition metal atoms, Ptx−yMy (M = Ti, V), where x = 2–6, y = 1, 2. Spin effects are studied and the global minimum structures are presented for the various spin multiplicities. It is found that varying spin can have significant effects on the pure Pt clusters, while spin has less effect for the doped clusters.Figure optionsDownload full-size imageDownload as PowerPoint slide
Co-reporter:Dora J. Borbón-González, Alessandro Fortunelli, Giovanni Barcaro, Luca Sementa, Roy L. Johnston, and Alvaro Posada-Amarillas
The Journal of Physical Chemistry A 2013 Volume 117(Issue 51) pp:14261-14266
Publication Date(Web):December 4, 2013
DOI:10.1021/jp410079t
In this work, we report finding dodecahedral core–shell structures as the putative global minima of Pt13M20 (M = Ag, Au, Cu, Pd) clusters by using the basin hopping method and the many-body Gupta model potential to model interatomic interactions. These nanoparticles consist of an icosahedral 13-atom platinum core encapsulated by a 20 metal-atom shell exhibiting a dodecahedral geometry (and Ih symmetry). The interaction between the icosahedral platinum core and the dodecahedral shell is analyzed in terms of the increase in volume of the icosahedral core, and the strength and stickiness of M–Pt and M–M interactions. Low-lying metastable isomers are also obtained. Local relaxations at the DFT level are performed to verify the energetic ordering and stability of the structures predicted by the Gupta potential finding that dodecahedral core–shell structures are indeed the putative global minima for Pt13Ag20 and Pt13Pd20, whereas decahedral structures are obtained as the minimum energy configurations for Pt13Au20 and Pt13Cu20 clusters.
Co-reporter:Ramli Ismail, Riccardo Ferrando, and Roy L. Johnston
The Journal of Physical Chemistry C 2013 Volume 117(Issue 1) pp:293-301
Publication Date(Web):December 11, 2012
DOI:10.1021/jp3093435
Bimetallic nanoparticles composed of palladium and gold are particularly interesting from the viewpoint of their catalytic properties, for example, for selective hydrogenation and alcohol oxidation. More accurate catalytic modeling is achieved by the inclusion of the substrate (e.g., metal oxides). In this work, the structures and chemical ordering (atomic segregation) of Pd–Au clusters supported on MgO(100) were studied using a combined empirical potential–density functional theory approach. The focus is on 30- and 40-atom clusters, including variation in the bimetallic composition. Consistent with the available experimental findings, Pd atoms preferentially bind to the substrate oxygen sites. Good cluster-substrate epitaxy is observed, but there is a strong dependence on the size and composition of the clusters.
Co-reporter:Mark T. Oakley, Emmanuel Oheix, Anna F. A. Peacock, and Roy L. Johnston
The Journal of Physical Chemistry B 2013 Volume 117(Issue 27) pp:8122-8134
Publication Date(Web):June 12, 2013
DOI:10.1021/jp4043039
We present a combined computational and experimental study of the energy landscapes of cyclic tetra-α/β-peptides. We have performed discrete path sampling calculations on a series of cyclic tetra-α/β-peptides to obtain the relative free energies and barriers to interconversion of their conformers. The most stable conformers of cyclo-[(β-Ala-Gly)2] contain all-trans peptide groups. The relative energies of the cis isomers and the cis–trans barriers are lower than in acyclic peptides but not as low as in the highly strained cyclic α-peptides. For cyclic tetra-α/β-peptides containing a single proline residue, of the type cyclo-[β-Ala-Xaa-β-Ala-Pro], the energy landscapes show that the most stable isomers containing cis and trans β-Ala-Pro have similar free energies and are separated by barriers of approximately 15 kcal mol–1. We show that the underlying energy landscapes of cyclo-[β-Ala-Lys-β-Ala-Pro] and cyclo-[β-Ala-Ala-β-Ala-Pro] are similar, allowing the substitution of the flexible side chain of Lys with Ala to reduce the computational demand of our calculations. However, the steric bulk of the Val side chain in cyclo-[β-Ala-Val-β-Ala-Pro] affects the conformations of the ring, leading to significant differences between its energy landscape and that of cyclo-[β-Ala-Ala-β-Ala-Pro]. We have synthesized the cyclic peptide cyclo-[β-Ala-Lys-β-Ala-Pro], and NMR spectroscopy shows the presence of conformers that interconvert slowly on the NMR time scale at temperatures up to 80 °C. Calculated circular dichroism (CD) spectra for the proposed major isomer of cyclo-[β-Ala-Ala-β-Ala-Pro] are in good agreement with the experimental spectra of cyclo-[β-Ala-Lys-β-Ala-Pro], suggesting that the Ala cyclic tetrapeptide is a viable model for the Lys analogue.
Co-reporter:Sven Heiles, Andrew J. Logsdail, Rolf Schäfer and Roy L. Johnston
Nanoscale 2012 vol. 4(Issue 4) pp:1109-1115
Publication Date(Web):19 Oct 2011
DOI:10.1039/C1NR11053E
A genetic algorithm (GA) coupled with density functional theory (DFT) calculations is used to perform global optimisations for all compositions of 8-atom Au–Ag bimetallic clusters. The performance of this novel GA-DFT approach for bimetallic nanoparticles is tested for structures reported in the literature. New global minimum structures for various compositions are predicted and the 2D–3D transition is located. Results are explained with the aid of an analysis of the electronic density of states. The chemical ordering of the predicted lowest energy isomers are explained via a detailed analysis of the charge separation and mixing energies of the bimetallic clusters. Finally, dielectric properties are computed and the composition and dimensionality dependence of the electronic polarizability and dipole moment is discussed, enabling predictions to be made for future electric beam deflection experiments.
Co-reporter:Paul C. Jennings, Bruno G. Pollet and Roy L. Johnston
Physical Chemistry Chemical Physics 2012 vol. 14(Issue 9) pp:3134-3139
Publication Date(Web):06 Jan 2012
DOI:10.1039/C2CP23430K
A theoretical investigation is presented of alloying platinum with titanium to form binary Pt–Ti nanoalloys as an alternative to the expensive pure platinum catalysts commonly used for Proton Exchange Membrane Fuel Cell cathode electrocatalysts. Density Functional Theory calculations are performed to investigate compositional effects on structural properties as well as Oxygen Reduction Reaction kinetics and poisoning effects. High symmetry A32–B6clusters are studied to investigate structural properties. From these structures binding energies of hydroxyl and carbon monoxide are studied on a range of sites on the surface of the clusters. Promising results are obtained suggesting that the bimetallic Pt–Ti nanoalloys may exhibit enhanced properties compared to pure platinum catalysts.
Co-reporter:Andrew J. Logsdail and Roy L. Johnston
RSC Advances 2012 vol. 2(Issue 13) pp:5863-5869
Publication Date(Web):20 Apr 2012
DOI:10.1039/C2RA20309J
In this work we study the stabilities of high-symmetry AuN, PdN and (AuPd)N clusters, for N < 1500, using mathematical constructs, a semi-empirical potential with two different parameter sets, and a quasi-Newtonian minimisation technique. For PdN clusters, both parameter sets tested result in preferences for icosahedral (Ih) structures for N < 1000 over other high-symmetry 12-vertex geometries; for AuN clusters we find a tendency towards face-centred cubic (FCC) structures at values of N lower than seen for PdN: parameter set I of Cleri and Rosato [Cleri and Rosato, Phys. Rev. B, 1993, 48, 22] gave a transition at N ≈ 650 to the I-Dh, whilst for parameter set II of Baletto et al. [Baletto et al., J. Chem. Phys., 2002, 116, 3856] this value was lower still. For (AuPd)N clusters we found that the preferred arrangement is (PdcoreAushell)N, with thin (monolayer) surface coverings of Au being most energetically favourable compared to the homogeneous clusters; however for parameter set II multiple layers of Au lead to energetic instability. (AucorePdshell)N clusters are not energetically favourable with thin coatings of Pd, however as the shell coating thickens so the stability improves. Ih structures are unfavourable compared to the Ino-decahedron and cuboctahedron for (AucorePdshell)N, whereas the FCC-type structures are strongly preferred for (PdcoreAushell)N. Overall, the strong tendency towards core–shell segregation is emphasised for parameter set I more than II, agreeing with previous work on smaller (AuPd)N clusters.
Co-reporter:Andrew J. Logsdail and Roy L. Johnston
The Journal of Physical Chemistry C 2012 Volume 116(Issue 44) pp:23616-23628
Publication Date(Web):October 18, 2012
DOI:10.1021/jp306000u
An investigation of the optical properties of off-centered core–shell segregated Au–M (M = Ag, Pd) nanoalloys is conducted using the discrete-dipole approximation (DDA). While the secondary metal M is found to dominate the optical spectra in core-centered spherical particles, displacement of the core within the Au–M structure results in an increase in maximum extinction intensity, at the wavelength λmax, illustrating the potential for controlled tunability. The greatest effect is seen when the core is touching the particle surface and the nonhomogeneity of the shell covering is maximized. The increase in intensity at λmax is attributed to the greater division of the dielectric materials and thus a stronger response from the dominant dielectric. The intensity of response is also found to be mildly dependent on the segregated particles' orientation relative to the angle of the incoming electromagnetic radiation, k. Calculations are performed for elongated elliptical nanorods, with aspect ratios of 2, where core positioning is also found to play an important role in the position of λmax. Changes to the spectra for M = Pd are slight; however, the movement of the elliptical core in the AucoreAgshell results in significant shifting of λmax. We conclude that our observations will have profound implications for the use of optical absorption spectra in characterizing nonsymmetrically segregated nanoalloys.
Co-reporter:Paul C. Jennings, Bruno G. Pollet, and Roy L. Johnston
The Journal of Physical Chemistry C 2012 Volume 116(Issue 29) pp:15241-15250
Publication Date(Web):June 26, 2012
DOI:10.1021/jp303577t
Theoretical investigations of Pt–Ti nanoparticles are presented using density functional theory (DFT) to study clusters up to sizes of 1.7 nm (201 atoms). Several compositions have been studied for varying sizes, and a projected density of states analysis has been performed. Changes in d-band properties have been correlated with differences in OH and CO binding energies with relation to changes in composition. It was found that a Pt-rich Pt–Ti alloy shows promise for potential use in a PEMFC with the alloy resulting in a downshift in d-center, compared to pure Pt clusters, which correlates with a weakening of the OH and CO adsorption energies. Furthermore, it was observed that varying the size of the cluster gives rise to changes in the d-band center with larger clusters typically having a more negative d-band center, although values obtained are not as negative as the d-band center for bulk Pt3Ti reported in the literature.
Co-reporter:Rafael Pacheco-Contreras, Maribel Dessens-Félix, Dora J. Borbón-González, L. Oliver Paz-Borbón, Roy L. Johnston, J. Christian Schön, and Alvaro Posada-Amarillas
The Journal of Physical Chemistry A 2012 Volume 116(Issue 21) pp:5235-5239
Publication Date(Web):May 4, 2012
DOI:10.1021/jp3023925
The threshold method is used to explore the potential energy surface of the Pt1Pd12 bimetallic cluster, defined by the Gupta semiempirical potential. A set of helical structures, which follow a Bernal tetrahelix pattern, correspond to local minima for the Pt1Pd12 cluster, characterizing the region of the energy landscape where these structures are present. Both right-handed and left-handed chiral forms were discovered in our searches. Energetic and structural details of each of the tetrahelices are reported as well as the corresponding transition probabilities between these structures and with respect to the icosahedron-shaped global minimum structure via a disconnectivity graph analysis.
Co-reporter:Mark T. Oakley, David J. Wales, and Roy L. Johnston
The Journal of Physical Chemistry B 2011 Volume 115(Issue 39) pp:11525-11529
Publication Date(Web):August 26, 2011
DOI:10.1021/jp207246m
The three-color (BLN) 69-residue model protein was designed to exhibit frustrated folding. We investigate the energy landscape of this protein using disconnectivity graphs and compare it to a Go̅ model, which is designed to reduce the frustration by removing all non-native attractive interactions. Finding the global minimum on a frustrated energy landscape is a good test of global optimization techniques, and we present calculations evaluating the performance of basin-hopping and genetic algorithms for this system. Comparisons are made with the widely studied 46-residue BLN protein. We show that the energy landscape of the 69-residue BLN protein contains several deep funnels, each of which corresponds to a different β-barrel structure.
Co-reporter:Ramli Ismail and Roy L. Johnston
Physical Chemistry Chemical Physics 2010 vol. 12(Issue 30) pp:8607-8619
Publication Date(Web):12 Jul 2010
DOI:10.1039/C004044D
The energetics, structures and segregation of Pd–Au nanoalloys (all compositions for 34- and 38-atoms) have been studied using a genetic algorithm global optimization technique with the Gupta empirical potential. Three modifications of the Pd–Au parameters have been studied: parameter set I in which all parameters (A, ξ, p, q and r0) in the Gupta potential are weighted in a symmetrical fashion; parameter set II (symmetric weighting of only the pair and many-body energy scaling parameters A and ξ); and parameter set III (antisymmetric weighting of A and ξ). Structural analysis reveals competition between a range of structural families; decahedra, polyicosahedra and truncated octahedra (for 34 atoms) and incomplete-icosahedra-Mackay, decahedra, polyicosahedra (low-symmetry), six-fold-polyicosahedra and a mixed octahedron–icosahedron (Oh–Ih) structure (for 38 atoms). It is shown that, by finely tuning the Gupta potential, it is possible to qualitatively reproduce the results observed at higher levels of theory (e.g. Density Functional Theory). There are four main types of chemical ordering which are observed: core–shell; spherical cap; ball-and-cup; and mixed. It is shown that the chemical ordering and the proportion of Pd–Au heteronuclear bonds in these clusters are strongly dependent on the potential parameters. Comparison of the results from parameter set III and two previously fitted potentials shows that the DFT-fit potential gives rise to similar results for energies, and lowest energy structures and homotops to those for parameter set III with wa = 0.8, but the exp-fit potential gives rise to qualitatively different results.
Co-reporter:Paul S. West ; Roy L. Johnston ; Giovanni Barcaro ;Alessandro Fortunelli
The Journal of Physical Chemistry C 2010 Volume 114(Issue 46) pp:19678-19686
Publication Date(Web):October 29, 2010
DOI:10.1021/jp108387x
The effect of alloying on ligand adsorption energies and how this can modify the segregation patterns of selected binary nanoalloys is studied via first-principles total energy calculations. A model setup is considered, in which high-symmetry 38-atom truncated-octahedral (TO) clusters with compositions A6B32 and B6A32 are used as substrates to bind a single CO molecule or H atom in centroid sites for the following (A,B) pairs: (Au,Pd), (Pd,Pt), and (Cu,Pt), and the relative changes in the energetics of the systems upon ligand coordination are analyzed. We find qualitative similarities but quantitative differences between the CO and H cases (as examples of reducing agents), and a wide variety of behavior for the three (A,B) pairs. In AuPd, Pd−CO bonding is not strongly affected by neighboring Au atoms but the PdcoreAusurface segregation pattern (favored for bare particles) is expected to be inverted in the presence of CO coordinating species. At the other extreme, in CuPt, both Pt−CO and Pt−H bonding is strongly enhanced by neighboring Cu atoms, but the predicted segregation pattern differs from that expected on the basis of results for extended systems due to finite-size effects.
Co-reporter:Andrew J. Logsdail ; Nikki J. Cookson ; Sarah L. Horswell ; Z. W. Wang ; Z. Y. Li
The Journal of Physical Chemistry C 2010 Volume 114(Issue 49) pp:21247-21251
Publication Date(Web):November 17, 2010
DOI:10.1021/jp108486a
We identify and report on UV−vis spectral features of AucorePdshell nanoparticles. Chains of conjoined nanospheres are formed with clear core/shell segregation and identified via experimental spectral red shifts of λmax and scanning transmission electron microscopy (STEM) imaging. Theoretical calculations performed using the discrete dipole approximation (DDA) validate our findings. The DDA simulation method is then used to delve further into possible spectroscopic implications of geometric and environmental changes. Structural configurations including extended chains of nanospheres, differing levels of nanosphere conjoinment, and increased Pd shell thickness are investigated, as well as variations of the surrounding dielectric medium.
Co-reporter:Dung T. Tran and Roy L. Johnston
Physical Chemistry Chemical Physics 2009 vol. 11(Issue 44) pp:10340-10349
Publication Date(Web):30 Sep 2009
DOI:10.1039/B912501A
Thirty-eight atom Cu–Au clusters have been studied because this is a magic size for a complete truncated octahedral cluster. The clusters are investigated using two approaches, at different levels of theory, which are complementary. The first is an empirical potential (EP) approach which is used together with a genetic algorithm (GA) to tackle the problems of global optimization—i.e., searching for lowest-lying energy structures. The second is an ab initio approach based on density functional theory (DFT) which is used to reoptimize the initial EP structures (both global minima and other low energy isomers). Structural distributions and energy landscapes, including calculations of electronic energy gaps for all compositions of Cu38−nAun, are investigated. The energy competition between different structural motifs and different configurations are studied at the DFT level. The analysis of mixing and segregation effects results in confirmation of the preference for Cu(core)–Au(shell) configurations at the DFT level. Charge transfer is calculated for different structural motifs of Cu19Au19 to study the role of this phenomenon in driving cluster configuration.
Co-reporter:Faye Pittaway, Lauro Oliver Paz-Borbón, Roy L. Johnston, Haydar Arslan, Riccardo Ferrando, Christine Mottet, Giovanni Barcaro and Alessandro Fortunelli
The Journal of Physical Chemistry C 2009 Volume 113(Issue 21) pp:9141-9152
Publication Date(Web):2017-2-22
DOI:10.1021/jp9006075
Global optimization of Pd−Au bimetallic clusters in the size range N = 2−50 has been performed using a genetic algorithm, coupled with the Gupta many-body empirical potential (EP) to model interatomic interactions. Three sets of EP parameters have been examined in this work: (a) an average of pure Pd and Au parameters, (b) experimental Pd−Au-fitted parameters, and (c) DFT-fitted parameters. Stability criteria, such as binding energy and second difference in energy, have been used to determine the lowest energy structures, that is, the global minima (GM). DFT local relaxations have been performed on all the “putative” GM structures for 1:1 compositions of (Pd−Au)N/2 up to N = 50 for the three sets of EP parameters. It is found that the average parameter set a leads to a PdcoreAushell segregation, whereas the fitted parameter sets b and c lead to more Pd−Au mixing. DFT reoptimization of the structures produced by potentials a, b, and c shows small differences in binding energies. In addition, 34- and 38-atom Pd−Au clusters were studied using these three Gupta potential parametrizations as a function of composition and analyzed in terms of their mixing energies and chemical order parameters. DFT relaxations were performed on the lowest mixing energy compositions, allowing us to have a clearer description of the energy landscape for all three EP parameter sets at these cluster sizes. For the compositions, Pd17Au17 and Pd19Au19, DFT calculations confirm that some degree of Au surface segregation is energetically preferred, though it is not necessarily complete PdcoreAushell segregation, as predicted by the average potential a.
Co-reporter:Lauro Oliver Paz-Borbón, Abhishek Gupta and Roy L. Johnston
Journal of Materials Chemistry A 2008 vol. 18(Issue 35) pp:4154-4164
Publication Date(Web):22 Jul 2008
DOI:10.1039/B805147J
The structures and chemical ordering (segregation properties) of Pd–Pt clusters (1 : 1 compositions for N = 2–20 atoms and all compositions for 34 atoms) have been studied using a combination of a genetic algorithm global optimization technique (GA) coupled with the Gupta semi-empirical potential and density functional theory (DFT) calculations. An initial DFT energetic analysis of small Pd–Pt clusters (N = 2–20) showed that their corresponding binding energies are slightly biased towards the stronger metal–metal bonding interactions (i.e. Pt–Pt). This led to a detailed analysis of Pd–Pt structural motifs and segregation effects, where the heteronuclear Pd–Pt parameters in the Gupta potential are derived as weighted averages of the Pd–Pd and Pt–Pt parameters, with the weighting factor (w) ranging from 0 (Pt-biased) to 1 (Pd-biased). The introduction of the weighting factor allowed us to identify three main types of segregation: core–shell; spherical cap; and ball-and-cup (intermediate between the first two types). The structural motifs predicted by the Gupta potential, as a function of composition and potential weighting factor, have been compared to our previous published Gupta and DFT calculations for 34-atom Pt–Pt clusters. From this study, we have found that a slightly Pd-biased weighting factor (w = 0.6) stabilises the mixed decahedral close packed structural motif, previously reported as the DFT global minimum in the region of most exothermic mixing for 34-atom Pd–Pt clusters. Our results show that by finely tuning the Gupta potential, one can qualitatively reproduce structural and chemical ordering patterns observed at higher levels of theory (e.g. DFT).
Co-reporter:Fuyi Chen, Roy L. Johnston
Acta Materialia 2008 Volume 56(Issue 10) pp:2374-2380
Publication Date(Web):June 2008
DOI:10.1016/j.actamat.2008.01.048
Abstract
The structural, optical and electronic properties of 13-atom Ag–Au nanoalloys are determined by a combination of global optimization using semi-empirical potentials and density functional theory calculations. A family of Au surface-segregated structures are found for core–shell AgnAu13−n (n = 1, 2, 3, 5, 7, 8, 9, 12) and hollow AgnAu13−n (n = 4, 6, 10, 11) clusters, whose stability is enhanced by directional charge transfer. The atomic ordering in core–shell structures is related to the electric dipole moment and odd-numbered surface Au-atom clusters have high moments. Their ferroelectric and ferromagnetic properties provide a potential approach for tailoring their surface plasmonic modes.
Co-reporter:Fuyi Chen and Roy L. Johnston
ACS Nano 2008 Volume 2(Issue 1) pp:165
Publication Date(Web):December 22, 2007
DOI:10.1021/nn700226y
Using a genetic algorithm global optimization approach combined with density functional theory calculations, a search has been made for the lowest energies of (AgAu)m nanoalloys with 20−150 atoms (diameters of 1.0–2.0 nm). A total of 31 decahedra, 35 icosahedra, and 2 close-packed motifs are identified in two icosahedral windows and one Marks-decahedral window. These structural motifs have twinned, capped, defective, and distorted atomic packing compared to classical clusters, such as the icosahedron. The magic numbers, atomic ordering, electronic structure, and melting behavior are further studied, and a new poly-nanocrystalline decahedral motif, Ag44Au44, is found to have high structural, electronic, and thermal stability. Our results show that alloying can lead to a remarkable stabilization of local order and provide a comprehensive model for the structures and properties of Ag−Au nanoalloys.Keywords: electronic and thermal stability; global optimization; nanoalloy; size; structure
Co-reporter:Lauro Oliver Paz-Borbón, Thomas V. Mortimer-Jones, Roy L. Johnston, Alvaro Posada-Amarillas, Giovanni Barcaro and Alessandro Fortunelli
Physical Chemistry Chemical Physics 2007 vol. 9(Issue 38) pp:5202-5208
Publication Date(Web):17 Jul 2007
DOI:10.1039/B707136A
The energetics of 98 atom bimetallic Pd–Pt clusters are studied using a combination of: a genetic algorithm technique (to explore vast areas of the configurational space); a basin-hopping atom-exchange routine (to search for lowest-energy homotops at fixed composition); and a shell optimisation approach (to search for high symmetry isomers). The interatomic interactions between Pd and Pt are modelled by the Gupta many-body empirical potential. For most compositions, the putative global minima are found to have structures based on defective Marks decahedra, but in the composition range from Pd46Pt52 to Pd63Pt35, the Leary tetrahedron (LT)—a structure previously identified for 98 atom Lennard-Jones clusters—is consistently found as the most stable structure. Based on the excess energy stability criterion, Pd56Pt42 represents the most stable cluster across the entire composition range. This structure, a Td-symmetry LT, exhibits multi-layer segregation with an innermost core of Pd atoms, an intermediate layer of Pt atoms and an outermost Pd surface shell (Pd–Pt–Pd). The stability of the Leary tetrahedron is compared against other low-energy competing structural motifs: the Marks decahedron (Dh-M), a “quasi” tetrahedron (a closed-packed structure) and two other closed-packed structures. The stability of LT structures is rationalized in terms of their spherical shape and the large number of nearest neighbours.
Co-reporter:Nicholas T. Wilson, Mark S. Bailey, Roy L. Johnston
Inorganica Chimica Acta 2006 Volume 359(Issue 11) pp:3649-3658
Publication Date(Web):1 August 2006
DOI:10.1016/j.ica.2006.02.029
Energy calculations have been carried out on high-symmetry cuboctahedral Ni–Al nanoalloy clusters, of varying composition, with the interatomic interactions modelled by the Gupta many-body potential. Relaxations of cuboctahedral fragments cut from the bulk lattice of Ni3Al, with 13–561 atoms, were undertaken, as were relaxations of high symmetry clusters with 55 and 147 atoms. The lowest energy isomers were found to be dominated by three factors: the tendency toward mixing due to the favourable energy of mixing, ΔmixE; the size difference between nickel and aluminium; and the higher cohesive and surface energy of nickel compared to aluminium. The latter two factors favour Al-segregation to the surface. The most stable Ni:Al composition approaches 3:1 for larger clusters.Energy calculations, using the Gupta many-body potential, have been carried out on cuboctahedral geometric shell Ni–Al clusters (nanoalloys) with up to 561 atoms. The competition between exothermic Ni–Al mixing and size and surface energy effects, and their effect on the atomic distribution of the Ni and Al atoms have been investigated.
Co-reporter:Ilker Demiroglu, Z. Y. Li, Laurent Piccolo and Roy L. Johnston
Catalysis Science & Technology (2011-Present) 2016 - vol. 6(Issue 18) pp:NaN6931-6931
Publication Date(Web):2016/06/30
DOI:10.1039/C6CY01107A
Density functional theory calculations are performed to investigate both mixing and adsorption properties of 38-atom and 79-atom Au–Rh nanoalloys at the nanoscale. The RhcoreAushell and RhballAucup isomers are found to be energetically favourable with respect to other isomers. The adsorption strengths of reactive species such as H2, O2 and CO are found to be greater on the Rh part than on the Au part of the nanoalloys and therefore a core–shell inversion is found to be feasible under a molecular environment. It is also found that underlying Rh atoms decrease the adsorption strength on the Au part whereas underlying Au atoms increase it on the Rh part of the nanoalloys. The strain, alloying and relaxation effects on adsorption strength are characterized using a sequential approach and their competing nature is demonstrated for the Au–Rh bimetallic system.
Co-reporter:Jiali Wang, Fuyi Chen, Yachao Jin and Roy L. Johnston
Journal of Materials Chemistry A 2016 - vol. 4(Issue 45) pp:NaN17837-17837
Publication Date(Web):2016/10/27
DOI:10.1039/C6TA07519C
Bimetallic AuNi nanodendrite catalysts have been prepared for the oxygen reduction reaction (ORR) in alkaline media by a facile electrodeposition and electrochemical dealloying method. The dealloyed AuNi catalyst consists of hierarchical dendrites with a high electrochemically active surface area. The half-wave potential (E1/2) of the dealloyed AuNi catalyst is 0.896 V vs. RHE, exhibiting about 67 and 27 mV positive shift relative to the commercial Pt/C and as-prepared (before dealloying) AuNi catalysts, respectively. Compared to the commercial Pt/C catalyst, the dealloyed AuNi achieves a 2.8-fold improvement in specific activity at 0.8 V vs. RHE and suffers less degradation of the ORR activity after 5000 potential cycles. The ORR catalyzed by the bimetallic AuNi catalyst proceeds through a four-electron pathway in basic solution. TEM and XPS characterizations indicate that the enhancement of ORR activity is attributed to the favorable morphology and electronic effect caused by the incorporation of Ni atoms into the Au substrate. Dealloyed AuNi hierarchical dendrites possess great application potential as cathode electrocatalysts in metal-air batteries and alkaline fuel cells due to the facile preparation, high ORR activity and long-term cycling durability.
Co-reporter:Heider A. Hussein, Jack B. A. Davis and Roy L. Johnston
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 37) pp:NaN26143-26143
Publication Date(Web):2016/09/12
DOI:10.1039/C6CP03958H
The Birmingham Parallel Genetic Algorithm (BPGA) has been adopted for the global optimization of free and MgO(100)-supported Pd, Au and AuPd nanocluster structures, over the size range N = 4–10. Structures were evaluated directly using density functional theory, which has allowed the identification of Pd, Au and AuPd global minima. The energetics, structures, and tendency of segregation have been evaluated by different stability criteria such as binding energy, excess energy, second difference in energy, and adsorption energy. The ability of the approach in searching for putative global minimum has been assessed against a systematic homotop search method, which shows a high degree of success.
Co-reporter:Mikail Aslan, Jack B. A. Davis and Roy L. Johnston
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 9) pp:NaN6682-6682
Publication Date(Web):2016/02/04
DOI:10.1039/C6CP00342G
The global optimisation of small bimetallic PdCo binary nanoalloys are systematically investigated using the Birmingham Cluster Genetic Algorithm (BCGA). The effect of size and composition on the structures, stability, magnetic and electronic properties including the binding energies, second finite difference energies and mixing energies of Pd–Co binary nanoalloys are discussed. A detailed analysis of Pd–Co structural motifs and segregation effects is also presented. The maximal mixing energy corresponds to Pd atom compositions for which the number of mixed Pd–Co bonds is maximised. Global minimum clusters are distinguished from transition states by vibrational frequency analysis. HOMO–LUMO gap, electric dipole moment and vibrational frequency analyses are made to enable correlation with future experiments.
Co-reporter:Dung T. Tran and Roy L. Johnston
Physical Chemistry Chemical Physics 2009 - vol. 11(Issue 44) pp:NaN10349-10349
Publication Date(Web):2009/09/30
DOI:10.1039/B912501A
Thirty-eight atom Cu–Au clusters have been studied because this is a magic size for a complete truncated octahedral cluster. The clusters are investigated using two approaches, at different levels of theory, which are complementary. The first is an empirical potential (EP) approach which is used together with a genetic algorithm (GA) to tackle the problems of global optimization—i.e., searching for lowest-lying energy structures. The second is an ab initio approach based on density functional theory (DFT) which is used to reoptimize the initial EP structures (both global minima and other low energy isomers). Structural distributions and energy landscapes, including calculations of electronic energy gaps for all compositions of Cu38−nAun, are investigated. The energy competition between different structural motifs and different configurations are studied at the DFT level. The analysis of mixing and segregation effects results in confirmation of the preference for Cu(core)–Au(shell) configurations at the DFT level. Charge transfer is calculated for different structural motifs of Cu19Au19 to study the role of this phenomenon in driving cluster configuration.
Co-reporter:Lauro Oliver Paz-Borbón, Abhishek Gupta and Roy L. Johnston
Journal of Materials Chemistry A 2008 - vol. 18(Issue 35) pp:NaN4164-4164
Publication Date(Web):2008/07/22
DOI:10.1039/B805147J
The structures and chemical ordering (segregation properties) of Pd–Pt clusters (1 : 1 compositions for N = 2–20 atoms and all compositions for 34 atoms) have been studied using a combination of a genetic algorithm global optimization technique (GA) coupled with the Gupta semi-empirical potential and density functional theory (DFT) calculations. An initial DFT energetic analysis of small Pd–Pt clusters (N = 2–20) showed that their corresponding binding energies are slightly biased towards the stronger metal–metal bonding interactions (i.e. Pt–Pt). This led to a detailed analysis of Pd–Pt structural motifs and segregation effects, where the heteronuclear Pd–Pt parameters in the Gupta potential are derived as weighted averages of the Pd–Pd and Pt–Pt parameters, with the weighting factor (w) ranging from 0 (Pt-biased) to 1 (Pd-biased). The introduction of the weighting factor allowed us to identify three main types of segregation: core–shell; spherical cap; and ball-and-cup (intermediate between the first two types). The structural motifs predicted by the Gupta potential, as a function of composition and potential weighting factor, have been compared to our previous published Gupta and DFT calculations for 34-atom Pt–Pt clusters. From this study, we have found that a slightly Pd-biased weighting factor (w = 0.6) stabilises the mixed decahedral close packed structural motif, previously reported as the DFT global minimum in the region of most exothermic mixing for 34-atom Pd–Pt clusters. Our results show that by finely tuning the Gupta potential, one can qualitatively reproduce structural and chemical ordering patterns observed at higher levels of theory (e.g. DFT).
Co-reporter:Paul C. Jennings, Bruno G. Pollet and Roy L. Johnston
Physical Chemistry Chemical Physics 2012 - vol. 14(Issue 9) pp:NaN3139-3139
Publication Date(Web):2012/01/06
DOI:10.1039/C2CP23430K
A theoretical investigation is presented of alloying platinum with titanium to form binary Pt–Ti nanoalloys as an alternative to the expensive pure platinum catalysts commonly used for Proton Exchange Membrane Fuel Cell cathode electrocatalysts. Density Functional Theory calculations are performed to investigate compositional effects on structural properties as well as Oxygen Reduction Reaction kinetics and poisoning effects. High symmetry A32–B6clusters are studied to investigate structural properties. From these structures binding energies of hydroxyl and carbon monoxide are studied on a range of sites on the surface of the clusters. Promising results are obtained suggesting that the bimetallic Pt–Ti nanoalloys may exhibit enhanced properties compared to pure platinum catalysts.
Co-reporter:Adnan Qaseem, Fuyi Chen, Xiaoqiang Wu and Roy L. Johnston
Catalysis Science & Technology (2011-Present) 2016 - vol. 6(Issue 10) pp:NaN3340-3340
Publication Date(Web):2016/03/04
DOI:10.1039/C5CY02270C
The oxygen reduction reaction (ORR) plays a crucial role in electrochemical energy conversion and storage devices such as alkaline fuel cells and metal–air batteries. These systems, which could employ non-platinum catalysts for oxygen reduction, are cheaper and stable alternatives to their expensive counterparts like proton exchange membrane fuel cells (PEMFCs) working on platinum based catalysts. Various binary and ternary silver nanoalloys have been reported to act as efficient electrocatalysts for the ORR in alkaline fuel cells and batteries. Herein, we present a critical review on the recent advances made in silver nanoalloy electrocatalysts for the ORR in alkaline media. The mechanism of ORR on nanoalloys is described; the effect of structure and composition of various silver nanoalloys (including Ag–Cu, Ag–Pd, Ag–Au, Ag–Co etc.) on their ORR activity and stability is discussed. The rational design of electrocatalysts in order to maximize the number of catalytically active sites on the surface of the electrocatalysts for the ORR is also reviewed. Finally, we provide insights into the remaining challenges and directions for future perspectives and research.
Co-reporter:Andrew J. Logsdail, Z. Y. Li and Roy L. Johnston
Physical Chemistry Chemical Physics 2013 - vol. 15(Issue 21) pp:NaN8400-8400
Publication Date(Web):2013/04/15
DOI:10.1039/C3CP50978H
The structural preferences of nanoparticles are important for understanding their chemical properties and potential applications, and remain widely debated. Based on recent experimental observations, we present calculations on the stability of high-symmetry AuN and PdN clusters of various structural motifs, performing a systematic search of faceting preferences using mathematical constructs, a semi-empirical potential with two different parameter sets, and a quasi-Newtonian minimisation technique. We have studied the preferred ratios of (100) and (111) faces for two experimentally observed nanostructures: (a) FCC crystals, comparing octahedra with 8 (111) faces to cuboctahedra where the vertices have been systematically removed (for N < 1500); and (b) Marks-decahedra, with differing “stellation” depths (for N < 6000). For PdN and AuN we see preference towards minimisation of (100) surfaces using the parameter sets of both Cleri and Rosato [Cleri and Rosato, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 22] and Baletto et al. [Baletto et al., J. Chem. Phys., 2002, 116, 3856]. Fully stellated Marks-decahedra are found to be unfavourable at large sizes, with truncated facets identified which are similar to recent experimental observations. We find however that these stellations are deeper in PdN particles than AuN. Truncated-octahedra are found to prefer much reduced (100) surfaces and increased (111) surface areas.
Co-reporter:Lauro Oliver Paz-Borbón, Thomas V. Mortimer-Jones, Roy L. Johnston, Alvaro Posada-Amarillas, Giovanni Barcaro and Alessandro Fortunelli
Physical Chemistry Chemical Physics 2007 - vol. 9(Issue 38) pp:NaN5208-5208
Publication Date(Web):2007/07/17
DOI:10.1039/B707136A
The energetics of 98 atom bimetallic Pd–Pt clusters are studied using a combination of: a genetic algorithm technique (to explore vast areas of the configurational space); a basin-hopping atom-exchange routine (to search for lowest-energy homotops at fixed composition); and a shell optimisation approach (to search for high symmetry isomers). The interatomic interactions between Pd and Pt are modelled by the Gupta many-body empirical potential. For most compositions, the putative global minima are found to have structures based on defective Marks decahedra, but in the composition range from Pd46Pt52 to Pd63Pt35, the Leary tetrahedron (LT)—a structure previously identified for 98 atom Lennard-Jones clusters—is consistently found as the most stable structure. Based on the excess energy stability criterion, Pd56Pt42 represents the most stable cluster across the entire composition range. This structure, a Td-symmetry LT, exhibits multi-layer segregation with an innermost core of Pd atoms, an intermediate layer of Pt atoms and an outermost Pd surface shell (Pd–Pt–Pd). The stability of the Leary tetrahedron is compared against other low-energy competing structural motifs: the Marks decahedron (Dh-M), a “quasi” tetrahedron (a closed-packed structure) and two other closed-packed structures. The stability of LT structures is rationalized in terms of their spherical shape and the large number of nearest neighbours.
Co-reporter:Riccardo Ferrando, Roy L. Johnston and Catherine Louis
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 42) pp:NaN27921-27921
Publication Date(Web):2015/08/12
DOI:10.1039/C5CP90142A
A graphical abstract is available for this content
Co-reporter:Xiaoqiang Wu, Fuyi Chen, Nan Zhang, Adnan Qaseem and Roy L. Johnston
Journal of Materials Chemistry A 2016 - vol. 4(Issue 9) pp:NaN3537-3537
Publication Date(Web):2016/01/29
DOI:10.1039/C5TA09266C
Highly active electrocatalysts with good long term stability are vital for the commercialization of metal air batteries and alkaline fuel cells which involve the oxygen reduction reaction (ORR) at the cathode end. Herein, we developed a pulsed laser deposition (PLD) technique for the precise fabrication of silver–copper metallic glass (AgCu-MG) electrocatalysts. This PLD technique provides excellent control over the surface microtopography along with high flexibility for the deposition of different compositions of silver–copper metallic glass electrocatalysts onto nickel foam. Among all investigated Ag-based catalysts, AgCu-MG catalysts exhibit high electrocatalytic activity with a half-wave potential of 0.67 V (vs. RHE) which can be in situ enhanced by dealloying treatment in N2 saturated 0.1 M KOH solution. In situ dealloying of the AgCu-MG provides exceptional ORR catalytic activity with a half-wave potential of 0.78 V (vs. RHE) at 1600 rpm, which is comparable to 0.81 V (vs. RHE) of commercial Pt/C-20%. The AgCu-MG electrocatalyst showed excellent long-term stability in rechargeable zinc–air batteries. After 1000 charge–discharge cycles at 20 mA cm−2, the discharge voltage of batteries was stable at 1.0 V demonstrating the potential application of AgCu-MG as an alternative to Pt/C-20% in zinc–air batteries and alkaline fuel cells.
Co-reporter:Christopher J. Heard and Roy L. Johnston
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 39) pp:NaN21048-21048
Publication Date(Web):2014/03/03
DOI:10.1039/C3CP55507K
The structures and optical response of helical clusters (“Bernal spirals”) with compositions Ag12Cu1+ and Ag1Cu12+ are calculated within Kohn–Sham density functional theory and the configuration interaction singles variant of time dependent density functional theory. The effects of dopant position within the cluster on the vertical excitation spectrum are investigated according to the underlying electronic structure of the major transitions. The roles of symmetry and geometry are investigated by calculating the optical response of helical, icosahedral and nanorod-like clusters of Ag13+, finding local structure to be significant in driving the resultant optical response at the subnanometre scale. Further, it is noted that helical clusters have optical properties which are quite distinct from those of nanorods of similar dimensions. The effect of multiple doping is studied by introducing copper atoms into the centre of the silver helix, over the composition range Ag13+ to Ag6Cu7+. There is a complex variation of the major plasmon-like peak over this range, attributed to subtle variations in the influence of the copper 3d band on the excitations and charge transfer for different sites within the cluster. This work suggests that coinage metal nanohelices have unusual, tunable electronic properties, which in addition to their inherent chirality makes them interesting systems to study for chiral catalysis and optoelectronics.
Co-reporter:Paul C. Jennings, Hristiyan A. Aleksandrov, Konstantin M. Neyman and Roy L. Johnston
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 48) pp:NaN26545-26545
Publication Date(Web):2014/07/15
DOI:10.1039/C4CP02147A
Density functional theory calculations are performed to investigate oxygen dissociation on 116-atom truncated octahedron platinum particles. This work builds on results presented previously [Jennings et al., Nanoscale, 2014, 6, 1153], where it was shown that shell flexibility played an important role in facilitating fast oxygen dissociation. In this study, through investigation of the larger particle size, it is shown that oxygen dissociation on the (111) facet of pure platinum species is still aided by shell flexibility at larger sizes. Only the hollow sites close to the edges of the (111) facet mediate oxygen dissociation; oxygen is bound too weakly at other hollow sites for dissociation to occur. Further studies are performed on the (100) facet, which is larger for the Pt116 particle than for either the Pt38 or Pt79 ones. Much higher dissociation barriers are found on the (100) facet compared to the (111) facet, where the bridge sites are favourable for oxygen dissociation.
Co-reporter:Ramli Ismail and Roy L. Johnston
Physical Chemistry Chemical Physics 2010 - vol. 12(Issue 30) pp:NaN8619-8619
Publication Date(Web):2010/07/12
DOI:10.1039/C004044D
The energetics, structures and segregation of Pd–Au nanoalloys (all compositions for 34- and 38-atoms) have been studied using a genetic algorithm global optimization technique with the Gupta empirical potential. Three modifications of the Pd–Au parameters have been studied: parameter set I in which all parameters (A, ξ, p, q and r0) in the Gupta potential are weighted in a symmetrical fashion; parameter set II (symmetric weighting of only the pair and many-body energy scaling parameters A and ξ); and parameter set III (antisymmetric weighting of A and ξ). Structural analysis reveals competition between a range of structural families; decahedra, polyicosahedra and truncated octahedra (for 34 atoms) and incomplete-icosahedra-Mackay, decahedra, polyicosahedra (low-symmetry), six-fold-polyicosahedra and a mixed octahedron–icosahedron (Oh–Ih) structure (for 38 atoms). It is shown that, by finely tuning the Gupta potential, it is possible to qualitatively reproduce the results observed at higher levels of theory (e.g. Density Functional Theory). There are four main types of chemical ordering which are observed: core–shell; spherical cap; ball-and-cup; and mixed. It is shown that the chemical ordering and the proportion of Pd–Au heteronuclear bonds in these clusters are strongly dependent on the potential parameters. Comparison of the results from parameter set III and two previously fitted potentials shows that the DFT-fit potential gives rise to similar results for energies, and lowest energy structures and homotops to those for parameter set III with wa = 0.8, but the exp-fit potential gives rise to qualitatively different results.
Co-reporter:A. Shayeghi, D. Götz, J. B. A. Davis, R. Schäfer and R. L. Johnston
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 3) pp:NaN2112-2112
Publication Date(Web):2014/12/01
DOI:10.1039/C4CP04323E
The Birmingham cluster genetic algorithm is a package that performs global optimisations for homo- and bimetallic clusters based on either first principles methods or empirical potentials. Here, we present a new parallel implementation of the code which employs a pool strategy in order to eliminate sequential steps and significantly improve performance. The new approach meets all requirements of an evolutionary algorithm and contains the main features of the previous implementation. The performance of the pool genetic algorithm is tested using the Gupta potential for the global optimisation of the Au10Pd10 cluster, which demonstrates the high efficiency of the method. The new implementation is also used for the global optimisation of the Au10 and Au20 clusters directly at the density functional theory level.






































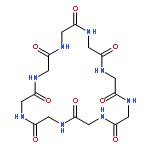
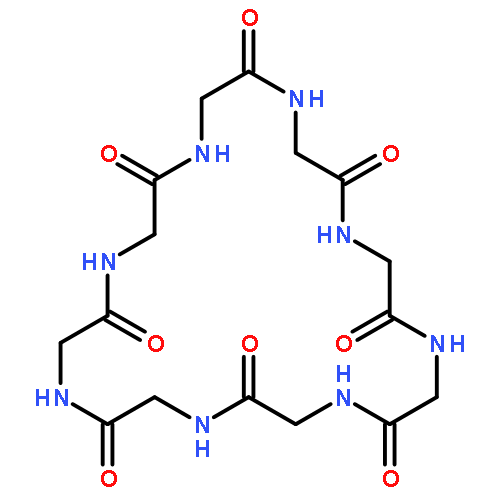































































































![Silane, [5-(1,1-dimethylethyl)-1,1-dioxido-2-thienyl]trimethyl-](http://img.cochemist.com/ccimg/241900/241808-85-5.png)
![Silane, [5-(1,1-dimethylethyl)-1,1-dioxido-2-thienyl]trimethyl-](http://img.cochemist.com/ccimg/241900/241808-85-5_b.png)
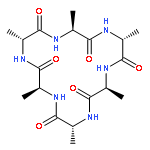





















![L-Lysine, N2-[(9H-fluoren-9-ylmethoxy)carbonyl]-, 2-propenyl ester](http://img.cochemist.com/ccimg/161600/161580-12-7.png)
![L-Lysine, N2-[(9H-fluoren-9-ylmethoxy)carbonyl]-, 2-propenyl ester](http://img.cochemist.com/ccimg/161600/161580-12-7_b.png)











![D-Alanine,N-[N-(N-D-alanyl-D-alanyl)-D-alanyl]- (9CI)](http://img.cochemist.com/ccimg/1000/926-78-3.png)
![D-Alanine,N-[N-(N-D-alanyl-D-alanyl)-D-alanyl]- (9CI)](http://img.cochemist.com/ccimg/1000/926-78-3_b.png)


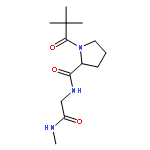
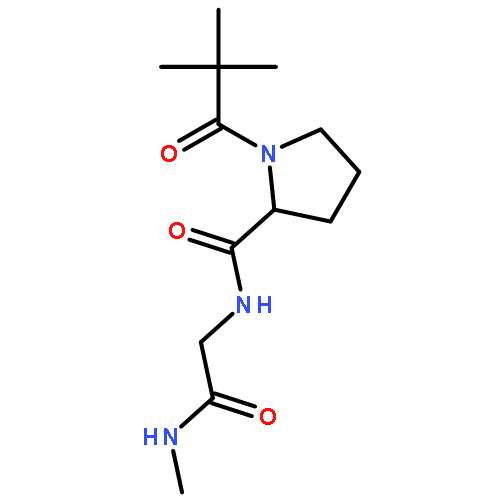







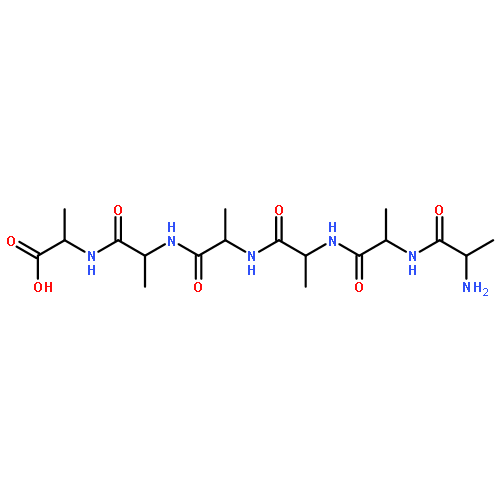





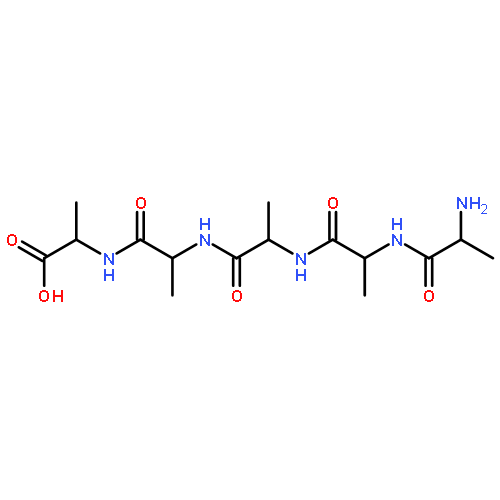










![2-ACETAMIDO-N-[2-[[2-(METHYLAMINO)-2-OXOETHYL]AMINO]-2-OXOETHYL]ACETAMIDE](http://img.cochemist.com/ccimg/30300/30264-19-8.png)
![2-ACETAMIDO-N-[2-[[2-(METHYLAMINO)-2-OXOETHYL]AMINO]-2-OXOETHYL]ACETAMIDE](http://img.cochemist.com/ccimg/30300/30264-19-8_b.png)