Co-reporter:Anna E. Allen and David W. C. MacMillan
Journal of the American Chemical Society March 30, 2011 Volume 133(Issue 12) pp:4260-4263
Publication Date(Web):March 9, 2011
DOI:10.1021/ja2008906
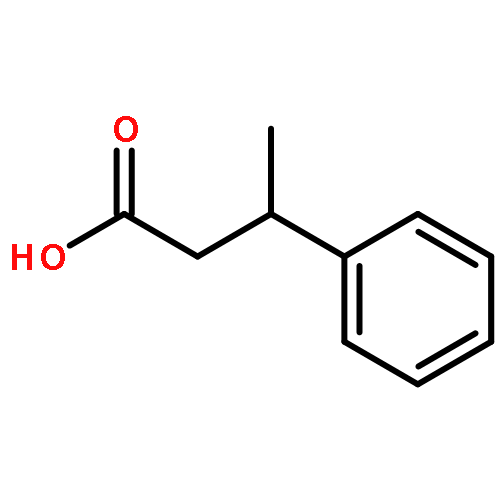
The enantioselective α-arylation of aldehydes has been accomplished using diaryliodonium salts and a combination of copper and organic catalysts. These mild catalytic conditions provide a new strategy for the enantioselective construction and retention of enolizable α-formyl benzylic stereocenters, a valuable synthon for the production of medicinal agents. As one example, this new asymmetric protocol has been applied to the rapid synthesis of (S)-ketoprofen, a commercially successful oral and topical analgesic.
Co-reporter:Chi “Chip” Le, Michael K. Wismer, Zhi-Cai Shi, Rui Zhang, Donald V. Conway, Guoqing Li, Petr Vachal, Ian W. Davies, and David W. C. MacMillan
ACS Central Science June 28, 2017 Volume 3(Issue 6) pp:647-647
Publication Date(Web):May 17, 2017
DOI:10.1021/acscentsci.7b00159
Photocatalysis for organic synthesis has experienced an exponential growth in the past 10 years. However, the variety of experimental procedures that have been reported to perform photon-based catalyst excitation has hampered the establishment of general protocols to convert visible light into chemical energy. To address this issue, we have designed an integrated photoreactor for enhanced photon capture and catalyst excitation. Moreover, the evaluation of this new reactor in eight photocatalytic transformations that are widely employed in medicinal chemistry settings has confirmed significant performance advantages of this optimized design while enabling a standardized protocol.
Co-reporter:Xiaheng Zhang and David W. C. MacMillan
Journal of the American Chemical Society August 23, 2017 Volume 139(Issue 33) pp:11353-11353
Publication Date(Web):August 6, 2017
DOI:10.1021/jacs.7b07078
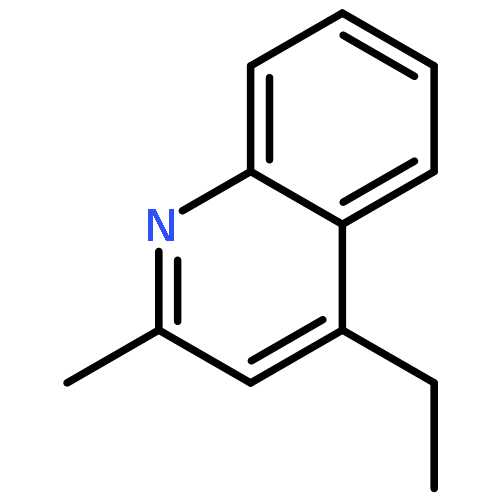
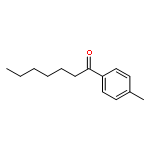
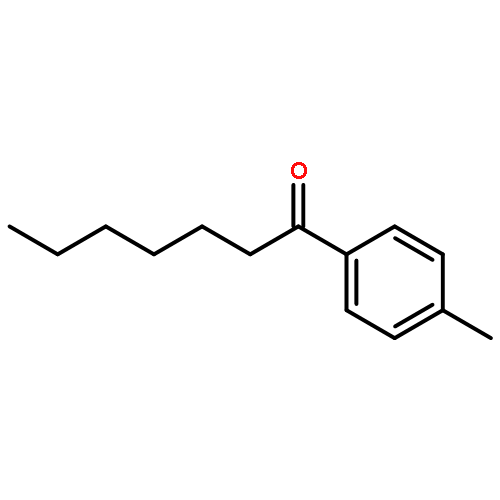
A mechanism that enables direct aldehyde C–H functionalization has been achieved via the synergistic merger of photoredox, nickel, and hydrogen atom transfer catalysis. This mild, operationally simple protocol transforms a wide variety of commercially available aldehydes, along with aryl or alkyl bromides, into the corresponding ketones in excellent yield. This C–H abstraction coupling technology has been successfully applied to the expedient synthesis of the medicinal agent haloperidol.
Co-reporter:Daniela M. Arias-Rotondo;James K. McCusker;Eric R. Welin;Chip Le
Science 2017 Volume 355(Issue 6323) pp:380-385
Publication Date(Web):27 Jan 2017
DOI:10.1126/science.aal2490
A nickel's worth of transferred energy
Traditional organic photochemistry often relies on sensitizers, molecules that efficiently absorb light and then transfer the energy to other compounds to spur reactivity. Welin et al. leveraged this approach to stimulate an organometallic nickel catalyst. They photoexcited an iridium complex with blue light. The ensuing energy transfer to nickel enabled C–O bond formation, coupling aryl halides with carboxylic acids. A similar approach could generate a variety of additional reactivity patterns through photosensitized transition metal catalysis.
Science, this issue p. 380
Co-reporter:Xiaheng Zhang and David W. C. MacMillan
Journal of the American Chemical Society 2016 Volume 138(Issue 42) pp:13862-13865
Publication Date(Web):October 9, 2016
DOI:10.1021/jacs.6b09533
Alkyl oxalates, prepared from their corresponding alcohols, are engaged for the first time as carbon radical fragments in metallaphotoredox catalysis. In this report, we demonstrate that alcohols, native organic functional groups, can be readily activated with simple oxalyl chloride to become radical precursors in a net redox-neutral Csp3–Csp2 cross-coupling with a broad range of aryl halides. This alcohol-activation coupling is successfully applied to the functionalization of a naturally occurring steroid and the expedient synthesis of a medicinally relevant drug lead.
Co-reporter:Chun Liu; E. Zachary Oblak; Mark N. Vander Wal; Andrew K. Dilger; Danielle K. Almstead
Journal of the American Chemical Society 2016 Volume 138(Issue 7) pp:2134-2137
Publication Date(Web):January 21, 2016
DOI:10.1021/jacs.5b13041
A generic activation mode for asymmetric LUMO-lowering catalysis has been developed using the long-established principles of oxy-allyl cation chemistry. Here, the enantioselective conversion of racemic α-tosyloxy ketones to optically enriched α-indolic carbonyls has been accomplished using a new amino alcohol catalyst in the presence of electron-rich indole nucleophiles. Kinetic studies reveal that the rate-determining step in this SN1 pathway is the catalyst-mediated α-tosyloxy ketone deprotonation step to form an enantiodiscriminant oxy-allyl cation prior to the stereodefining nucleophilic addition event.
Co-reporter:Patricia Zhang; Chi “Chip” Le
Journal of the American Chemical Society 2016 Volume 138(Issue 26) pp:8084-8087
Publication Date(Web):June 6, 2016
DOI:10.1021/jacs.6b04818
A strategy for cross-electrophile coupling has been developed via the merger of photoredox and transition metal catalysis. In this report, we demonstrate the use of commercially available tris(trimethylsilyl)silane with metallaphotoredox catalysis to efficiently couple alkyl bromides with aryl or heteroaryl bromides in excellent yields. We hypothesize that a photocatalytically generated silyl radical species can perform halogen-atom abstraction to activate alkyl halides as nucleophilic cross-coupling partners. This protocol allows the use of mild yet robust conditions to construct Csp3–Csp2 bonds generically via a unique cross-coupling pathway.
Co-reporter:Zhiwei Zuo; Huan Cong; Wei Li; Junwon Choi; Gregory C. Fu
Journal of the American Chemical Society 2016 Volume 138(Issue 6) pp:1832-1835
Publication Date(Web):February 5, 2016
DOI:10.1021/jacs.5b13211
An asymmetric decarboxylative Csp3–Csp2 cross-coupling has been achieved via the synergistic merger of photoredox and nickel catalysis. This mild, operationally simple protocol transforms a wide variety of naturally abundant α-amino acids and readily available aryl halides into valuable chiral benzylic amines in high enantiomeric excess, thereby producing motifs found in pharmacologically active agents.
Co-reporter:Emily B. Corcoran;Michael T. Pirnot;Shishi Lin;Spencer D. Dreher;Daniel A. DiRocco;Ian W. Davies;Stephen L. Buchwald
Science 2016 Volume 353(Issue 6296) pp:279-283
Publication Date(Web):15 Jul 2016
DOI:10.1126/science.aag0209
A light approach to C-N bond formation
The need to form C-N bonds arises frequently in drug discovery research. One versatile approach involves the attachment of the C and N fragments to a Pd catalyst. This approach needs a bulky ligand to “crowd” the fragments together off the metal center. Corcoran et al. present a complementary approach that uses Ni in place of Pd. Instead of the bulky ligand, they used a light-activated cocatalyst that strips an electron from the Ni to accelerate the bond formation. A screen involving elaborately substituted reagents confirmed the utility of this approach in cases that challenge the traditional Pd coupling.
Science, this issue p. 279
Co-reporter:Megan H. Shaw, Jack Twilton, and David W. C. MacMillan
The Journal of Organic Chemistry 2016 Volume 81(Issue 16) pp:6898-6926
Publication Date(Web):August 1, 2016
DOI:10.1021/acs.joc.6b01449
In recent years, photoredox catalysis has come to the forefront in organic chemistry as a powerful strategy for the activation of small molecules. In a general sense, these approaches rely on the ability of metal complexes and organic dyes to convert visible light into chemical energy by engaging in single-electron transfer with organic substrates, thereby generating reactive intermediates. In this Perspective, we highlight the unique ability of photoredox catalysis to expedite the development of completely new reaction mechanisms, with particular emphasis placed on multicatalytic strategies that enable the construction of challenging carbon–carbon and carbon–heteroatom bonds.
Co-reporter:Megan H. Shaw;Valerie W. Shurtleff;Jack A. Terrett;James D. Cuthbertson
Science 2016 Volume 352(Issue 6291) pp:1304-1308
Publication Date(Web):10 Jun 2016
DOI:10.1126/science.aaf6635
A tip of the HAT to C-C bond formation
Iridium and nickel are already a proven team for forging carbon-carbon bonds. The iridium harvests blue light from a simple light-emitting diode and orchestrates the coupling by the nickel. Shaw et al. now add a third player to the team, a hydrogen atom transfer (HAT) catalyst (see the Perspective by Fruit). Together, the trio of catalysts can link bromo- or chloroaryl rings directly to C-H sites adjacent to nitrogen or oxygen, with no need for prior modification. The reaction is highly selective across a broad range of substrates.
Science, this issue p. 1304; see also p. 1277
Co-reporter:Chi “Chip” Le
Journal of the American Chemical Society 2015 Volume 137(Issue 37) pp:11938-11941
Publication Date(Web):September 2, 2015
DOI:10.1021/jacs.5b08304
In this study we demonstrate that molecular fragments, which can be readily coupled via a simple, in situ RO—C═OR bond-forming reaction, can subsequently undergo metal insertion–decarboxylation–recombination to generate Csp2–Csp3 bonds when subjected to metallaphotoredox catalysis. In this embodiment the conversion of a wide variety of mixed anhydrides (formed in situ from carboxylic acids and acyl chlorides) to fragment-coupled ketones is accomplished in good to high yield. A three-step synthesis of the medicinal agent edivoxetine is also described using this new decarboxylation–recombination protocol.
Co-reporter:Christopher C. Nawrat; Christopher R. Jamison; Yuriy Slutskyy; David W. C. MacMillan;Larry E. Overman
Journal of the American Chemical Society 2015 Volume 137(Issue 35) pp:11270-11273
Publication Date(Web):August 31, 2015
DOI:10.1021/jacs.5b07678
Alkyl oxalates are new bench-stable alcohol-activating groups for radical generation under visible light photoredox conditions. Using these precursors, the first net redox-neutral coupling of tertiary and secondary alcohols with electron-deficient alkenes is achieved.
Co-reporter:Sandrine Ventre; Filip R. Petronijevic
Journal of the American Chemical Society 2015 Volume 137(Issue 17) pp:5654-5657
Publication Date(Web):April 16, 2015
DOI:10.1021/jacs.5b02244
The direct conversion of aliphatic carboxylic acids to the corresponding alkyl fluorides has been achieved via visible light-promoted photoredox catalysis. This operationally simple, redox-neutral fluorination method is amenable to a wide variety of carboxylic acids. Photon-induced oxidation of carboxylates leads to the formation of carboxyl radicals, which upon rapid CO2-extrusion and F• transfer from a fluorinating reagent yield the desired fluoroalkanes with high efficiency. Experimental evidence indicates that an oxidative quenching pathway is operable in this broadly applicable fluorination protocol.
Co-reporter:Jenna L. Jeffrey; Filip R. Petronijević
Journal of the American Chemical Society 2015 Volume 137(Issue 26) pp:8404-8407
Publication Date(Web):June 15, 2015
DOI:10.1021/jacs.5b05376
A direct β-coupling of cyclic ketones with imines has been accomplished via the synergistic combination of photoredox catalysis and organocatalysis. Transient β-enaminyl radicals derived from ketones via enamine and oxidative photoredox catalysis readily combine with persistent α-amino radicals in a highly selective hetero radical–radical coupling. This novel pathway to γ-aminoketones is predicated upon the use of DABCO as both a base and an electron transfer agent. This protocol also formally allows for the direct synthesis of β-Mannich products via a chemoselective three-component coupling of aryl aldehydes, amines, and ketones.
Co-reporter:Dr. Lingling Chu;Jeffrey M. Lipshultz ;Dr. David W. C. MacMillan
Angewandte Chemie International Edition 2015 Volume 54( Issue 27) pp:7929-7933
Publication Date(Web):
DOI:10.1002/anie.201501908
Abstract
The direct decarboxylative arylation of α-oxo acids has been achieved by synergistic visible-light-mediated photoredox and nickel catalysis. This method offers rapid entry to aryl and alkyl ketone architectures from simple α-oxo acid precursors via an acyl radical intermediate. Significant substrate scope is observed with respect to both the oxo acid and arene coupling partners. This mild decarboxylative arylation can also be utilized to efficiently access medicinal agents, as demonstrated by the rapid synthesis of fenofibrate.
Co-reporter:Eric R. Welin;Dr. Alexer A. Warkentin;Dr. Jay C. Conrad ;Dr. David W. C. MacMillan
Angewandte Chemie International Edition 2015 Volume 54( Issue 33) pp:9668-9672
Publication Date(Web):
DOI:10.1002/anie.201503789
Abstract
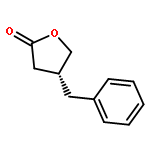
The combination of photoredox catalysis and enamine catalysis has enabled the development of an enantioselective α-cyanoalkylation of aldehydes. This synergistic catalysis protocol allows for the coupling of two highly versatile yet orthogonal functionalities, allowing rapid diversification of the oxonitrile products to a wide array of medicinally relevant derivatives and heterocycles. This methodology has also been applied to the total synthesis of the lignan natural product (−)-bursehernin.
Co-reporter:Dr. Jian Jin ;Dr. David W. C. MacMillan
Angewandte Chemie 2015 Volume 127( Issue 5) pp:1585-1589
Publication Date(Web):
DOI:10.1002/ange.201410432
Abstract
The direct α-arylation of cyclic and acyclic ethers with heteroarenes has been accomplished through the design of a photoredox-mediated CH functionalization pathway. Transiently generated α-oxyalkyl radicals, produced from a variety of widely available ethers through hydrogen atom transfer (HAT), were coupled with a range of electron-deficient heteroarenes in a Minisci-type mechanism. This mild, visible-light-driven protocol allows direct access to medicinal pharmacophores of broad utility using feedstock substrates and a commercial photocatalyst.
Co-reporter:Dr. Lingling Chu;Jeffrey M. Lipshultz ;Dr. David W. C. MacMillan
Angewandte Chemie 2015 Volume 127( Issue 27) pp:8040-8044
Publication Date(Web):
DOI:10.1002/ange.201501908
Abstract
The direct decarboxylative arylation of α-oxo acids has been achieved by synergistic visible-light-mediated photoredox and nickel catalysis. This method offers rapid entry to aryl and alkyl ketone architectures from simple α-oxo acid precursors via an acyl radical intermediate. Significant substrate scope is observed with respect to both the oxo acid and arene coupling partners. This mild decarboxylative arylation can also be utilized to efficiently access medicinal agents, as demonstrated by the rapid synthesis of fenofibrate.
Co-reporter:Eric R. Welin;Dr. Alexer A. Warkentin;Dr. Jay C. Conrad ;Dr. David W. C. MacMillan
Angewandte Chemie 2015 Volume 127( Issue 33) pp:9804-9808
Publication Date(Web):
DOI:10.1002/ange.201503789
Abstract
The combination of photoredox catalysis and enamine catalysis has enabled the development of an enantioselective α-cyanoalkylation of aldehydes. This synergistic catalysis protocol allows for the coupling of two highly versatile yet orthogonal functionalities, allowing rapid diversification of the oxonitrile products to a wide array of medicinally relevant derivatives and heterocycles. This methodology has also been applied to the total synthesis of the lignan natural product (−)-bursehernin.
Co-reporter:Dr. Jian Jin ;Dr. David W. C. MacMillan
Angewandte Chemie International Edition 2015 Volume 54( Issue 5) pp:1565-1569
Publication Date(Web):
DOI:10.1002/anie.201410432
Abstract
The direct α-arylation of cyclic and acyclic ethers with heteroarenes has been accomplished through the design of a photoredox-mediated CH functionalization pathway. Transiently generated α-oxyalkyl radicals, produced from a variety of widely available ethers through hydrogen atom transfer (HAT), were coupled with a range of electron-deficient heteroarenes in a Minisci-type mechanism. This mild, visible-light-driven protocol allows direct access to medicinal pharmacophores of broad utility using feedstock substrates and a commercial photocatalyst.
Co-reporter:Jenna L. Jeffrey;Jack A. Terrett
Science 2015 Vol 349(6255) pp:1532-1536
Publication Date(Web):25 Sep 2015
DOI:10.1126/science.aac8555
A trio helps activate C-H bonds in alcohols
Enzymes can accelerate chemical reactions by activating specific portions of a molecule through well-placed H bonds. Jeffrey et al. showcase the power of H-bonding in a synthetic context. Here, reactivity at the C centers of alcohols is selectively induced by using an H-bonding catalyst to bind the hydroxyl group of the alcohol. The adjacent C-H bonds now become susceptible to a reaction accelerated by another pair of catalysts. In combination, the trio of catalysts promotes C-C bond formation at the alcohol C within an array of competing sites.
Science, this issue p. 1532
Co-reporter:Dominik Hager
Journal of the American Chemical Society 2014 Volume 136(Issue 49) pp:16986-16989
Publication Date(Web):November 17, 2014
DOI:10.1021/ja5102695
The photoredox-mediated coupling of benzylic ethers with Schiff bases has been accomplished. Direct benzylic C–H activation by a combination of a thiol catalyst with an iridium photocatalyst and subsequent radical–radical coupling with secondary aldimines affords a variety of β-amino ether products in good to excellent yields. Mechanistic studies suggest that a reductive quenching pathway of the photocatalyst is operable.
Co-reporter:Lingling Chu ; Chisa Ohta ; Zhiwei Zuo
Journal of the American Chemical Society 2014 Volume 136(Issue 31) pp:10886-10889
Publication Date(Web):July 17, 2014
DOI:10.1021/ja505964r
The direct application of carboxylic acids as a traceless activation group for radical Michael additions has been accomplished via visible light-mediated photoredox catalysis. Photon-induced oxidation of a broad series of carboxylic acids, including hydrocarbon-substituted, α-oxy, and α-amino acids, provides a versatile CO2-extrusion platform to generate Michael donors without the requirement for organometallic activation or propagation. A diverse array of Michael acceptors is amenable to this new conjugate addition strategy. An application of this technology to a three-step synthesis of the medicinal agent pregabalin (commercialized by Pfizer under the trade name Lyrica) is also presented.
Co-reporter:Adam Noble
Journal of the American Chemical Society 2014 Volume 136(Issue 33) pp:11602-11605
Publication Date(Web):July 14, 2014
DOI:10.1021/ja506094d
A new coupling protocol has been developed that allows the union of vinyl sulfones with photoredox-generated α-amino radicals to provide allylic amines of broad diversity. Direct C–H vinylations of N-aryl tertiary amines, as well as decarboxylative vinylations of N-Boc α-amino acids, proceed in high yield and with excellent olefin geometry control. The utility of this new allyl amine forming reaction has been demonstrated via the syntheses of several natural products and a number of established pharmacophores.
Co-reporter:Jack A. Terrett ; Michael D. Clift
Journal of the American Chemical Society 2014 Volume 136(Issue 19) pp:6858-6861
Publication Date(Web):April 22, 2014
DOI:10.1021/ja502639e
Direct β-alkylation of saturated aldehydes has been accomplished by synergistically combining photoredox catalysis and organocatalysis. Photon-induced enamine oxidation provides an activated β-enaminyl radical intermediate, which readily combines with a wide range of Michael acceptors to produce β-alkyl aldehydes in a highly efficient manner. Furthermore, this redox-neutral, atom-economical C–H functionalization protocol can be achieved both inter- and intramolecularly. Mechanistic studies by various spectroscopic methods suggest that a reductive quenching pathway is operable.
Co-reporter:Zhiwei Zuo
Journal of the American Chemical Society 2014 Volume 136(Issue 14) pp:5257-5260
Publication Date(Web):March 12, 2014
DOI:10.1021/ja501621q
The direct decarboxylative arylation of α-amino acids has been achieved via visible light-mediated photoredox catalysis. This method offers rapid entry to prevalent benzylic amine architectures from an abundant biomass, specifically α-amino acid precursors. Significant substrate scope is observed with respect to both the amino acid and arene components.
Co-reporter:Manuel Peifer ; Raphaëlle Berger ; Valerie W. Shurtleff ; Jay C. Conrad
Journal of the American Chemical Society 2014 Volume 136(Issue 16) pp:5900-5903
Publication Date(Web):March 26, 2014
DOI:10.1021/ja502205q
An efficient route towards biologically relevant pentose derivatives is described. The de novo synthetic strategy features an enantioselective α-oxidation reaction enabled by a chiral amine in conjunction with copper(II) catalysis. A subsequent Mukaiyama aldol coupling allows for the incorporation of a wide array of modular two-carbon fragments. Lactone intermediates accessed via this route provide a useful platform for elaboration, as demonstrated by the preparation of a variety of C-nucleosides and fluorinated pentoses. Finally, this work has facilitated expedient syntheses of pharmaceutically active compounds currently in clinical use.
Co-reporter:Adam Noble; Stefan J. McCarver
Journal of the American Chemical Society 2014 Volume 137(Issue 2) pp:624-627
Publication Date(Web):December 18, 2014
DOI:10.1021/ja511913h
Decarboxylative cross-coupling of alkyl carboxylic acids with vinyl halides has been accomplished through the synergistic merger of photoredox and nickel catalysis. This new methodology has been successfully applied to a variety of α-oxy and α-amino acids, as well as simple hydrocarbon-substituted acids. Diverse vinyl iodides and bromides give rise to vinylation products in high efficiency under mild, operationally simple reaction conditions.
Co-reporter:Christopher K. Prier and David W. C. MacMillan
Chemical Science 2014 vol. 5(Issue 11) pp:4173-4178
Publication Date(Web):04 Aug 2014
DOI:10.1039/C4SC02155J
The direct α-heteroarylation of tertiary amines has been accomplished via photoredox catalysis to generate valuable benzylic amine pharmacophores. A variety of five- and six-membered chloroheteroarenes are shown to function as viable coupling partners for the α-arylation of a diverse range of cyclic and acyclic amines. Evidence is provided for a homolytic aromatic substitution mechanism, in which a catalytically-generated α-amino radical undergoes direct addition to an electrophilic chloroarene.
Co-reporter:Zhiwei Zuo;Derek T. Ahneman;Lingling Chu;Jack A. Terrett;Abigail G. Doyle
Science 2014 Volume 345(Issue 6195) pp:437-440
Publication Date(Web):25 Jul 2014
DOI:10.1126/science.1255525
A bright outlook for carbon coupling
In contemporary organic chemistry, it is straightforward to forge bonds between unsaturated carbons (i.e., carbons already engaged in double bonds) using cross-coupling catalysis. The protocol runs into some trouble, however, if one or both starting carbon centers are saturated (purely single-bonded). Tellis et al. and Zuo et al. independently found that combining a second, light-activated catalyst with a nickel cross-coupling catalyst could achieve selective coupling of saturated and unsaturated reagents (see the Perspective by Lloyd-Jones and Ball). Their methods rely on single-electron transfer from the light-activated catalyst to the saturated carbon, thereby enhancing its reactivity more effectively than the twoelectron mechanisms prevailing in traditional protocols.
Science, this issue p. 433, p. 437; see also p. 381
Co-reporter:Christopher K. Prier, Danica A. Rankic, and David W. C. MacMillan
Chemical Reviews 2013 Volume 113(Issue 7) pp:5322
Publication Date(Web):March 19, 2013
DOI:10.1021/cr300503r
Co-reporter:Robert J. Comito ; Fernanda G. Finelli
Journal of the American Chemical Society 2013 Volume 135(Issue 25) pp:9358-9361
Publication Date(Web):June 7, 2013
DOI:10.1021/ja4047312
A highly selective method for the synthesis of asymmetrically substituted carbocycles and heterocycles from unactivated aldehyde–olefin precursors has been achieved via enantioselective SOMO-catalysis. Addition of a catalytically generated enamine radical cation across a pendent olefin serves to establish a general asymmetric strategy toward the production of a wide range of formyl-substituted rings with alkene transposition. Conceptually, this novel mechanism allows direct access to “homo-ene”-type products.
Co-reporter:Giuseppe Cecere ; Christian M. König ; Jennifer L. Alleva
Journal of the American Chemical Society 2013 Volume 135(Issue 31) pp:11521-11524
Publication Date(Web):July 19, 2013
DOI:10.1021/ja406181e
The direct, asymmetric α-amination of aldehydes has been accomplished via a combination of photoredox and organocatalysis. Photon-generated N-centered radicals undergo enantioselective α-addition to catalytically formed chiral enamines to directly produce stable α-amino aldehyde adducts bearing synthetically useful amine substitution patterns. Incorporation of a photolabile group on the amine precursor obviates the need to employ a photoredox catalyst in this transformation. Importantly, this photoinduced transformation allows direct and enantioselective access to α-amino aldehyde products that do not require postreaction manipulation.
Co-reporter:Jason M. Stevens
Journal of the American Chemical Society 2013 Volume 135(Issue 32) pp:11756-11759
Publication Date(Web):July 28, 2013
DOI:10.1021/ja406356c
The enantioselective α-alkenylation of aldehydes has been accomplished using boronic acids via the synergistic combination of copper and chiral amine catalysis. The merger of two highly utilized and robust catalytic systems has allowed for the development of a mild and operationally trivial protocol for the direct formation of α-formyl olefins employing common building blocks for organic synthesis.
Co-reporter:Filip R. Petronijević ; Manuel Nappi
Journal of the American Chemical Society 2013 Volume 135(Issue 49) pp:18323-18326
Publication Date(Web):November 15, 2013
DOI:10.1021/ja410478a
The direct β-coupling of cyclic ketones with aryl ketones has been achieved via the synergistic combination of photoredox catalysis and organocatalysis. Diaryl oxymethyl or aryl–alkyl oxymethyl radicals, transiently generated via single-electron reduction of ketone precursors, readily merge with β-enaminyl radical species, generated by photon-induced enamine oxidation, to produce γ-hydroxyketone adducts. Experimental evidence indicates that two discrete reaction pathways can be operable in this process depending upon the nature of the ketyl radical precursor and the photocatalyst.
Co-reporter:Katrine Qvortrup ; Danica A. Rankic
Journal of the American Chemical Society 2013 Volume 136(Issue 2) pp:626-629
Publication Date(Web):December 16, 2013
DOI:10.1021/ja411596q
Direct C–H functionalization and arylation of benzyl ethers has been accomplished via photoredox organocatalysis. The productive merger of a thiol catalyst and a commercially available iridium photoredox catalyst in the presence of household light directly affords benzylic arylation products in good to excellent yield. The utility of this methodology is further demonstrated in direct arylation of 2,5-dihydrofuran to form a single regioisomer.
Co-reporter:Ryan W. Evans ; Jason R. Zbieg ; Shaolin Zhu ; Wei Li
Journal of the American Chemical Society 2013 Volume 135(Issue 43) pp:16074-16077
Publication Date(Web):October 9, 2013
DOI:10.1021/ja4096472
The direct α-amination of ketones, esters, and aldehydes has been accomplished via copper catalysis. In the presence of catalytic copper(II) bromide, a diverse range of carbonyl and amine substrates undergo fragment coupling to produce synthetically useful α-amino-substituted motifs. The transformation is proposed to proceed via a catalytically generated α-bromo carbonyl species; nucleophilic displacement of the bromide by the amine then delivers the α-amino carbonyl adduct while the catalyst is reconstituted. The practical value of this transformation is highlighted through one-step syntheses of two high-profile pharmaceutical agents, Plavix and amfepramone.
Co-reporter:Benjamin D. Horning
Journal of the American Chemical Society 2013 Volume 135(Issue 17) pp:6442-6445
Publication Date(Web):April 15, 2013
DOI:10.1021/ja402933s
A concise and highly enantioselective total synthesis of the akuammiline alkaloid (−)-vincorine has been accomplished. A key element of the synthesis is a stereoselective organocatalytic Diels–Alder, iminium cyclization cascade sequence, which serves to construct the tetracyclic alkaloid core architecture in one step from simple achiral precursors. The challenging seven-membered azepanyl ring system is installed by way of a single electron-mediated cyclization event initiated from an acyl telluride precursor. The total synthesis of (−)-vincorine is achieved in nine steps and 9% overall yield from commercially available starting materials.
Co-reporter:Mark N. Vander Wal, Andrew K. Dilger and David W. C. MacMillan
Chemical Science 2013 vol. 4(Issue 8) pp:3075-3079
Publication Date(Web):11 Jun 2013
DOI:10.1039/C3SC51266E
Oxy-allyl cations have been known as transient electrophilic species since they were first proposed as intermediates in the Favorskii rearrangement in 1894. Since that time, they also have been used as a mode of activation for [4 + 3] cycloadditions in a variety of natural product syntheses. In this manuscript, we describe a method for the interception of oxy-allyl cations with a diverse range of common nucleophiles, thereby demonstrating the value of this intermediate as a generic mode of activation. This simple, mild, room temperature protocol allows for the formation of a variety of high value carbon–carbon and carbon–heteroatom bonds that are readily incorporated within a series of cyclic and acyclic ketone systems. Initial efforts into the development of an enantioselective catalytic variant are also described.
Co-reporter:Brian N. Laforteza;Mark Pickworth ; David W. C. MacMillan
Angewandte Chemie International Edition 2013 Volume 52( Issue 43) pp:11269-11272
Publication Date(Web):
DOI:10.1002/anie.201305171
Co-reporter:Michael T. Pirnot;David B. C. Martin;Danica A. Rankic
Science 2013 Volume 339(Issue 6127) pp:1593-1596
Publication Date(Web):29 Mar 2013
DOI:10.1126/science.1232993
Beta Bonding
Carbonyl compounds, which incorporate carbon-oxygen double bonds, are among the most productively reactive molecules in synthetic as well as biochemical contexts. The carbon directly bonded to oxygen is rendered highly electrophilic, while the adjacent (α) carbons are easily deprotonated to undergo further transformations. In contrast, the β carbons that are one step further along the chain tend to be relatively inert. Pirnot et al. (p. 1593) now show that a dual catalyst system—consisting of an amine and a photoactive metal complex—can activate the β carbons of carbonyl compounds to couple with aryl substrates.
Co-reporter:Brian N. Laforteza;Mark Pickworth ; David W. C. MacMillan
Angewandte Chemie 2013 Volume 125( Issue 43) pp:11479-11482
Publication Date(Web):
DOI:10.1002/ange.201305171
Co-reporter:Eduardas Skucas
Journal of the American Chemical Society 2012 Volume 134(Issue 22) pp:9090-9093
Publication Date(Web):May 22, 2012
DOI:10.1021/ja303116v
The enantioselective α-vinylation of aldehydes using vinyl iodonium triflate salts has been accomplished via the synergistic combination of copper and chiral amine catalysis. These mild catalytic conditions provide a direct route for the enantioselective construction of enolizable α-formyl vinylic stereocenters without racemization or olefin transposition. These high-value coupling adducts are readily converted into a variety of useful olefin synthons.
Co-reporter:Shaolin Zhu
Journal of the American Chemical Society 2012 Volume 134(Issue 26) pp:10815-10818
Publication Date(Web):June 21, 2012
DOI:10.1021/ja305100g
An enantioselective arylation–cyclization cascade has been accomplished using a combination of diaryliodonium salts and asymmetric copper catalysis. These mild catalytic conditions provide a new strategy for the enantioselective construction of pyrroloindolines, an important alkaloid structural motif that is commonly found among biologically active natural products.
Co-reporter:Nathan T. Jui ; Jeffrey A. O. Garber ; Fernanda Gadini Finelli
Journal of the American Chemical Society 2012 Volume 134(Issue 28) pp:11400-11403
Publication Date(Web):July 5, 2012
DOI:10.1021/ja305076b
A new method to rapidly generate pyrrolidines via a SOMO-activated enantioselective (3 + 2) coupling of aldehydes and conjugated olefins has been accomplished. A radical-polar crossover mechanism is proposed wherein olefin addition to a transient enamine radical cation and oxidation of the resulting radical furnish a cationic intermediate which is vulnerable to nucleophilic addition of a tethered amine group. A range of olefins, including styrenes and dienes, are shown to provide stereochemically complex pyrrolidine products with high chemical efficiency and enantiocontrol.
Co-reporter:Anna E. Allen and David W. C. MacMillan
Chemical Science 2012 vol. 3(Issue 3) pp:633-658
Publication Date(Web):25 Jan 2012
DOI:10.1039/C2SC00907B
Synergistic catalysis is a synthetic strategy wherein both the nucleophile and the electrophile are simultaneously activated by two separate and distinct catalysts to afford a single chemical transformation. This powerful catalysis strategy leads to several benefits, specifically synergistic catalysis can (i) introduce new, previously unattainable chemical transformations, (ii) improve the efficiency of existing transformations, and (iii) create or improve catalytic enantioselectivity where stereocontrol was previously absent or challenging. This perspective aims to highlight these benefits using many of the successful examples of synergistic catalysis found in the literature.
Co-reporter:Scott P. Simonovich, Jeffrey F. Van Humbeck and David W. C. MacMillan
Chemical Science 2012 vol. 3(Issue 1) pp:58-61
Publication Date(Web):22 Sep 2011
DOI:10.1039/C1SC00556A
A new enantioselective α-oxidation of aldehydes has been accomplished using TEMPO and a synergistic combination of copper and organic catalysis. Expanding upon recently reported mechanistic studies, these mild catalytic conditions provide stable aldehyde products bearing a wide array of electronically and sterically diverse substructures. The utility of these oxidized products is highlighted by subsequent derivatization to a variety of common chiral synthons, without loss in enantiopurity.
Co-reporter:Piotr Kwiatkowski ; Teresa D. Beeson ; Jay C. Conrad
Journal of the American Chemical Society 2011 Volume 133(Issue 6) pp:1738-1741
Publication Date(Web):January 19, 2011
DOI:10.1021/ja111163u
The first highly enantioselective α-fluorination of ketones using organocatalysis has been accomplished. The long-standing problem of enantioselective ketone α-fluorination via enamine activation has been overcome via high-throughput evaluation of a new library of amine catalysts. The optimal system, a primary amine functionalized Cinchona alkaloid, allows the direct and asymmetric α-fluorination of a variety of carbo- and heterocyclic substrates. Furthermore, this protocol also provides diastereo-, regio-, and chemoselective catalyst control in fluorinations involving complex carbonyl systems.
Co-reporter:James S. Harvey ; Scott P. Simonovich ; Christopher R. Jamison
Journal of the American Chemical Society 2011 Volume 133(Issue 35) pp:13782-13785
Publication Date(Web):August 17, 2011
DOI:10.1021/ja206050b
The enantioselective α-arylation of both lactones and acyl oxazolidones has been accomplished using a combination of diaryliodonium salts and copper catalysis. These mild catalytic conditions provide a new strategy for the enantioselective construction and retention of enolizable α-carbonyl benzylic stereocenters, a valuable synthon for the production of medicinal agents.
Co-reporter:Phong V. Pham, Kate Ashton and David W. C. MacMillan
Chemical Science 2011 vol. 2(Issue 8) pp:1470-1473
Publication Date(Web):19 May 2011
DOI:10.1039/C1SC00176K
The intramolecular asymmetric cyclization of aldehydes has been accomplished using singly occupied molecular orbital (SOMO) catalysis. Selective oxidation of chiral enamines (formed by the condensation of an aldehyde and a secondary amine catalyst) leads to the formation of a 3π-electron radical species. These chiral SOMO-activated radical cations undergo enantioselective cyclization with an array of pendent allylsilanes thus efficiently providing a new approach to the construction of five-, six- and seven-membered carbocycles and heterocycles.
Co-reporter:Robert R. Knowles, Joseph Carpenter, Simon B. Blakey, Akio Kayano, Ian K. Mangion, Christopher J. Sinz and David W. C. MacMillan
Chemical Science 2011 vol. 2(Issue 2) pp:308-311
Publication Date(Web):24 Dec 2010
DOI:10.1039/C0SC00577K
A total synthesis of the marine natural product diazonamide A (1) has been accomplished. This work features a highly stereoselective synthesis of the C(10) quaternary center and the central furanoindoline core enabled by an iminium-catalyzed alkylation–cyclization cascade. Additionally, a magnesium-mediated intramolecular macroaldolization and a palladium-catalyzed tandem borylation/annulation were developed to enable the closure of the two 12-membered macrocycles of diazonamide A. This synthesis involves 20 steps in its longest linear sequence and proceeds in 1.8% overall yield.
Co-reporter:Phong V. Pham;David A. Nagib ; David W. C. MacMillan
Angewandte Chemie 2011 Volume 123( Issue 27) pp:6243-6246
Publication Date(Web):
DOI:10.1002/ange.201101861
Co-reporter:Phong V. Pham;David A. Nagib ; David W. C. MacMillan
Angewandte Chemie International Edition 2011 Volume 50( Issue 27) pp:6119-6122
Publication Date(Web):
DOI:10.1002/anie.201101861
Co-reporter:Andrew McNally;Christopher K. Prier
Science 2011 Volume 334(Issue 6059) pp:1114-1117
Publication Date(Web):25 Nov 2011
DOI:10.1126/science.1213920
An unanticipated photocatalytic carbon-carbon bond-forming reaction emerged from screening many unusual reagent combinations.
Co-reporter:Sebastian Rendler
Journal of the American Chemical Society 2010 Volume 132(Issue 14) pp:5027-5029
Publication Date(Web):March 24, 2010
DOI:10.1021/ja100185p
The first organocatalytic enantioselective radical polycyclization has been accomplished using singly occupied molecular orbital (SOMO) catalysis. The presented strategy relies on a selective single-electron oxidation of chiral enamines formed by condensation of polyenals with an imidazolidinone catalyst employing a suitable copper(II) oxidant. The reaction proceeds under mildly acidic conditions at room temperature and shows compatibility with an array of electron-poor as well as electron-rich functional groups. Upon termination by radical arylation followed by subsequent oxidation and rearomatization, a range of polycyclic aldehydes were accessed (12 examples, 54−77% yield, 85−93% ee). The enantioselective formation of up to six new carbocycles in a single catalyst-controlled cascade is described. Evidence for a radical-based cascade mechanism is indicated by a series of experimental results.
Co-reporter:Anna E. Allen
Journal of the American Chemical Society 2010 Volume 132(Issue 14) pp:4986-4987
Publication Date(Web):March 18, 2010
DOI:10.1021/ja100748y
An enantioselective organocatalytic α-trifluoromethylation of aldehydes has been accomplished using a commercially available, electrophilic trifluoromethyl source. The merging of Lewis acid and organocatalysis provides a new strategy for the enantioselective construction of trifluoromethyl stereogenicity, an important chiral synthon for pharmaceutical, materials, and agrochemical applications. This mild and operationally simple protocol allows rapid access to enantioenriched α-trifluoromethylated aldehydes through a nonphotolytic pathway.
Co-reporter:Joann M. Um ; Osvaldo Gutierrez ; Franziska Schoenebeck ; K. N. Houk
Journal of the American Chemical Society 2010 Volume 132(Issue 17) pp:6001-6005
Publication Date(Web):April 13, 2010
DOI:10.1021/ja9063074
The intramolecular α-arylation of aldehydes via organo-SOMO catalysis was investigated using density functional theory (B3LYP and M06-2X functionals). The geometries, spin densities, Mulliken charges, and molecular orbitals of the reacting enamine radical cations were analyzed, and the nature of the resulting cyclized radical cation intermediates was characterized. In agreement with experimental observations, the calculated 1,3-disubstituted aromatic system shows ortho selectivity, while the 1,3,4-trisubstituted systems show para, meta (instead of ortho, meta) selectivity. The selectivity change for the trisubstituted rings is attributed to a distortion of the ortho substituents in the ortho, meta cyclization transition structures that causes a destabilization of these isomers and therefore results in selectivity for the para, meta product.
Co-reporter:Nathan T. Jui ; Esther C. Y. Lee
Journal of the American Chemical Society 2010 Volume 132(Issue 29) pp:10015-10017
Publication Date(Web):July 1, 2010
DOI:10.1021/ja104313x
A highly selective, radical-mediated (4 + 2) coupling reaction of aldehydes and conjugated olefins has been achieved through asymmetric SOMO-catalysis. A radical-polar crossover mechanism is proposed wherein olefin addition to a transient enamine radical cation and oxidation of the resulting radical furnishes a cation which is vulnerable to nucleophilic addition. A range of aromatic aldehydes are shown to couple with styrenes and dienes to provide cyclic products with high chemical efficiency, regioselectivity, and stereoselectivity.
Co-reporter:Jeffrey F. Van Humbeck ; Scott P. Simonovich ; Robert R. Knowles
Journal of the American Chemical Society 2010 Volume 132(Issue 29) pp:10012-10014
Publication Date(Web):July 7, 2010
DOI:10.1021/ja1043006
The mechanism of a recently reported aldehyde α-oxyamination reaction has been studied using a combination of kinetic, spectrometric, and spectrophotometric techniques. Most crucially, the use of a validated cyclopropane-based radical-clock substrate has demonstrated that carbon−oxygen bond formation occurs predominantly through an enamine activation manifold. The mechanistic details reported herein indicate that, as has been proposed for previously studied alcohol oxidations, complexation between TEMPO and a simple metal salt leads to electrophilic ionic reactivity.
Co-reporter:Hui-Wen Shih ; Mark N. Vander Wal ; Rebecca L. Grange
Journal of the American Chemical Society 2010 Volume 132(Issue 39) pp:13600-13603
Publication Date(Web):September 10, 2010
DOI:10.1021/ja106593m
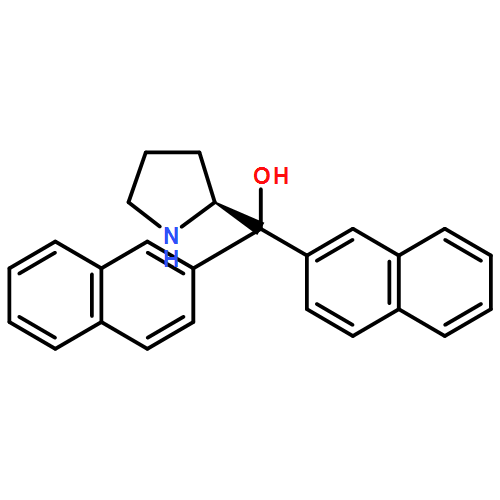
The first enantioselective aldehyde α-benzylation using electron-deficient aryl and heteroaryl substrates has been accomplished. The productive merger of a chiral imidazolidinone organocatalyst and a commercially available iridium photoredox catalyst in the presence of household fluorescent light directly affords the desired homobenzylic stereogenicity in good to excellent yield and enantioselectivity. The utility of this methodology has been demonstrated via rapid access to an enantioenriched drug target for angiogenesis suppression.
Co-reporter:Maud Reiter, Staffan Torssell, Sandra Lee and David W. C. MacMillan
Chemical Science 2010 vol. 1(Issue 1) pp:37-42
Publication Date(Web):21 May 2010
DOI:10.1039/C0SC00204F
The frondosins are a family of marine sesquiterpenes isolated from the sponge Dysidea frondosa that exhibit biological activities ranging from anti-inflammatory properties to potential application in anticancer and HIV therapy. Herein, a concise enantioselective total synthesis of (+)-frondosin B is described which requires a total of three chemical steps. The enantioselective conjugate addition of a benzofuran-derived boronic acid to crotonaldehyde in the presence of an imidazolidinone organocatalyst builds the critical stereogenic center of frondosin B in the first operation, while the remaining two ring systems of this natural product are installed in the two subsequent steps. A combination of X-ray crystallographic data, deuterium labeling, and chemical correlation studies provides further evidence as to the correct absolute stereochemical assignment of (+)-frondosin B.
Co-reporter:Eric N. Jacobsen
PNAS 2010 Volume 107 (Issue 48 ) pp:20618-20619
Publication Date(Web):2010-11-30
DOI:10.1073/pnas.1016087107
The use of small-molecule organic catalysts in organic synthesis has flourished over the past decade. Examples of defining
concepts and cutting-edge results are provided in the papers in this Special Feature.
Co-reporter:Anthony Mastracchio;Abbas M. Walji;Alexander A. Warkentin
PNAS 2010 Volume 107 (Issue 48 ) pp:20648-20651
Publication Date(Web):2010-11-30
DOI:10.1073/pnas.1002845107
The first enantioselective organocatalytic α-allylation of cyclic ketones has been accomplished via singly occupied molecular
orbital catalysis. Geometrically constrained radical cations, forged from the one-electron oxidation of transiently generated
enamines, readily undergo allylic alkylation with a variety of commercially available allyl silanes. A reasonable latitude
in both the ketone and allyl silane components is readily accommodated in this new transformation. Moreover, three new oxidatively
stable imidazolidinone catalysts have been developed that allow cyclic ketones to successfully participate in this transformation.
The new catalyst platform has also been exploited in the first catalytic enantioselective α-enolation and α-carbooxidation
of ketones.
Co-reporter:Spencer B. Jones ; Bryon Simmons
Journal of the American Chemical Society 2009 Volume 131(Issue 38) pp:13606-13607
Publication Date(Web):September 2, 2009
DOI:10.1021/ja906472m
An enantioselective total synthesis of the Strychnos alkaloid (+)-minfiensine has been accomplished. Prominent features of this synthesis include (i) a new enantioselective organocatalytic Diels−Alder/amine cyclization sequence to build the central tetracyclic pyrroloindoline framework in four steps from commercial materials and (ii) a 6-exo-dig radical cyclization to forge the final piperidinyl ring system. This total synthesis of (+)-minfiensine was completed in nine chemical steps and 21% overall yield.
Co-reporter:David A. Nagib ; Mark E. Scott
Journal of the American Chemical Society 2009 Volume 131(Issue 31) pp:10875-10877
Publication Date(Web):July 20, 2009
DOI:10.1021/ja9053338
The first enantioselective, organocatalytic α-trifluoromethylation and α-perfluoroalkylation of aldehydes have been accomplished using a readily available iridium photocatalyst and a chiral imidazolidinone catalyst. A range of α-trifluoromethyl and α-perfluoroalkyl aldehydes were obtained from commercially available perfluoroalkyl halides with high efficiency and enantioselectivity. The resulting α-trifluoromethyl aldehydes were subsequently shown to be versatile precursors for the construction of a variety of enantioenriched trifluoromethylated building blocks.
Co-reporter:Jonathan E. Wilson ; Anthony D. Casarez
Journal of the American Chemical Society 2009 Volume 131(Issue 32) pp:11332-11334
Publication Date(Web):July 24, 2009
DOI:10.1021/ja904504j
The first enantioselective organocatalytic α-nitroalkylation of aldehydes has been accomplished. The aforementioned process involves the oxidative coupling of an enamine intermediate, generated transiently via condensation of an amine catalyst with an aldehyde, with a silyl nitronate to produce a β-nitroaldehyde. Two methods, one that furnishes the syn β-nitroaldehyde and a second that provides access to the anti isomer, have been developed. Data are presented to support a hypothesis that explains this phenomenon in terms of a silyl group-controlled change in mechanism. Finally, a three-step procedure for the synthesis of both syn- and anti-α,β-disubstituted β-amino acids is presented.
Co-reporter:Jay C. Conrad ; Jongrock Kong ; Brian N. Laforteza
Journal of the American Chemical Society 2009 Volume 131(Issue 33) pp:11640-11641
Publication Date(Web):July 29, 2009
DOI:10.1021/ja9026902
The intramolecular α-arylation of aldehydes has been accomplished using singly occupied molecular orbital (SOMO) catalysis. Selective oxidation of chiral enamines (formed by the condensation of an aldehyde and a secondary amine catalyst) leads to the formation of a 3π-electron radical species. These chiral SOMO-activated radical cations undergo enantioselective reaction with an array of pendent electron-rich aromatics and heterocycles thus efficiently providing cyclic α-aryl aldehyde products (10 examples: ≥70% yield and ≥90% ee). In accordance with our radical mechanism, when there is a choice between arylation at the ortho or para position of anisole substrates, we find that arylation proceeds selectively at the ortho position.
Co-reporter:Bryon Simmons;AbbasM. Walji Dr. ;DavidW.C. MacMillan
Angewandte Chemie International Edition 2009 Volume 48( Issue 24) pp:4349-4353
Publication Date(Web):
DOI:10.1002/anie.200900220
Co-reporter:Muriel Amatore Dr.;TeresaD. Beeson;SeanP. Brown ;DavidW.C. MacMillan
Angewandte Chemie International Edition 2009 Volume 48( Issue 28) pp:5121-5124
Publication Date(Web):
DOI:10.1002/anie.200901855
Co-reporter:Bryon Simmons;AbbasM. Walji Dr. ;DavidW.C. MacMillan
Angewandte Chemie 2009 Volume 121( Issue 24) pp:4413-4417
Publication Date(Web):
DOI:10.1002/ange.200900220
Co-reporter:Muriel Amatore Dr.;TeresaD. Beeson;SeanP. Brown ;DavidW.C. MacMillan
Angewandte Chemie 2009 Volume 121( Issue 28) pp:5223-5226
Publication Date(Web):
DOI:10.1002/ange.200901855
Co-reporter:Christopher J. Borths, Diane E. Carrera, David W.C. MacMillan
Tetrahedron 2009 65(33) pp: 6746-6753
Publication Date(Web):
DOI:10.1016/j.tet.2009.06.066
Co-reporter:Nick A. Paras, Bryon Simmons, David W.C. MacMillan
Tetrahedron 2009 65(16) pp: 3232-3238
Publication Date(Web):
DOI:10.1016/j.tet.2008.12.054
Co-reporter:Joseph Carpenter;AlanB. Northrup;deMichael Chung Dr.;JohnJ.M. Wiener;Sung-Gon Kim Dr.;DavidW.C. MacMillan
Angewandte Chemie 2008 Volume 120( Issue 19) pp:3624-3628
Publication Date(Web):
DOI:10.1002/ange.200800086
Co-reporter:Joseph Carpenter;AlanB. Northrup;deMichael Chung Dr.;JohnJ.M. Wiener;Sung-Gon Kim Dr.;DavidW.C. MacMillan
Angewandte Chemie International Edition 2008 Volume 47( Issue 19) pp:3568-3572
Publication Date(Web):
DOI:10.1002/anie.200800086
Co-reporter:David W. C. MacMillan;David A. Nicewicz
Science 2008 Volume 322(Issue 5898) pp:77-80
Publication Date(Web):03 Oct 2008
DOI:10.1126/science.1161976
Abstract
Photoredox catalysis and organocatalysis represent two powerful fields of molecule activation that have found widespread application in the areas of inorganic and organic chemistry, respectively. We merged these two catalysis fields to solve problems in asymmetric chemical synthesis. Specifically, the enantioselective intermolecular α-alkylation of aldehydes has been accomplished using an interwoven activation pathway that combines both the photoredox catalyst Ru(bpy)3Cl2 (where bpy is 2,2′-bipyridine) and an imidazolidinone organocatalyst. This broadly applicable, yet previously elusive, alkylation reaction is now highly enantioselective and operationally trivial.
Co-reporter:Teresa D. Beeson;Anthony Mastracchio;Jun-Bae Hong;Kate Ashton
Science 2007 Volume 316(Issue 5824) pp:582-585
Publication Date(Web):27 Apr 2007
DOI:10.1126/science. 1142696
Abstract
The asymmetric α-addition of relatively nonpolar hydrocarbon substrates, such as allyl and aryl groups, to aldehydes and ketones remains a largely unsolved problem in organic synthesis, despite the wide potential utility of direct routes to such products. We reasoned that well-established chiral amine catalysis, which activates aldehydes toward electrophile addition by enamine formation, could be expanded to this important reaction class by applying a single-electron oxidant to create a transient radical species from the enamine. We demonstrated the concept of singly occupied molecular orbital (SOMO) activation with a highly selective α-allylation of aldehydes, and we here present preliminary results for enantioselective heteroarylations and cyclization/halogenation cascades.
Co-reporter:Anna E. Allen
Journal of the American Chemical Society () pp:
Publication Date(Web):March 9, 2011
DOI:10.1021/ja2008906
The enantioselective α-arylation of aldehydes has been accomplished using diaryliodonium salts and a combination of copper and organic catalysts. These mild catalytic conditions provide a new strategy for the enantioselective construction and retention of enolizable α-formyl benzylic stereocenters, a valuable synthon for the production of medicinal agents. As one example, this new asymmetric protocol has been applied to the rapid synthesis of (S)-ketoprofen, a commercially successful oral and topical analgesic.
Co-reporter:Anna E. Allen and David W. C. MacMillan
Chemical Science (2010-Present) 2012 - vol. 3(Issue 3) pp:NaN658-658
Publication Date(Web):2012/01/25
DOI:10.1039/C2SC00907B
Synergistic catalysis is a synthetic strategy wherein both the nucleophile and the electrophile are simultaneously activated by two separate and distinct catalysts to afford a single chemical transformation. This powerful catalysis strategy leads to several benefits, specifically synergistic catalysis can (i) introduce new, previously unattainable chemical transformations, (ii) improve the efficiency of existing transformations, and (iii) create or improve catalytic enantioselectivity where stereocontrol was previously absent or challenging. This perspective aims to highlight these benefits using many of the successful examples of synergistic catalysis found in the literature.
Co-reporter:Mark N. Vander Wal, Andrew K. Dilger and David W. C. MacMillan
Chemical Science (2010-Present) 2013 - vol. 4(Issue 8) pp:NaN3079-3079
Publication Date(Web):2013/06/11
DOI:10.1039/C3SC51266E
Oxy-allyl cations have been known as transient electrophilic species since they were first proposed as intermediates in the Favorskii rearrangement in 1894. Since that time, they also have been used as a mode of activation for [4 + 3] cycloadditions in a variety of natural product syntheses. In this manuscript, we describe a method for the interception of oxy-allyl cations with a diverse range of common nucleophiles, thereby demonstrating the value of this intermediate as a generic mode of activation. This simple, mild, room temperature protocol allows for the formation of a variety of high value carbon–carbon and carbon–heteroatom bonds that are readily incorporated within a series of cyclic and acyclic ketone systems. Initial efforts into the development of an enantioselective catalytic variant are also described.
Co-reporter:Christopher K. Prier and David W. C. MacMillan
Chemical Science (2010-Present) 2014 - vol. 5(Issue 11) pp:NaN4178-4178
Publication Date(Web):2014/08/04
DOI:10.1039/C4SC02155J
The direct α-heteroarylation of tertiary amines has been accomplished via photoredox catalysis to generate valuable benzylic amine pharmacophores. A variety of five- and six-membered chloroheteroarenes are shown to function as viable coupling partners for the α-arylation of a diverse range of cyclic and acyclic amines. Evidence is provided for a homolytic aromatic substitution mechanism, in which a catalytically-generated α-amino radical undergoes direct addition to an electrophilic chloroarene.
Co-reporter:Phong V. Pham, Kate Ashton and David W. C. MacMillan
Chemical Science (2010-Present) 2011 - vol. 2(Issue 8) pp:NaN1473-1473
Publication Date(Web):2011/05/19
DOI:10.1039/C1SC00176K
The intramolecular asymmetric cyclization of aldehydes has been accomplished using singly occupied molecular orbital (SOMO) catalysis. Selective oxidation of chiral enamines (formed by the condensation of an aldehyde and a secondary amine catalyst) leads to the formation of a 3π-electron radical species. These chiral SOMO-activated radical cations undergo enantioselective cyclization with an array of pendent allylsilanes thus efficiently providing a new approach to the construction of five-, six- and seven-membered carbocycles and heterocycles.
Co-reporter:Scott P. Simonovich, Jeffrey F. Van Humbeck and David W. C. MacMillan
Chemical Science (2010-Present) 2012 - vol. 3(Issue 1) pp:NaN61-61
Publication Date(Web):2011/09/22
DOI:10.1039/C1SC00556A
A new enantioselective α-oxidation of aldehydes has been accomplished using TEMPO and a synergistic combination of copper and organic catalysis. Expanding upon recently reported mechanistic studies, these mild catalytic conditions provide stable aldehyde products bearing a wide array of electronically and sterically diverse substructures. The utility of these oxidized products is highlighted by subsequent derivatization to a variety of common chiral synthons, without loss in enantiopurity.
Co-reporter:Robert R. Knowles, Joseph Carpenter, Simon B. Blakey, Akio Kayano, Ian K. Mangion, Christopher J. Sinz and David W. C. MacMillan
Chemical Science (2010-Present) 2011 - vol. 2(Issue 2) pp:NaN311-311
Publication Date(Web):2010/12/24
DOI:10.1039/C0SC00577K
A total synthesis of the marine natural product diazonamide A (1) has been accomplished. This work features a highly stereoselective synthesis of the C(10) quaternary center and the central furanoindoline core enabled by an iminium-catalyzed alkylation–cyclization cascade. Additionally, a magnesium-mediated intramolecular macroaldolization and a palladium-catalyzed tandem borylation/annulation were developed to enable the closure of the two 12-membered macrocycles of diazonamide A. This synthesis involves 20 steps in its longest linear sequence and proceeds in 1.8% overall yield.
Co-reporter:Maud Reiter, Staffan Torssell, Sandra Lee and David W. C. MacMillan
Chemical Science (2010-Present) 2010 - vol. 1(Issue 1) pp:NaN42-42
Publication Date(Web):2010/05/21
DOI:10.1039/C0SC00204F
The frondosins are a family of marine sesquiterpenes isolated from the sponge Dysidea frondosa that exhibit biological activities ranging from anti-inflammatory properties to potential application in anticancer and HIV therapy. Herein, a concise enantioselective total synthesis of (+)-frondosin B is described which requires a total of three chemical steps. The enantioselective conjugate addition of a benzofuran-derived boronic acid to crotonaldehyde in the presence of an imidazolidinone organocatalyst builds the critical stereogenic center of frondosin B in the first operation, while the remaining two ring systems of this natural product are installed in the two subsequent steps. A combination of X-ray crystallographic data, deuterium labeling, and chemical correlation studies provides further evidence as to the correct absolute stereochemical assignment of (+)-frondosin B.
.jpg)




























![SILANE, TRIS(1-METHYLETHYL)[(2-METHYL-1H-INDEN-3-YL)OXY]-](http://img.cochemist.com/ccimg/620700/620606-68-0.png)
![SILANE, TRIS(1-METHYLETHYL)[(2-METHYL-1H-INDEN-3-YL)OXY]-](http://img.cochemist.com/ccimg/620700/620606-68-0_b.png)



![tert-Butyl 2-chloro-1H-benzo[d]imidazole-1-carboxylate](http://img.cochemist.com/ccimg/214200/214147-60-1.png)
![tert-Butyl 2-chloro-1H-benzo[d]imidazole-1-carboxylate](http://img.cochemist.com/ccimg/214200/214147-60-1_b.png)














![Thieno[3,2-c]pyridine-5(4H)-aceticacid, a-(2-chlorophenyl)-6,7-dihydro-,methyl ester](http://img.cochemist.com/ccimg/90100/90055-48-4.png)
![Thieno[3,2-c]pyridine-5(4H)-aceticacid, a-(2-chlorophenyl)-6,7-dihydro-,methyl ester](http://img.cochemist.com/ccimg/90100/90055-48-4_b.png)
![3-PENTANONE, 2-[[(4-METHYLPHENYL)SULFONYL]OXY]-](http://img.cochemist.com/ccimg/81500/81447-33-8.png)
![3-PENTANONE, 2-[[(4-METHYLPHENYL)SULFONYL]OXY]-](http://img.cochemist.com/ccimg/81500/81447-33-8_b.png)










![CYCLOPENTADECANONE, 2-[[(4-METHYLPHENYL)SULFONYL]OXY]-](http://img.cochemist.com/ccimg/17400/17344-56-8.png)
![CYCLOPENTADECANONE, 2-[[(4-METHYLPHENYL)SULFONYL]OXY]-](http://img.cochemist.com/ccimg/17400/17344-56-8_b.png)





![6H,13aH-3a,5a-Ethano-1H-indolizino[8,1-cd]carbazole-5-carboxylicacid, 2,3,4,5,11,12-hexahydro-, methyl ester, (3aR,5R,5aR,10bR,13aS)-](http://img.cochemist.com/ccimg/600/559-51-3.png)
![6H,13aH-3a,5a-Ethano-1H-indolizino[8,1-cd]carbazole-5-carboxylicacid, 2,3,4,5,11,12-hexahydro-, methyl ester, (3aR,5R,5aR,10bR,13aS)-](http://img.cochemist.com/ccimg/600/559-51-3_b.png)






![Tricyclo[3.3.1.13,7]decane-1-acetaldehyde](http://img.cochemist.com/ccimg/18300/18220-83-2.png)
![Tricyclo[3.3.1.13,7]decane-1-acetaldehyde](http://img.cochemist.com/ccimg/18300/18220-83-2_b.png)









![Acetic acid, [[(1,1-dimethylethyl)dimethylsilyl]oxy]-, 1-methylethyl ester](http://img.cochemist.com/ccimg/150900/150811-52-2.png)
![Acetic acid, [[(1,1-dimethylethyl)dimethylsilyl]oxy]-, 1-methylethyl ester](http://img.cochemist.com/ccimg/150900/150811-52-2_b.png)


![Benzene, [(2E)-3-iodo-2-propenyl]-](http://img.cochemist.com/ccimg/141700/141694-63-5.png)
![Benzene, [(2E)-3-iodo-2-propenyl]-](http://img.cochemist.com/ccimg/141700/141694-63-5_b.png)
![Cyclopentanecarboxylicacid, 2-[[(1,1-dimethylethoxy)carbonyl]amino]-, (1R,2S)-rel-](http://img.cochemist.com/ccimg/136400/136315-70-3.png)
![Cyclopentanecarboxylicacid, 2-[[(1,1-dimethylethoxy)carbonyl]amino]-, (1R,2S)-rel-](http://img.cochemist.com/ccimg/136400/136315-70-3_b.png)







![Cyclopentanone, 2-[[(4-methylphenyl)sulfonyl]oxy]-](http://img.cochemist.com/ccimg/125800/125774-14-3.png)
![Cyclopentanone, 2-[[(4-methylphenyl)sulfonyl]oxy]-](http://img.cochemist.com/ccimg/125800/125774-14-3_b.png)




![Acetaldehyde, [(4-methoxyphenyl)methoxy]-](http://img.cochemist.com/ccimg/121300/121289-23-4.png)
![Acetaldehyde, [(4-methoxyphenyl)methoxy]-](http://img.cochemist.com/ccimg/121300/121289-23-4_b.png)
![Ethanone, 1-[4-(1-phenylethoxy)phenyl]-](http://img.cochemist.com/ccimg/117400/117321-40-1.png)
![Ethanone, 1-[4-(1-phenylethoxy)phenyl]-](http://img.cochemist.com/ccimg/117400/117321-40-1_b.png)

![Morpholine, 4-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/113900/113845-69-5.png)
![Morpholine, 4-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/113900/113845-69-5_b.png)



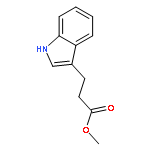
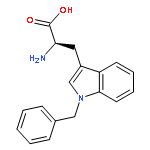
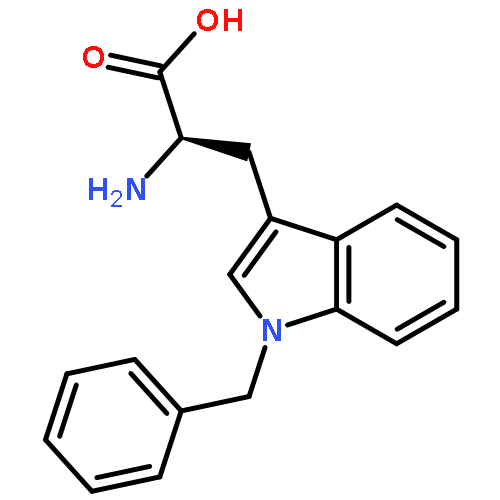
![L-Tryptophan, N-[(1,1-dimethylethoxy)carbonyl]-1-methyl-](http://img.cochemist.com/ccimg/110000/109927-44-8.png)
![L-Tryptophan, N-[(1,1-dimethylethoxy)carbonyl]-1-methyl-](http://img.cochemist.com/ccimg/110000/109927-44-8_b.png)
![Benzenesulfonamide,4-methyl-N-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/107500/107491-54-3.png)
![Benzenesulfonamide,4-methyl-N-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/107500/107491-54-3_b.png)

![2-Chloro-1H-imidazo[4,5-b]pyridine](http://img.cochemist.com/ccimg/104700/104685-82-7.png)
![2-Chloro-1H-imidazo[4,5-b]pyridine](http://img.cochemist.com/ccimg/104700/104685-82-7_b.png)
![Silane, (1,1-dimethylethyl)[(4-methoxyphenyl)methoxy]dimethyl-](http://img.cochemist.com/ccimg/101900/101803-60-5.png)
![Silane, (1,1-dimethylethyl)[(4-methoxyphenyl)methoxy]dimethyl-](http://img.cochemist.com/ccimg/101900/101803-60-5_b.png)
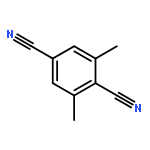
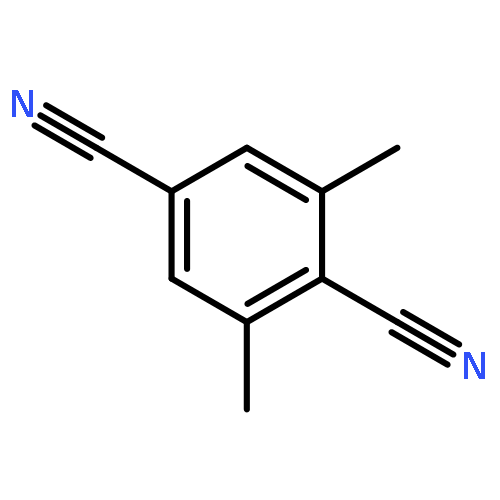


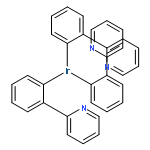
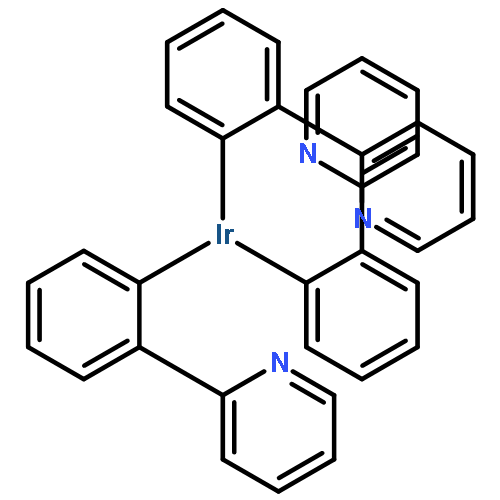


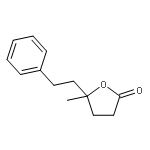
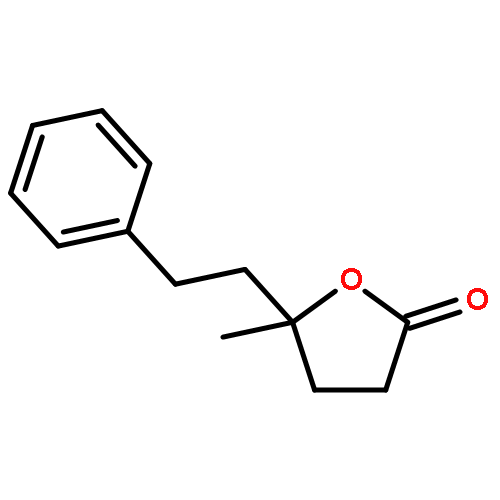
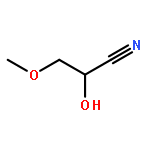





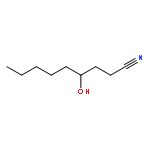


![Benzene, [[(2-methoxyethoxy)methoxy]methyl]-](http://img.cochemist.com/ccimg/88800/88738-41-4.png)
![Benzene, [[(2-methoxyethoxy)methoxy]methyl]-](http://img.cochemist.com/ccimg/88800/88738-41-4_b.png)
![Benzene, 1-methoxy-4-[(1E)-2-(phenylsulfonyl)ethenyl]-](http://img.cochemist.com/ccimg/87900/87891-80-3.png)
![Benzene, 1-methoxy-4-[(1E)-2-(phenylsulfonyl)ethenyl]-](http://img.cochemist.com/ccimg/87900/87891-80-3_b.png)

![1H-Pyrrolo[1,2-a]indole, 2,3-dihydro-9-(phenylmethyl)-](http://img.cochemist.com/ccimg/87700/87699-02-3.png)
![1H-Pyrrolo[1,2-a]indole, 2,3-dihydro-9-(phenylmethyl)-](http://img.cochemist.com/ccimg/87700/87699-02-3_b.png)
![Benzamide, N-[2-[(trimethylsilyl)oxy]-4-pyrimidinyl]-](http://img.cochemist.com/ccimg/85800/85743-99-3.png)
![Benzamide, N-[2-[(trimethylsilyl)oxy]-4-pyrimidinyl]-](http://img.cochemist.com/ccimg/85800/85743-99-3_b.png)
![Isoquinoline,5-[(2-methyl-1-piperazinyl)sulfonyl]-](http://img.cochemist.com/ccimg/84500/84477-87-2.png)
![Isoquinoline,5-[(2-methyl-1-piperazinyl)sulfonyl]-](http://img.cochemist.com/ccimg/84500/84477-87-2_b.png)







![8-bromo-5,6-dihydro-5-methyl-6-oxo-4H-Imidazo[1,5-a][1,4]benzodiazepine-3-carboxylic acid ethyl ester](http://img.cochemist.com/ccimg/78800/78756-37-3.png)
![8-bromo-5,6-dihydro-5-methyl-6-oxo-4H-Imidazo[1,5-a][1,4]benzodiazepine-3-carboxylic acid ethyl ester](http://img.cochemist.com/ccimg/78800/78756-37-3_b.png)





![Benzene, [(2-phenyl-1-propenyl)sulfonyl]-, (E)-](/data/chemimg/1287200/64329-88-0.png)
![Benzene, [(2-phenyl-1-propenyl)sulfonyl]-, (E)-](/data/chemimg/1287200/64329-88-0_b.png)







![[1,1'-Biphenyl]-4-aceticacid, methyl ester](http://img.cochemist.com/ccimg/59800/59793-29-2.png)
![[1,1'-Biphenyl]-4-aceticacid, methyl ester](http://img.cochemist.com/ccimg/59800/59793-29-2_b.png)



![1-[2-(BENZENESULFONYL)ETHENYL]NAPHTHALENE](http://img.cochemist.com/ccimg/56400/56361-22-9.png)
![1-[2-(BENZENESULFONYL)ETHENYL]NAPHTHALENE](http://img.cochemist.com/ccimg/56400/56361-22-9_b.png)










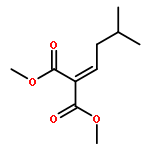
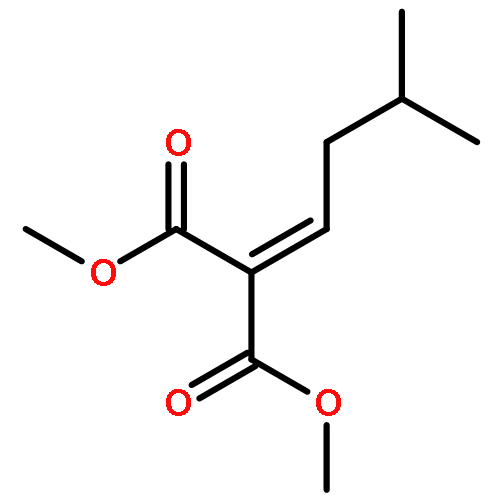


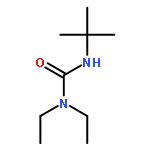
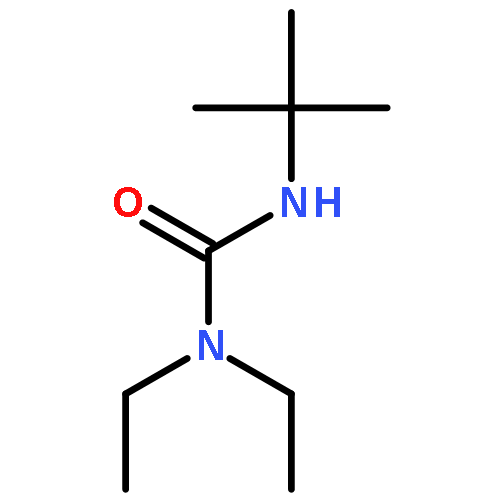



![Benzene, [(1-phenylethenyl)sulfonyl]-](http://img.cochemist.com/ccimg/49900/49833-39-8.png)
![Benzene, [(1-phenylethenyl)sulfonyl]-](http://img.cochemist.com/ccimg/49900/49833-39-8_b.png)
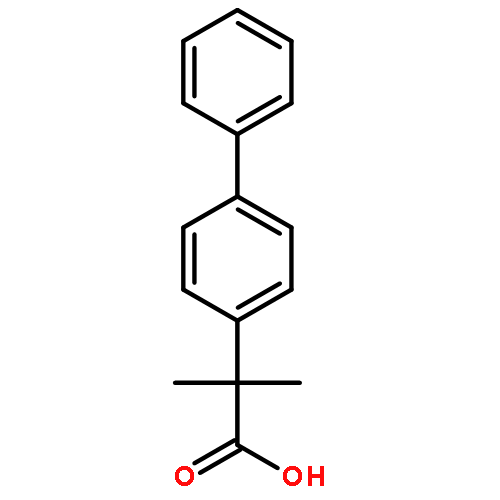
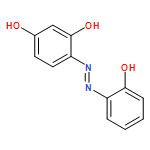
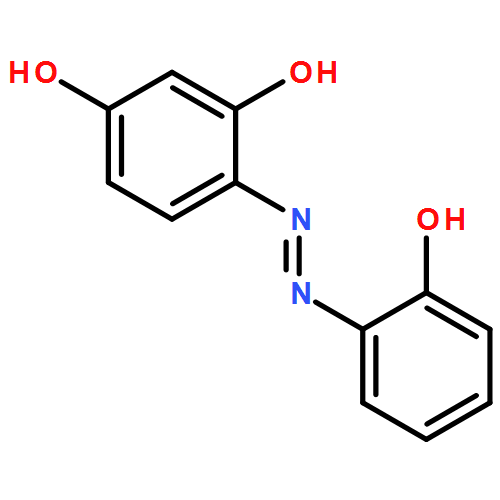
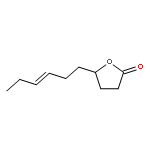
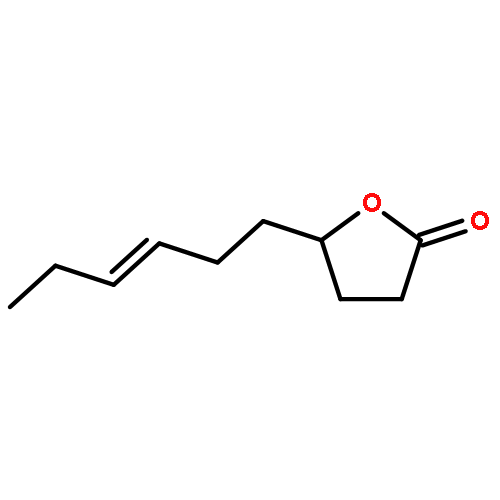
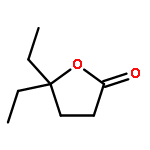
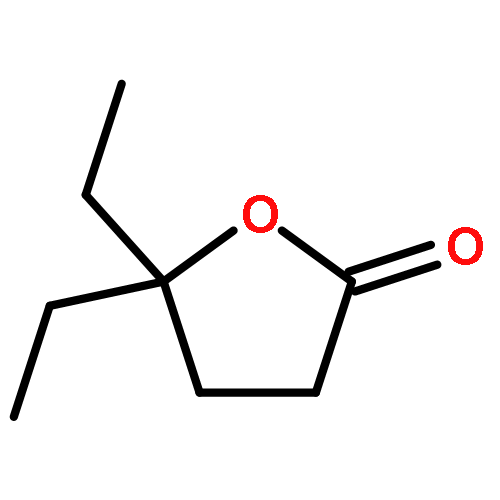
![1-OXASPIRO[4.6]UNDECAN-2-ONE](http://img.cochemist.com/ccimg/41800/41732-91-6.png)
![1-OXASPIRO[4.6]UNDECAN-2-ONE](http://img.cochemist.com/ccimg/41800/41732-91-6_b.png)
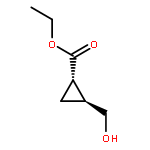

![Benzoic acid, 4-tricyclo[3.3.1.13,7]dec-1-yl-, methyl ester](http://img.cochemist.com/ccimg/38200/38197-55-6.png)
![Benzoic acid, 4-tricyclo[3.3.1.13,7]dec-1-yl-, methyl ester](http://img.cochemist.com/ccimg/38200/38197-55-6_b.png)






![1-Oxaspiro[4.4]nonan-2-one](http://img.cochemist.com/ccimg/33500/33448-80-5.png)
![1-Oxaspiro[4.4]nonan-2-one](http://img.cochemist.com/ccimg/33500/33448-80-5_b.png)









![(5s,8r,9s,10s,13s,14s,17r)-10,13-dimethylspiro[2,4,5,6,7,8,9,11,12,14,15,16-dodecahydro-1h-cyclopenta[a]phenanthrene-17,5'-oxolane]-2',3-dione](http://img.cochemist.com/ccimg/26400/26302-67-0.png)
![(5s,8r,9s,10s,13s,14s,17r)-10,13-dimethylspiro[2,4,5,6,7,8,9,11,12,14,15,16-dodecahydro-1h-cyclopenta[a]phenanthrene-17,5'-oxolane]-2',3-dione](http://img.cochemist.com/ccimg/26400/26302-67-0_b.png)
![Benzene, 1,1'-[(phenylsulfonyl)ethenylidene]bis-](http://img.cochemist.com/ccimg/26200/26189-62-8.png)
![Benzene, 1,1'-[(phenylsulfonyl)ethenylidene]bis-](http://img.cochemist.com/ccimg/26200/26189-62-8_b.png)












![N-(Trimethylsilyl)-2-[(trimethylsilyl)oxy]pyrimidin-4-amine](http://img.cochemist.com/ccimg/18100/18037-10-0.png)
![N-(Trimethylsilyl)-2-[(trimethylsilyl)oxy]pyrimidin-4-amine](http://img.cochemist.com/ccimg/18100/18037-10-0_b.png)









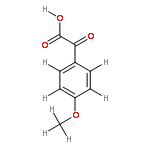
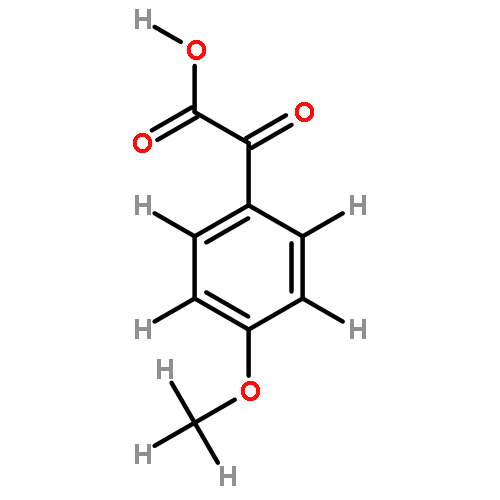




![[1,1'-Biphenyl]-4-aceticacid, a-methyl-](http://img.cochemist.com/ccimg/6400/6341-72-6.png)
![[1,1'-Biphenyl]-4-aceticacid, a-methyl-](http://img.cochemist.com/ccimg/6400/6341-72-6_b.png)
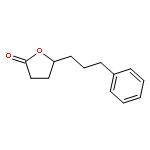
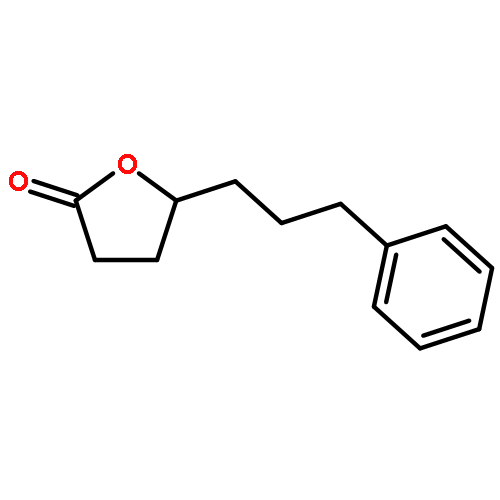


![methyl 5-(diethylcarbamoyl)-4-methyl-2-{[phenyl(phenylsulfanyl)acetyl]amino}thiophene-3-carboxylate](http://img.cochemist.com/ccimg/6200/6125-76-4.png)
![methyl 5-(diethylcarbamoyl)-4-methyl-2-{[phenyl(phenylsulfanyl)acetyl]amino}thiophene-3-carboxylate](http://img.cochemist.com/ccimg/6200/6125-76-4_b.png)

![(2E)-1-(3,4-dimethoxyphenyl)-3-[2-(3,4-dimethoxyphenyl)-4-(2-methylphenyl)-4H-chromen-3-yl]prop-2-en-1-one](http://img.cochemist.com/ccimg/5800/5736-95-8.png)
![(2E)-1-(3,4-dimethoxyphenyl)-3-[2-(3,4-dimethoxyphenyl)-4-(2-methylphenyl)-4H-chromen-3-yl]prop-2-en-1-one](http://img.cochemist.com/ccimg/5800/5736-95-8_b.png)



![[(phenylethynyl)sulfonyl]benzene](http://img.cochemist.com/ccimg/5400/5324-64-1.png)
![[(phenylethynyl)sulfonyl]benzene](http://img.cochemist.com/ccimg/5400/5324-64-1_b.png)
































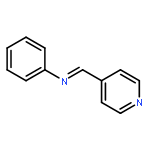
![Benzenamine, N-[(4-methylphenyl)methylene]-, (E)-](http://img.cochemist.com/ccimg/1700/1613-98-5.png)
![Benzenamine, N-[(4-methylphenyl)methylene]-, (E)-](http://img.cochemist.com/ccimg/1700/1613-98-5_b.png)
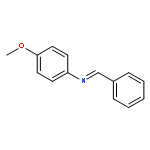
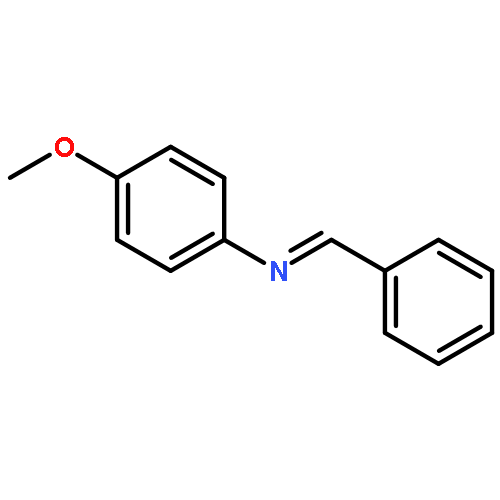
![Benzenamine, N-[(4-methoxyphenyl)methylene]-, (E)-](http://img.cochemist.com/ccimg/1700/1613-96-3.png)
![Benzenamine, N-[(4-methoxyphenyl)methylene]-, (E)-](http://img.cochemist.com/ccimg/1700/1613-96-3_b.png)








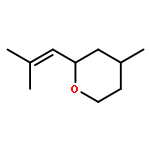
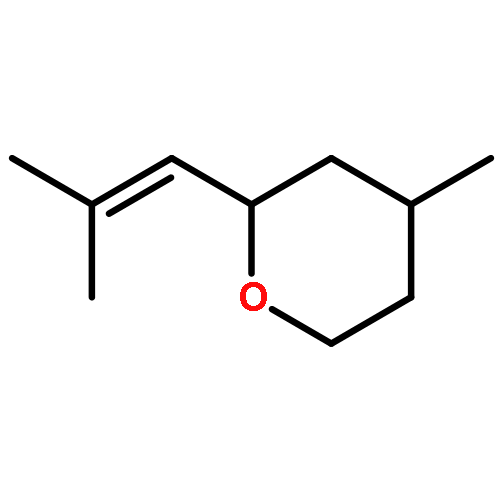
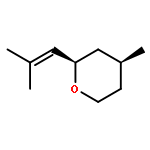
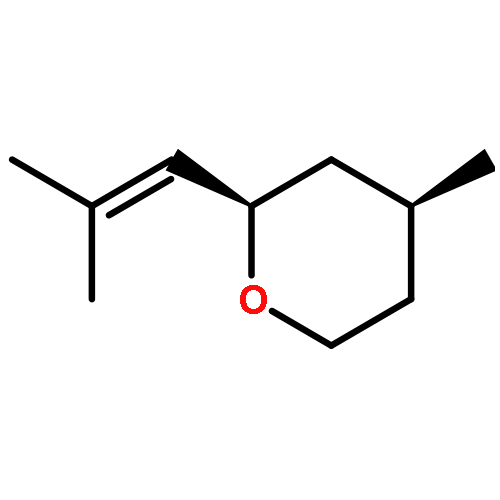






![1-Oxaspiro[4.5]decan-2-one](http://img.cochemist.com/ccimg/700/699-61-6.png)
![1-Oxaspiro[4.5]decan-2-one](http://img.cochemist.com/ccimg/700/699-61-6_b.png)


![Pyridine, 3-[[[(1,1-dimethylethyl)dimethylsilyl]oxy]methyl]-](http://img.cochemist.com/ccimg/401900/401815-75-6.png)
![Pyridine, 3-[[[(1,1-dimethylethyl)dimethylsilyl]oxy]methyl]-](http://img.cochemist.com/ccimg/401900/401815-75-6_b.png)



![Benzene, [[2-(1-methylcyclohexyl)ethyl]sulfonyl]-](http://img.cochemist.com/ccimg/145700/145642-37-1.png)
![Benzene, [[2-(1-methylcyclohexyl)ethyl]sulfonyl]-](http://img.cochemist.com/ccimg/145700/145642-37-1_b.png)