Co-reporter:Toshifumi Takeuchi; Paul T. Schumacker
Journal of the American Chemical Society 2014 Volume 137(Issue 2) pp:564-567
Publication Date(Web):December 3, 2014
DOI:10.1021/ja5101257
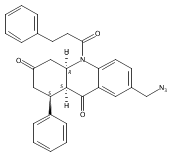
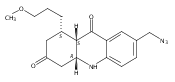
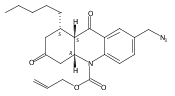
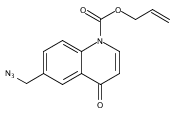
Development of cell-permeable small molecules that target enzymes involved in energy metabolism remains important yet challenging. We describe here the discovery of a new class of compounds with a nutrient-dependent cytotoxicity profile that arises from pharmacological inhibition of fumarate hydratase (also known as fumarase). This finding was enabled by a high-throughput screen of a diverse chemical library in a panel of human cancer cell lines cultured under different growth conditions, followed by subsequent structure–activity optimization and target identification. While the highest cytotoxicity was observed under low glucose concentrations, the antiproliferative activities and inhibition of oxygen consumption rates in cells were distinctly different from those displayed by typical inhibitors of mitochondrial oxidative phosphorylation. The use of a photoaffinity labeling strategy identified fumarate hydratase as the principal pharmacological target. Final biochemical studies confirmed dose-dependent, competitive inhibition of this enzyme in vitro, which was fully consistent with the initially observed growth inhibitory activity. Our work demonstrates how the phenotypic observations combined with a successful target identification strategy can yield a useful class of pharmacological inhibitors of an enzyme involved in the operation of tricarboxylic acid cycle.
Co-reporter:Jaime R. Cabrera-Pardo;David I. Chai
Advanced Synthesis & Catalysis 2013 Volume 355( Issue 13) pp:2495-2498
Publication Date(Web):
DOI:10.1002/adsc.201300443
Co-reporter:Song Liu, John S. Scotti, and Sergey A. Kozmin
The Journal of Organic Chemistry 2013 Volume 78(Issue 17) pp:8645-8654
Publication Date(Web):August 12, 2013
DOI:10.1021/jo401262v
We have developed a synthetic strategy that mimics the diversity-generating power of monoterpenoid indole alkaloid biosynthesis. Our general approach goes beyond diversification of a single natural product-like substructure and enables production of a highly diverse collection of small molecules. The reaction sequence begins with rapid and highly modular assembly of the tetracyclic indoloquinolizidine core, which can be chemoselectively processed into several additional skeletally diverse structural frameworks. The general utility of this approach was demonstrated by parallel synthesis of two representative chemical libraries containing 847 compounds with favorable physicochemical properties to enable its subsequent broad pharmacological evaluation.
Co-reporter:Dr. Timothy J. Montavon;Dr. Yunus E. Türkmen;Noumaan A. Shamsi;Christopher Miller;Chintan S. Sumaria;Dr. Viresh H. Rawal;Dr. Sergey A. Kozmin
Angewandte Chemie International Edition 2013 Volume 52( Issue 51) pp:13576-13579
Publication Date(Web):
DOI:10.1002/anie.201305711
Co-reporter:Yunus E. Türkmen ; Timothy J. Montavon ; Sergey A. Kozmin ;Viresh H. Rawal
Journal of the American Chemical Society 2012 Volume 134(Issue 22) pp:9062-9065
Publication Date(Web):May 19, 2012
DOI:10.1021/ja302537j
A highly effective silver-catalyzed formal inverse electron-demand Diels–Alder reaction of 1,2-diazines and siloxy alkynes has been developed. The reactions provide ready access to a wide range of siloxy naphthalenes and anthracenes, which are formed in good to high yields, under mild reaction conditions, using low catalyst loadings.
Co-reporter:Jiayue Cui, David I. Chai, Christopher Miller, Jason Hao, Christopher Thomas, JingQi Wang, Karl A. Scheidt, and Sergey A. Kozmin
The Journal of Organic Chemistry 2012 Volume 77(Issue 17) pp:7435-7470
Publication Date(Web):August 3, 2012
DOI:10.1021/jo301061r
We describe a unified synthetic strategy for efficient assembly of four new heterocyclic libraries. The synthesis began by creating a range of structurally diverse pyrrolidinones or piperidinones. Such compounds were obtained in a simple one-flask operation starting with readily available amines, ketoesters, and unsaturated anhydrides. The use of tetrahydropyran-containing ketoesters, which were rapidly assembled by our Prins cyclization protocol, enabled efficient fusion of pyran and piperidinone cores. A newly developed Au(I)-catalyzed cycloisomerization of alkyne-containing enamides further expanded heterocyclic diversity by providing rapid entry into a wide range of bicyclic and tricyclic dienamides. The final stage of the process entailed diversification of each of the initially produced carboxylic acids using a fully automated platform for amide synthesis, which delivered 1872 compounds in high diastereomeric and chemical purity.
Co-reporter:Sergey V. Pronin ; Anthony Martinez ; Konstantin Kuznedelov ; Konstantin Severinov ; Howard A. Shuman
Journal of the American Chemical Society 2011 Volume 133(Issue 31) pp:12172-12184
Publication Date(Web):June 29, 2011
DOI:10.1021/ja2041964
Inhibition of bacterial transcription represents an effective and clinically validated anti-infective chemotherapeutic strategy. We describe the evolution of our approach to the streptolydigin class of antibiotics that target bacterial RNA polymerases (RNAPs). This effort resulted in the synthesis and biological evaluation of streptolydigin, streptolydiginone, streptolic acid, and a series of new streptolydigin-based agents. Subsequent biochemical evaluation of RNAP inhibition demonstrated that the presence of both streptolic acid and tetramic acid subunits was required for activity of this class of antibiotics. In addition, we identified 10,11-dihydrostreptolydigin as a new RNAP-targeting agent, which was assembled with high synthetic efficiency of 15 steps in the longest linear sequence. Dihydrostreptolydigin inhibited three representative bacterial RNAPs and displayed in vitro antibacterial activity against S. salivarius. The overall increase in synthetic efficiency combined with substantial antibacterial activity of this fully synthetic antibiotic demonstrates the power of organic synthesis in enabling design and comprehensive in vitro pharmacological evaluation of new chemical agents that target bacterial transcription.
Co-reporter:Olesya A. Ulanovskaya, Jiayue Cui, Stephen J. Kron, Sergey A. Kozmin
Chemistry & Biology 2011 Volume 18(Issue 2) pp:222-230
Publication Date(Web):25 February 2011
DOI:10.1016/j.chembiol.2010.12.015
Oxidative phosphorylation (OXPHOS) and glycolysis are the two main pathways that control energy metabolism of a cell. The Warburg effect, in which glycolysis remains active even under aerobic conditions, is considered a key driver for cancer cell proliferation, malignancy, metastasis, and therapeutic resistance. To target aerobic glycolysis, we exploited the complementary roles of OXPHOS and glycolysis in ATP synthesis as the basis for a chemical genetic screen, enabling rapid identification of novel small-molecule inhibitors of facilitative glucose transport. Blocking mitochondrial electron transport with antimycin A or leucascandrolide A had little effect on highly glycolytic A549 lung carcinoma cells, but adding known glycolytic inhibitors 2-deoxy-D-glucose, iodoacetate or cytochalasin B, rapidly depleted intracellular ATP, displaying chemical synthetic lethality. Based on this principle, we exposed antimycin A-treated A549 cells to a newly synthesized 955 member diverse scaffold small-molecule library, screening for compounds that rapidly depleted ATP levels. Two compounds potently suppressed ATP synthesis, induced G1 cell-cycle arrest and inhibited lactate production. Pathway analysis revealed that these novel probes inhibited GLUT family of facilitative transmembrane transporters but, unlike cytochalasin B, had no effect on the actin cytoskeleton. Our work illustrated the utility of a pairwise chemical genetic screen for discovery of novel chemical probes, which would be useful not only to study the system-level organization of energy metabolism but could also facilitate development of drugs targeting upregulation of aerobic glycolysis in cancer.Graphical AbstractFigure optionsDownload full-size imageDownload high-quality image (97 K)Download as PowerPoint slideHighlights► Development of cellular screen for rapid identification of inhibitors of aerobic glycolysis ► Synthesis of a chemical library of cyclic and bicyclic pyrrolidinones ► Identification of two small-molecule inhibitors of glucose transport ► Biochemical characterization of chemical inhibition of facilitative glucose transport
Co-reporter:Jiayue Cui;Olesya A. Ulanovskaya;Jason Hao;Joseph Dundas;Jie Liang
PNAS 2011 Volume 108 (Issue 17 ) pp:6763-6768
Publication Date(Web):2011-04-26
DOI:10.1073/pnas.1015253108
We have developed an efficient strategy to a skeletally diverse chemical library, which entailed a sequence of enyne cycloisomerization,
[4 + 2] cycloaddition, alkene dihydroxylation, and diol carbamylation. Using this approach, only 16 readily available building
blocks were needed to produce a representative 191-member library, which displayed broad distribution of molecular shapes
and excellent physicochemical properties. This library further enabled identification of a small molecule, which effectively
suppressed glycolytic production of ATP and lactate in CHO-K1 cell line, representing a potential lead for the development
of a new class of glycolytic inhibitors.
Co-reporter:Syed Alipayam Rizvi ; Song Liu ; Zhonglei Chen ; Colleen Skau ; Matthew Pytynia ; David R. Kovar ; Steven J. Chmura
Journal of the American Chemical Society 2010 Volume 132(Issue 21) pp:7288-7290
Publication Date(Web):May 10, 2010
DOI:10.1021/ja101811x
We describe structure-based design and chemical synthesis of a simplified analog of bistramide A, which potently and reversibly binds monomeric actin with a Kd of 9.0 nM, depolymerizes filamentous actin in vitro and in A549 (nonsmall cell lung cancer) cells, inhibits growth of cancer cell lines in vitro at submicromolar concentrations, and significantly suppresses proliferation of A549 cells in a nude mice tumor xenograft model in terms of both tumor growth delay and average tumor volume. This study provides a conceptual framework for the design and development of new antiproliferative compounds that target cytoskeletal organization of cancer cells in vivo by a combination of reversible G-actin binding and effective F-actin severing.
Co-reporter:Sergey V. Pronin and Sergey A. Kozmin
Journal of the American Chemical Society 2010 Volume 132(Issue 41) pp:14394-14396
Publication Date(Web):September 29, 2010
DOI:10.1021/ja107190w
Streptolydigin is a highly potent, broad-spectrum antibiotic produced by Streptomyces lydicus, which inhibits bacterial RNA polymerase. We describe the first synthesis of streptolydigin, which was assembled in a highly convergent and fully stereocontrolled fashion with a longest linear sequence of 24 steps starting from commercially available precursors. The assembly process entailed preparation of fully elaborated streptolic and ydiginic subunits of the natural product, followed by a highly efficient union in a three-step one-pot procedure, which included Dieckmann cyclization with a concomitant imide opening, Horner−Wadsworth−Emmons olefination, and desilylation.
Co-reporter:Jianwei Sun;ValerieA. Keller;S. Todd Meyer ;SergeyA. Kozmin
Advanced Synthesis & Catalysis 2010 Volume 352( Issue 5) pp:839-842
Publication Date(Web):
DOI:10.1002/adsc.200900835
Abstract
We describe the development of a silver-catalyzed carbonyl olefination employing electron-rich siloxyalkynes. This process constitutes an efficient synthesis of trisubstituted unsaturated esters, and represents an alternative to the widely utilized Horner–Wadsworth–Emmons reaction. Excellent diastereoselectivities are observed for a range of aldehydes using either 1-siloxy-1-propyne or 1-siloxy-1-hexyne. This mild catalytic process also enables chemoselective olefination of aldehydes in the presence of either ester or ketone functionality. Furthermore, since no by-products are generated, this catalytic process is perfectly suited for development of sequential reactions that can be carried out in a single flask.
Co-reporter:Jiayue Cui;Kenji Matsumoto;Cindy Y. Wang;Marcus E. Peter
ChemBioChem 2010 Volume 11( Issue 9) pp:1224-1227
Publication Date(Web):
DOI:10.1002/cbic.201000193
Co-reporter:Hajoong Lee, Masato Suzuki, Jiayue Cui and Sergey A. Kozmin
The Journal of Organic Chemistry 2010 Volume 75(Issue 5) pp:1756-1759
Publication Date(Web):February 9, 2010
DOI:10.1021/jo9025447
We describe the assembly of a 960-member library of tricyclic 2,3-dihydro-4-quinolones using a combination of solution-phase high-throughput organic synthesis and parallel chromatographic purification. The library was produced with high efficiency and complete chemo- and diastereoselectivity by diversification of an azide-bearing quinolone via a sequence of [4 + 2] cycloadditions, N-acylations, and reductive aminations. The azide-functionalization of this library is designed to facilitate subsequent preparation of fluorescent or affinity probes, as well as small-molecule/surface conjugation.
Co-reporter:Kenji Matsumoto ;SergeyA. Kozmin
Advanced Synthesis & Catalysis 2008 Volume 350( Issue 4) pp:557-560
Publication Date(Web):
DOI:10.1002/adsc.200700537
Abstract
Routiennocin is a member of a family of polycyclic pyrrole ether antibiotics that simultaneously uncouple oxidative phosphorylation and inhibit ATPase as a result of selective complexation of divalent metal ions. We describe a concise synthesis of routiennocin with the longest linear sequence of 8 steps. Our synthesis features a unique bi-directional strategy, which entails a sequential ring-opening/cross metathesis of a highly strained cyclopropenone acetal. This approach enables rapid and highly convergent assembly of the fully extended polyketide subunit of this natural product from readily available homoallylic alcohol precursors.
Co-reporter:Syed Alipayam Rizvi;Valerie A. Keller;David S. Courson;Ronald S. Rock
PNAS 2008 Volume 105 (Issue 11 ) pp:4088-4092
Publication Date(Web):2008-03-18
DOI:10.1073/pnas.0710727105
This study provides comprehensive characterization of the mode of action of bistramide A and identifies structural requirements
of bistramide-based compounds that are responsible for severing actin filaments and inhibiting growth of cancer cells in vitro and in vivo. We rationally designed and assembled a series of structural analogs of the natural product, including a fluorescently labeled
conjugate. We used TIRF microscopy to directly observe actin filament severing by this series of small molecules, which established
that the combination of the spiroketal and the amide subunits was sufficient to enable rapid actin filament disassembly in vitro. In addition, we demonstrated that the enone subunit of bistramide A is responsible for covalent modification of the protein
in vitro and in A549 cells, resulting in further increase in the cytotoxicity of the natural product. Our results demonstrate that
bistramide A elicits its potent antiproliferative activity by a dual mechanism of action, which entails both severing of actin
filaments and covalent sequestration of monomeric actin in the cell.
Co-reporter:Jasmina Marjanovic;Sergey A. Kozmin Dr.
Angewandte Chemie International Edition 2007 Volume 46(Issue 46) pp:
Publication Date(Web):11 OCT 2007
DOI:10.1002/anie.200702440
Lock it in: A temporary silicon-based configurational lock (see scheme) has enabled an efficient and fully diastereoselective assembly of the spiroketal subunit of spirofungin A. This complex natural product was synthesized in 20 steps, including a rapid polyketide assembly based on the ring-opening metathesis of a cyclopropenone acetal. It was established that spirofungin A elicited notable antiproliferative activity against several human cancer cell lines and selectively inhibited isoleucyl-tRNA synthetase in vitro.
Co-reporter:Jasmina Marjanovic;Sergey A. Kozmin Dr.
Angewandte Chemie 2007 Volume 119(Issue 46) pp:
Publication Date(Web):11 OCT 2007
DOI:10.1002/ange.200702440
Durch die vorübergehende Fixierung der Konfiguration mithilfe einer Silylverknüpfung (siehe Schema) gelingt der effiziente, vollständig diastereoselektive Aufbau der Spiroketal-Einheit von Spirofungin A. Die 20-stufige Synthese des komplexen Naturstoffs umfasste auch den schnellen Aufbau eines Polyketids durch Ringöffnungsmetathese eines Cyclopropenonacetals. Spirofungin A zeigt eine beachtliche Proliferationshemmung gegen einige humane Krebszelllinien und inhibiert in vitro selektiv die Isoleucyl-tRNA-Synthetase.
Co-reporter:Liming Zhang;Jianwei Sun;Sergey A. Kozmin
Advanced Synthesis & Catalysis 2006 Volume 348(Issue 16-17) pp:
Publication Date(Web):27 NOV 2006
DOI:10.1002/adsc.200600368
This account provides a comprehensive overview of the development of gold and platinum catalysis of the enyne cycloisomerization. The use of these soft, alkynophilic metals enables mild, chemoselective and efficient transformations of a variety of readily available acyclic enynes to a wide range of synthetically useful carbocyclic and heterocyclic products. The review is organized according to diverse structural types of enynes that undergo skeletal cycloisomerizations. The account begins with an overview of transformations of primarily 1,6-enynes to 1-alkenylcyclopentenes, bicyclo[4.1.0]heptenes, methylenecycloalkenes, bicyclo[4.3.0]nonadienes and bicyclo[3.2.0]heptenes. This section is followed by the discussion of cycloisomerizations of 1,5-enynes, which enable a rapid access to a range of other cyclic products, including bicyclo[3.1.0]hexenes, cyclohexadienes, heterobicycloalkenes, methylenecyclopentenes, naphthalenes and methyleneindenes. In addition, the [3,3] rearrangement of 1,5-enynes provides efficient access to the corresponding allenes. The account concludes with an overview of the most recent studies on gold- and platinum-catalyzed cycloisomerizations of 1,4- and 1,3-enynes. Due to the rapidly increasing interest in this area during the past three to five years, we believe that this review provides a timely and comprehensive discussion of the development gold- and platinum-catalyzed cycloisomerization starting from the initial pioneering investigations to the latest advances in the field. A significant emphasis is placed on the mechanistic discussion of the observed manifolds of skeletal reorganizations.
Co-reporter:Jianwei Sun Dr.
Angewandte Chemie 2006 Volume 118(Issue 30) pp:
Publication Date(Web):29 JUN 2006
DOI:10.1002/ange.200601276
Ein neuer katalytischer Prozess: Die Silber-katalysierte Hydroaminierung von Siloxyalkinen mit sekundären Amiden liefert mit hoher Effizienz und ausgezeichneter Diastereoselektivität Silylketenaminale (siehe Schema), darunter auch solche, die mit üblichen Silylierungsmethoden nicht zugänglich sind. Einer schnellen und reversiblen Silber-Alkin-Komplexierung folgt als geschwindigkeitsbestimmender Schritt eine C-N-Bindungsbildung.
Co-reporter:Jianwei Sun Dr.
Angewandte Chemie International Edition 2006 Volume 45(Issue 30) pp:
Publication Date(Web):29 JUN 2006
DOI:10.1002/anie.200601276
New catalytic process: The silver-catalyzed hydroamination of siloxy alkynes with secondary amides furnishes silyl ketene aminals with high efficiency and excellent diastereoselectivity (see scheme), including some that are unavailable by conventional silylation methods. The reaction comprises a fast and reversible silver–alkyne complexation, followed by a rate-determining CN bond-forming step.
Co-reporter:Sergey A. Kozmin Dr.;Ying Wang Dr.
Angewandte Chemie International Edition 2003 Volume 42(Issue 8) pp:
Publication Date(Web):21 FEB 2003
DOI:10.1002/anie.200390238
An operationally simple and general technique for multiparallel organic synthesis is based on the three-dimensional delivery of reactants to an array of interconnected reaction wells. This technology can be readily adapted to the design of substantially large arrays, or multiple copies of interconnected arrays, thus allowing the necessary flexibility for the synthesis and screening of a wide range of chemical libraries.
Co-reporter:Dong Liu Dr.
Angewandte Chemie International Edition 2001 Volume 40(Issue 24) pp:
Publication Date(Web):18 DEC 2001
DOI:10.1002/1521-3773(20011217)40:24<4757::AID-ANIE4757>3.0.CO;2-S
A versatile precursor for the assembly of a range of polyol-containing fragments is the silacyclic alcohol 2 that results from the highly enantioselective, catalytic isomerization of diphenylsilacyclopentene oxide (1). The use of this precusor is illustrated with the efficient and highly diastereoselective assembly of acyclic tetraol motifs.
Co-reporter:Michael P. Schramm;D. Srinivasa Reddy Dr.
Angewandte Chemie International Edition 2001 Volume 40(Issue 22) pp:
Publication Date(Web):16 NOV 2001
DOI:10.1002/1521-3773(20011119)40:22<4274::AID-ANIE4274>3.0.CO;2-#
Mechanistically intriguing participation of siloxyalkynes occurs in the intramolecular Ru-catalyzed metathesis with terminal alkenes (see scheme; Ms=methanesulfonyl). Combined with efficient protodesilylation, this process resulted in the development of a new method for the synthesis of highly functionalized enones starting from readily accessible acyclic precursors. Heterocyclic and polycyclic compounds were prepared efficiently, which illustrates the generality of this novel method.
Co-reporter:Michael P. Schramm;D. Srinivasa Reddy Dr.
Angewandte Chemie 2001 Volume 113(Issue 22) pp:
Publication Date(Web):15 NOV 2001
DOI:10.1002/1521-3757(20011119)113:22<4404::AID-ANGE4404>3.0.CO;2-B
Eine mechanistisch interessante Beteiligung von Siloxyalkinen wurde bei der intramolekularen Ru-katalysierten Metathese mit terminalen Alkenen beobachtet (siehe Schema; Ms=Methansulfonyl). Mit einer anschließenden Protodesilylierung kombiniert, stellt diese Reaktion eine neue Methode für die Synthese hoch funktionalisierter Enone ausgehend von einfachen acyclischen Vorstufen dar. Die allgemeine Anwendbarkeit der Methode wurde durch die Synthese einer Reihe von heterocyclischen und polycyclischen Verbindungen demonstriert.
Co-reporter:Dong Liu Dr.
Angewandte Chemie 2001 Volume 113(Issue 24) pp:
Publication Date(Web):18 DEC 2001
DOI:10.1002/1521-3757(20011217)113:24<4893::AID-ANGE4893>3.0.CO;2-D
Eine vielseitige Vorstufe für den Aufbau von Polyolen ist das Silacyclopentenol 2, das durch hoch enantioselektive katalytische Isomerisierung des entsprechenden Epoxids 1 erhalten wird. Der präparative Nutzen von 2 wird anhand des effizienten und hoch diastereoselektiven Aufbaus acyclischer Tetraol-Motive demonstriert.
.jpg)
![1,4-Decadien-3-one, 10-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-6-methyl-7-(phenylmethoxy)-, (4Z,6S,7R)-](/data/chemimg/187000/1228458-58-9.png)
![1,4-Decadien-3-one, 10-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-6-methyl-7-(phenylmethoxy)-, (4Z,6S,7R)-](/data/chemimg/187000/1228458-58-9_b.png)
![6,9-Hexadecadien-8-one, 1-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-5,15-dimethyl-4,12,16-tris(phenylmethoxy)-, (4R,5S,6Z,9E,12R,15S)-](/data/chemimg/187000/1228458-57-8.png)
![6,9-Hexadecadien-8-one, 1-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-5,15-dimethyl-4,12,16-tris(phenylmethoxy)-, (4R,5S,6Z,9E,12R,15S)-](/data/chemimg/187000/1228458-57-8_b.png)
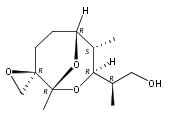
![1,7-Dioxaspiro[5.5]undecane-2-butanol, 8-(3-hydroxypropyl)-β,9-dimethyl-, (βS,2S,6S,8R,9S)-](/data/chemimg/187000/1228458-56-7.png)
![1,7-Dioxaspiro[5.5]undecane-2-butanol, 8-(3-hydroxypropyl)-β,9-dimethyl-, (βS,2S,6S,8R,9S)-](/data/chemimg/187000/1228458-56-7_b.png)
![1,4-Decadien-3-one, 10-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-6-methyl-7-(phenylmethoxy)-, (4E,6S,7R)-](/data/chemimg/187000/1228458-55-6.png)
![1,4-Decadien-3-one, 10-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-6-methyl-7-(phenylmethoxy)-, (4E,6S,7R)-](/data/chemimg/187000/1228458-55-6_b.png)
![Butanamide, 3-hydroxy-N-[3-[(2R,3S,6S,8S)-8-[(3S,4E,6S)-6-hydroxy-3,5-dimethyl-4-hepten-1-yl]-3-methyl-1,7-dioxaspiro[5.5]undec-2-yl]propyl]-4-[(3-methoxy-1-oxopropyl)amino]-2-methyl-, (2S,3R)-](/data/chemimg/187000/1228458-54-5.png)
![Butanamide, 3-hydroxy-N-[3-[(2R,3S,6S,8S)-8-[(3S,4E,6S)-6-hydroxy-3,5-dimethyl-4-hepten-1-yl]-3-methyl-1,7-dioxaspiro[5.5]undec-2-yl]propyl]-4-[(3-methoxy-1-oxopropyl)amino]-2-methyl-, (2S,3R)-](/data/chemimg/187000/1228458-54-5_b.png)
![6,9-Hexadecadien-8-one, 1-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-5,15-dimethyl-4,12,16-tris(phenylmethoxy)-, (4R,5S,6E,9E,12R,15S)-](/data/chemimg/186700/1027371-66-9.png)
![6,9-Hexadecadien-8-one, 1-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-5,15-dimethyl-4,12,16-tris(phenylmethoxy)-, (4R,5S,6E,9E,12R,15S)-](/data/chemimg/186700/1027371-66-9_b.png)
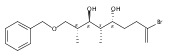
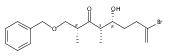
![Benzene, [[[(1R,2S)-1-[3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]propyl]-2-methyl-3-buten-1-yl]oxy]methyl]-](/data/chemimg/186700/1027371-63-6.png)
![Benzene, [[[(1R,2S)-1-[3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]propyl]-2-methyl-3-buten-1-yl]oxy]methyl]-](/data/chemimg/186700/1027371-63-6_b.png)
![Silane,(1,1-dimethylethyl)[(2,2-dimethyl-4-methylene-4H-1,3-dioxin-6-yl)oxy]dimethyl-](http://img.cochemist.com/ccimg/588800/588731-92-4.png)
![Silane,(1,1-dimethylethyl)[(2,2-dimethyl-4-methylene-4H-1,3-dioxin-6-yl)oxy]dimethyl-](http://img.cochemist.com/ccimg/588800/588731-92-4_b.png)
![CARBANIDE;COBALT;3-NITROSO-N-[2-(2H-PYRIDIN-1-ID-2-YL)ETHYL]BUT-2-EN-2-AMINE;3-N-(2-PIPERIDIN-2-YLETHYL)BUTANE-2,3-DIAMINE](http://img.cochemist.com/ccimg/7300/7229-28-9.png)
![CARBANIDE;COBALT;3-NITROSO-N-[2-(2H-PYRIDIN-1-ID-2-YL)ETHYL]BUT-2-EN-2-AMINE;3-N-(2-PIPERIDIN-2-YLETHYL)BUTANE-2,3-DIAMINE](http://img.cochemist.com/ccimg/7300/7229-28-9_b.png)
![Butanoic acid, 2(or3)-methyl-,(2R,3S,6S,7R,8R)-3-[[3-(formylamino)-2-hydroxybenzoyl]amino]-8-hexyl-2,6-dimethyl-4,9-dioxo-1,5-dioxonan-7-ylester (9CI)](http://img.cochemist.com/ccimg/700/642-15-9.png)
![Butanoic acid, 2(or3)-methyl-,(2R,3S,6S,7R,8R)-3-[[3-(formylamino)-2-hydroxybenzoyl]amino]-8-hexyl-2,6-dimethyl-4,9-dioxo-1,5-dioxonan-7-ylester (9CI)](http://img.cochemist.com/ccimg/700/642-15-9_b.png)
![2-PYRROLIDINEACETAMIDE, 4-[6-(1,4-DIMETHYLSPIRO[2,9-DIOXABICYCLO[3.3.1]NON-6-ENE-8,2'-OXIRAN-3-YL)-1-HYDROXY-4-METHYL-2,4-HEPTADIENYLIDENE]-N,.ALPHA.-DIMETHYL-3,5-DIOXO-1-(TETRAHYDRO-5-HYDROXY-6-METHYL-2H-PYRAN-2-YL)-](http://img.cochemist.com/ccimg/7300/7229-50-7.png)
![2-PYRROLIDINEACETAMIDE, 4-[6-(1,4-DIMETHYLSPIRO[2,9-DIOXABICYCLO[3.3.1]NON-6-ENE-8,2'-OXIRAN-3-YL)-1-HYDROXY-4-METHYL-2,4-HEPTADIENYLIDENE]-N,.ALPHA.-DIMETHYL-3,5-DIOXO-1-(TETRAHYDRO-5-HYDROXY-6-METHYL-2H-PYRAN-2-YL)-](http://img.cochemist.com/ccimg/7300/7229-50-7_b.png)


![Methanone, [4-(aminomethyl)phenyl]phenyl-](http://img.cochemist.com/ccimg/94400/94341-55-6.png)
![Methanone, [4-(aminomethyl)phenyl]phenyl-](http://img.cochemist.com/ccimg/94400/94341-55-6_b.png)



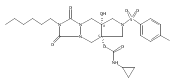

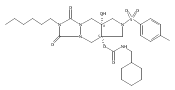

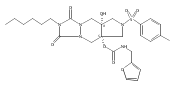
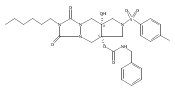
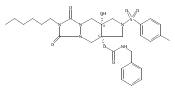
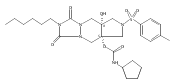
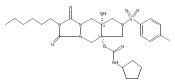

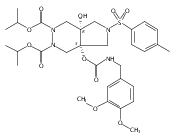









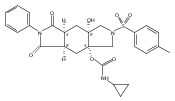

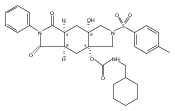


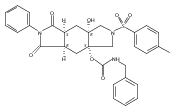





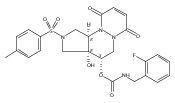


























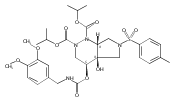
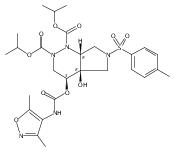
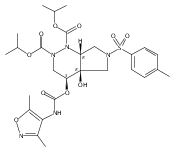

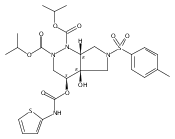


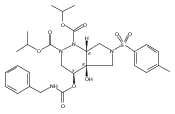

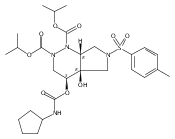
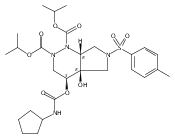
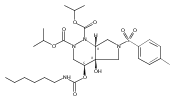





















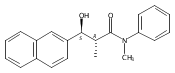


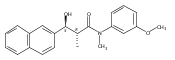



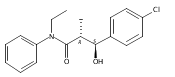


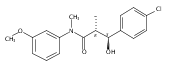










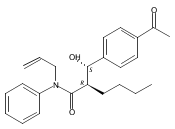


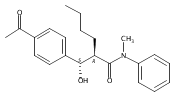

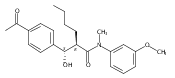
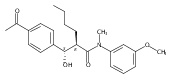



























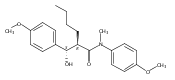
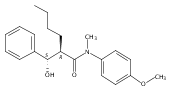
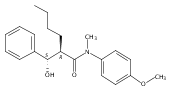
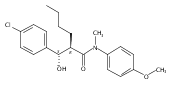



























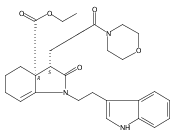












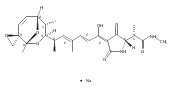

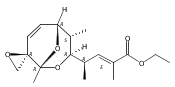








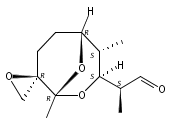
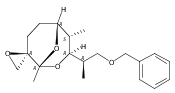
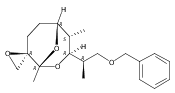
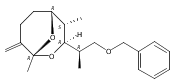









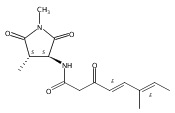
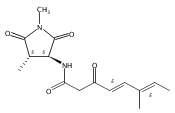
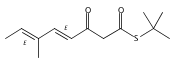
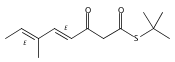
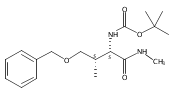

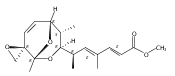

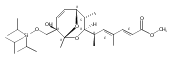
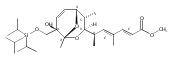





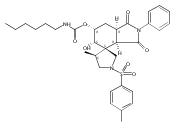


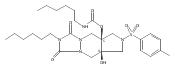


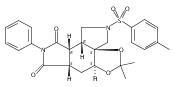


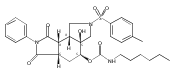












































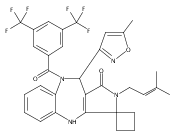
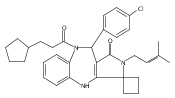
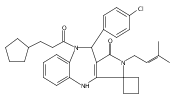
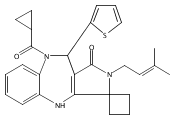




































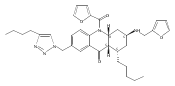
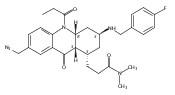


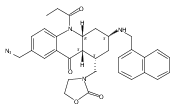


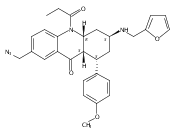










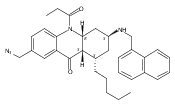

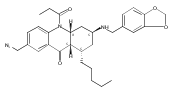
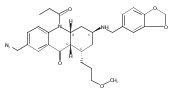



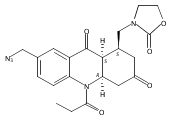




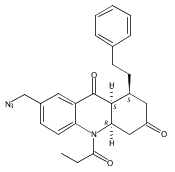

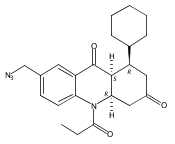


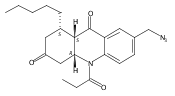

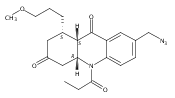

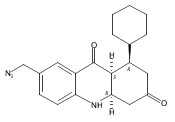



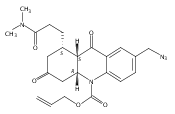
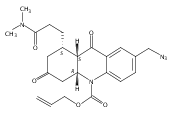

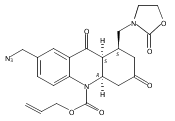






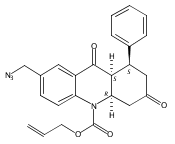
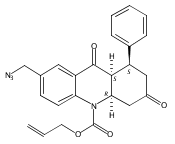

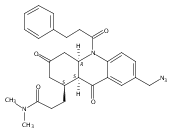
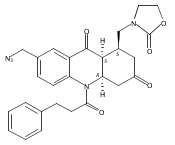


















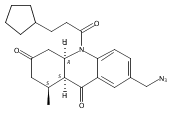
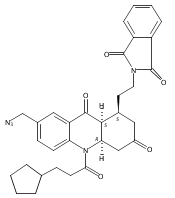
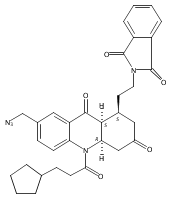
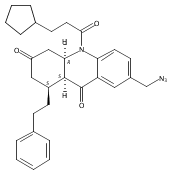










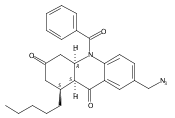
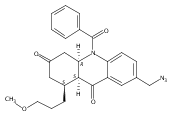
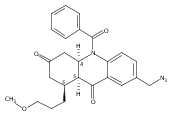

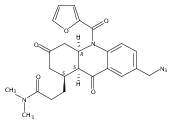




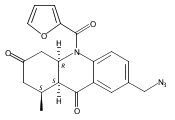



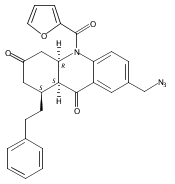
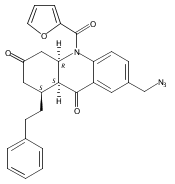
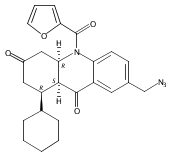
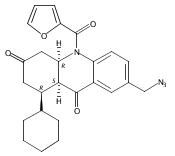
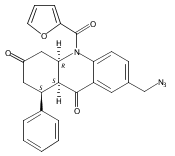

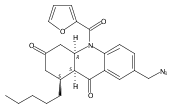
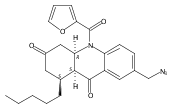
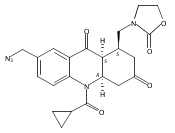
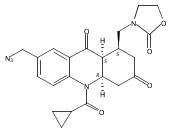

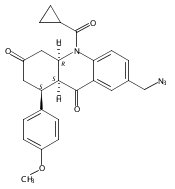
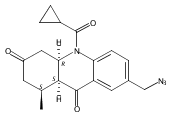

































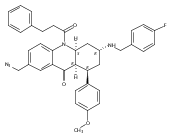



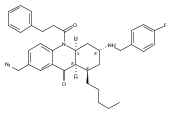
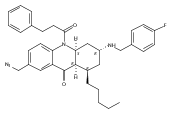
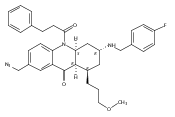





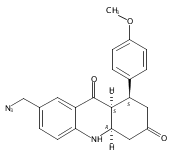
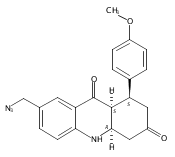
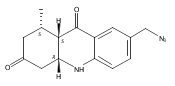
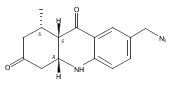











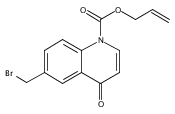



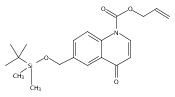

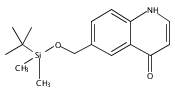
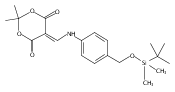



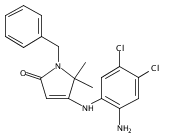
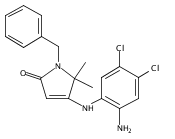
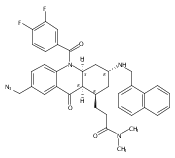
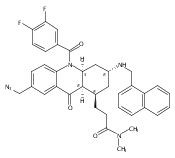












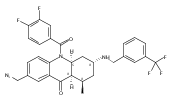

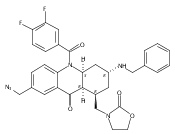

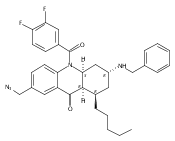




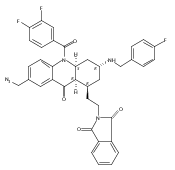
















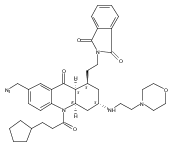

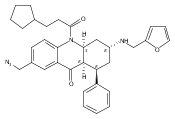
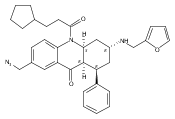
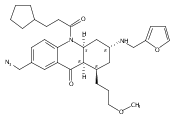





































































































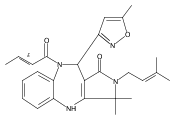
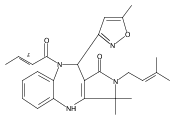

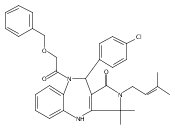


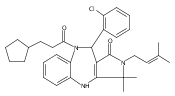
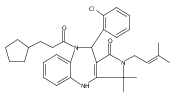

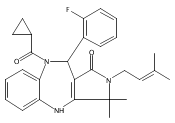


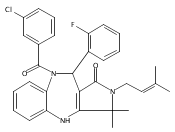
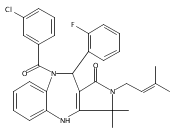





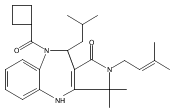
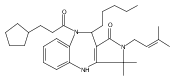

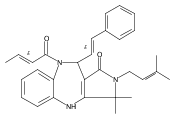

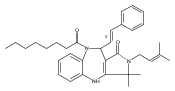



















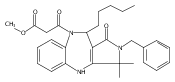
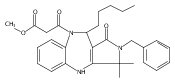
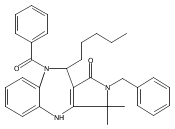

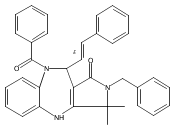


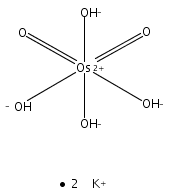







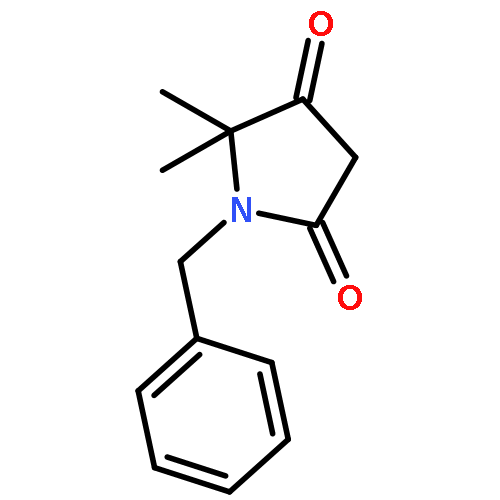









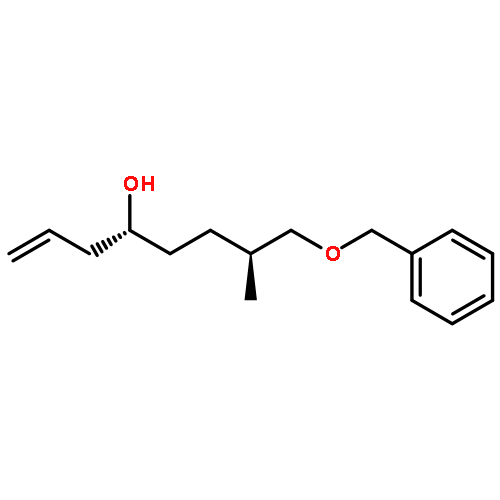









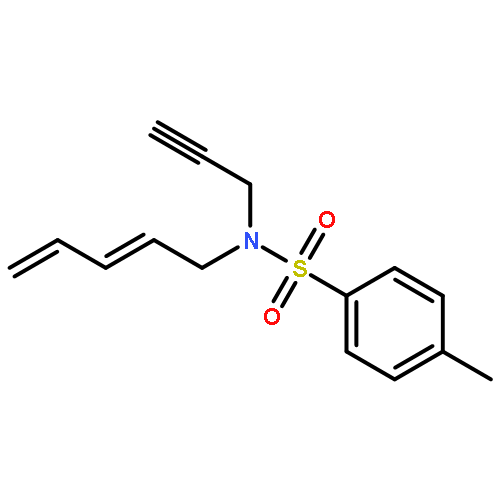


























![Silane, (1,1-dimethylethyl)dimethyl[(4-nitrophenyl)methoxy]-](http://img.cochemist.com/ccimg/135700/135605-97-9.png)
![Silane, (1,1-dimethylethyl)dimethyl[(4-nitrophenyl)methoxy]-](http://img.cochemist.com/ccimg/135700/135605-97-9_b.png)


![Benzenamine, 4-[[[(1,1-dimethylethyl)dimethylsilyl]oxy]methyl]-](/data/chemimg/116700/131230-76-7.png)
![Benzenamine, 4-[[[(1,1-dimethylethyl)dimethylsilyl]oxy]methyl]-](/data/chemimg/116700/131230-76-7_b.png)







![4,8-Dioxaspiro[2.5]oct-1-ene, 6,6-dimethyl-](http://img.cochemist.com/ccimg/61000/60935-22-0.png)
![4,8-Dioxaspiro[2.5]oct-1-ene, 6,6-dimethyl-](http://img.cochemist.com/ccimg/61000/60935-22-0_b.png)


























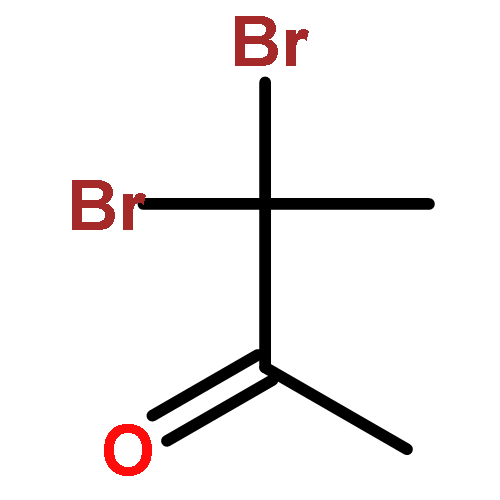



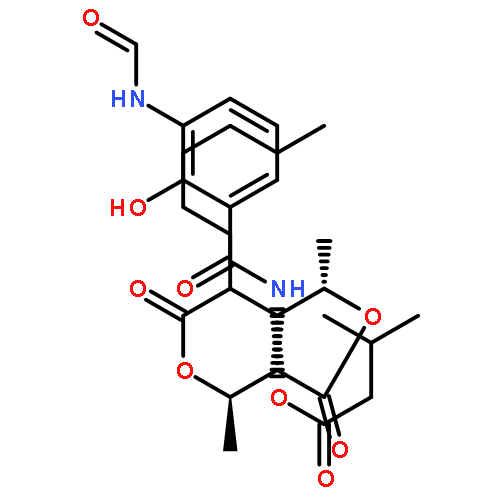
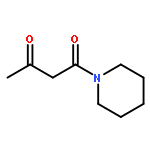
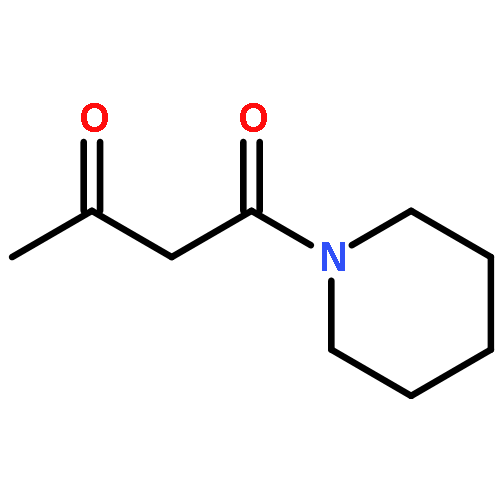



![Benzenesulfonamide,N-[(cyclohexylamino)carbonyl]-4-methyl-](http://img.cochemist.com/ccimg/700/664-95-9.png)
![Benzenesulfonamide,N-[(cyclohexylamino)carbonyl]-4-methyl-](http://img.cochemist.com/ccimg/700/664-95-9_b.png)


![Thieno[3,4-d]pyridazine](http://img.cochemist.com/ccimg/300/270-84-8.png)
![Thieno[3,4-d]pyridazine](http://img.cochemist.com/ccimg/300/270-84-8_b.png)
![Pyrido[2,3-d]pyridazine](http://img.cochemist.com/ccimg/300/253-73-6.png)
![Pyrido[2,3-d]pyridazine](http://img.cochemist.com/ccimg/300/253-73-6_b.png)









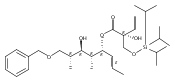

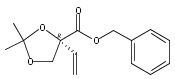
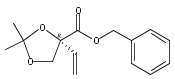
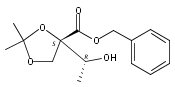



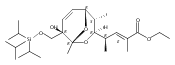
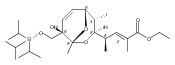
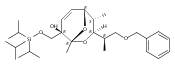
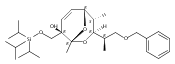











![(Acetonitrile)[(2-biphenyl)di-tert-butylphosphine]gold(I) hexafluoroantimonate](http://img.cochemist.com/ccimg/866700/866641-66-9.png)
![(Acetonitrile)[(2-biphenyl)di-tert-butylphosphine]gold(I) hexafluoroantimonate](http://img.cochemist.com/ccimg/866700/866641-66-9_b.png)
















![Benzo[g]phthalazine](http://img.cochemist.com/ccimg/300/260-35-5.png)
![Benzo[g]phthalazine](http://img.cochemist.com/ccimg/300/260-35-5_b.png)








![Silane, tris(1-methylethyl)[(phenylethynyl)oxy]-](http://img.cochemist.com/ccimg/96900/96845-72-6.png)
![Silane, tris(1-methylethyl)[(phenylethynyl)oxy]-](http://img.cochemist.com/ccimg/96900/96845-72-6_b.png)

![Silane, [(cyclohexylethynyl)oxy]tris(1-methylethyl)-](http://img.cochemist.com/ccimg/104900/104875-64-1.png)
![Silane, [(cyclohexylethynyl)oxy]tris(1-methylethyl)-](http://img.cochemist.com/ccimg/104900/104875-64-1_b.png)

