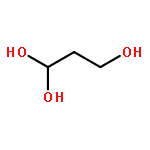
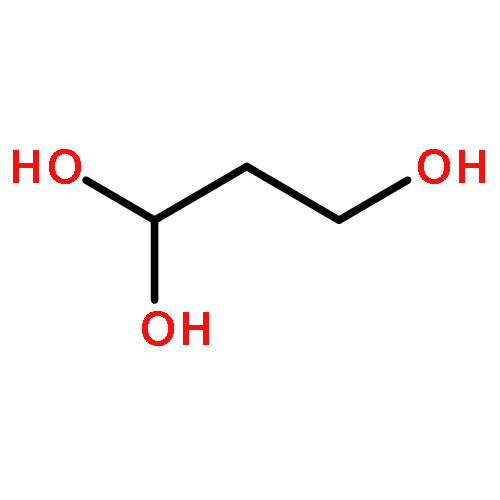
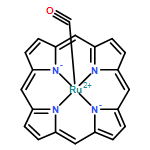
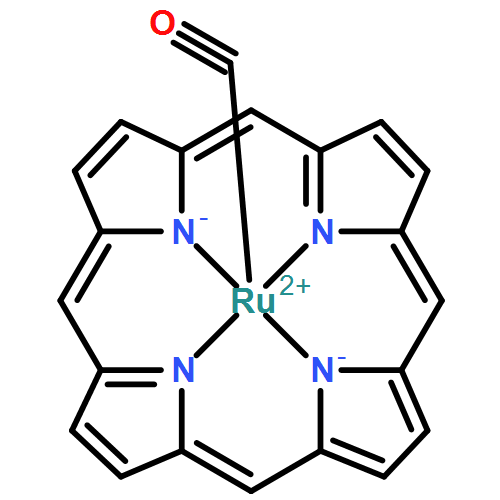
Co-reporter:Xufeng Lin;Yanyan Xi;David L. Phillips;Wenyue Guo
Journal of Physical Organic Chemistry 2016 Volume 29( Issue 3) pp:134-144
Publication Date(Web):
DOI:10.1002/poc.3509
The support effect is an important issue in heterogeneous catalysis. A systematic density functional theory computational study is reported here to better understand the C–H bond activation steps in the reaction between C2H6 and a model silica-supported Ni3O3 cluster, as well as that between C2H6 and an unsupported Ni3O3 cluster. Two mechanisms, namely, a radical mechanism (denoted as mechanism A) and a concerted mechanism (denoted as mechanism B) were examined. Both of these mechanisms contain two steps. For the C–H bond activation taking place via mechanism A, the involvement of the model silica support does not change the most favorable pathway significantly; however, it does result in a modest increase in the reaction barrier and the overall Gibbs energy change. For the C–H bond activation taking place via mechanism B, the involvement of the model silica support leads to an increase in the reaction barrier in the first step. The product of this step has a noticeable difference in the structures for the Ni3O3 moiety in the unsupported and model silica-supported systems. The result of charge analysis shows that there is no noticeable charge transfer between the silica support and Ni3O3 when they are in the starting reactants, while there is an electron withdrawal from Ni3O3 by the silica support when they are in transition states, intermediates, or products. The results here provide deeper insights into the support effect on the C–H bond activation of lower alkanes on supported transition metal catalysts. Copyright © 2015 John Wiley & Sons, Ltd.
Co-reporter:Yanyan Xi, Bili Chen, Xufeng Lin, Hui Fu, Chuangye Wang
Computational and Theoretical Chemistry 2016 Volume 1076() pp:65-73
Publication Date(Web):15 January 2016
DOI:10.1016/j.comptc.2015.12.006
•Support effect of silica on the CH4 activation on NixOx clusters was investigated.•Involvement of silica increases the reaction barrier of the CH bond activation.•Involvement of silica increases the reaction free energy of the CH bond activation.•The CH4 activation occurs on different PES at the presence and absence of silica.Supported transition metal catalysts belong to an important family of catalyst in chemical industry, making the support effect to be a important issue in fundamental research. A theoretical computation using density functional theory (DFT) was performed to investigate the support effect of a silica model on the initial step of methane activation on NixOx (where, x = 2, 3) clusters. The behavior of four reactions were studied by exploring their potential energy surfaces (PES), namely, (i) CH4 reacting with an unsupported Ni2O2 cluster, (ii) with a silica-supported Ni2O2 cluster, (iii) with an unsupported Ni3O3 cluster, and (iv) with an silica-supported Ni3O3 cluster. For each of these four reactions, only a radical mechanism was investigated, and the PESs with different spin states were explored. A spin transition process is involved for the reactions on the unsupported NixOx clusters, and no spin transition is required for the case of supported NixOx clusters. The reaction barrier as well as the reaction free energy is increased with the involvement of the model silica support. These results provide a deeper insight into the support effect on the CH bond activation of small alkanes in general and of methane in particular on supported transition metal catalysts.
Co-reporter:Xufeng Lin, Bili Chen, Yanyan Xi, Chuangye Wang, Hui Fu
Computational and Theoretical Chemistry 2016 1080() pp: 1-9
Publication Date(Web):15 March 2016
DOI:10.1016/j.comptc.2016.01.020
•The substituent effect was studied for the reaction behavior of Ru–nitrene complex.•Substitution of the PhF5 groups leads to a lowered reaction barrier.•Substitution of the PhF5 groups increases the reaction rate by 2 orders of magnitude.•Increased electron deficiency of the nitrene results in the increased reaction rate.The substituent effect of the pentafluorophenyl (PhF5) groups on the Ru-porphyrin complex-catalyzed intramolecular reaction was investigated with density function theory calculations. The interested reaction steps are the unsubstituted and substituted Ru-porphyrin complexes-mediated nitrene insertion into one of the CH bonds on a certain sulfamate ester. For both of the unsubstituted and substituted reactant complexes, the singlet state has a close energy to the triplet state. The substitution of the PhF5 groups leads to a lowered reaction barrier on both of the singlet and triplet potential energy surfaces (PESs). The barrier lowering corresponds to an increased reaction rate by 2 orders of magnitude on both PESs. The singlet PES has a predominant contribution to the overall reaction rate compared to the triplet one for both of the unsubstituted and substituted cases. The results from the natural bond orbital analysis show that the PhF5 groups are highly negative in charge, and the group substituting leads to an increased electron deficiency of the nitrene N atom. This increase can account for the increase of the reaction rate on both of the singlet and triplet PESs.
Co-reporter:Luyao Xu, Xufeng Lin, Yanyan Xi, Xiaoming Lu, Chuangye Wang, Chenguang Liu
Materials Research Bulletin 2014 59() pp: 254-260
Publication Date(Web):
DOI:10.1016/j.materresbull.2014.07.023
Co-reporter:Xufeng Lin, Yanyan Xi, Guodong Zhang, David Lee Phillips, and Wenyue Guo
Organometallics 2014 Volume 33(Issue 9) pp:2172-2181
Publication Date(Web):April 30, 2014
DOI:10.1021/om400996f
Density functional theory calculations were utilized to study the reaction mechanisms of nonoxidative coupling of methane (NOCM) occurring on a silica-supported single-site tantalum (Ta) catalyst. Two catalytic cycles, namely, catalytic cycles A (CCA) and B (CCB), as well as other competing pathways, were investigated by exploring the potential energy surfaces for the reactions of interest. The supported methyltantalum [(≡SiO3)2Ta–CH3] and tantalum hydride [(≡SiO3)2Ta–H] catalyzed the reaction of NOCM through CCA and CCB, respectively. CCA and CCB comprise five and six elementary steps, respectively. The two rate-determining states for both catalytic cycles were elucidated. The turnover number of methane conversion catalyzed by the supported methyltantalum was about 105 larger than that catalyzed by the supported tantalum hydride. This large difference indicates that the former species is predominantly responsible for the conversion of methane to ethane.
Co-reporter:Xufeng Lin, Yuanyuan Qu, Yanhong Lv, Yanyan Xi, David Lee Phillips and Chenguang Liu
Physical Chemistry Chemical Physics 2013 vol. 15(Issue 8) pp:2967-2982
Publication Date(Web):03 Jan 2013
DOI:10.1039/C2CP43644B
The dehydration mechanisms for glucose in β-pyranose (BP) and in open-chain (OC) forms, catalyzed by acids homogeneously and heterogeneously, were investigated using density functional and two-layer ONIOM calculations. The first dehydration reaction and competing reaction pathways are the main focus of the present study. The energetics of five dehydration and two isomerization pathways were examined for the protonated form of BP in acidic aqueous solutions and the most favorable pathway of these was found to be the dehydration at the anomeric site. No dehydration pathway of OC glucose is favored over its isomerization to BP or to fructose. The relative ease of dehydration over isomerization depends on the selection of the reaction media for the protonated form of BP. These two reaction pathways catalyzed by a surface Brönsted acid site were then examined and the isomerization pathway was found to be more favorable than dehydration at the anomeric site on a surface acid site. These mechanistic insights provide an important guide for the catalyst design/selection of the reaction media for glucose dehydration.
Co-reporter:Xufeng Lin, Yanhong Lv, Yuanyuan Qu, Guodong Zhang, Yanyan Xi, David L. Phillips and Chenguang Liu
Physical Chemistry Chemical Physics 2013 vol. 15(Issue 46) pp:20120-20133
Publication Date(Web):10 Oct 2013
DOI:10.1039/C3CP53915F
The catalytic activity and the acrolein selectivity for liquid phase glycerol dehydration on β zeolites (HNa-β-k) were found to be dependent on the reaction temperature as well as on the amount of acid sites on the zeolites. An increase in the reaction temperature favors the acrolein selectivity. The acrolein selectivity increases with the Na+/H+ ratio and the glycerol conversion decreases with it so that a maximum acrolein yield is obtained when a certain amount of acidic sites are replaced by non-active Na+ sites. The computational results indicate that 3-hydoxylpropanal (HPA) is an important intermediate that determines the final product selectivity. The relative rates of the different reaction pathways for HPA can be affected by the amount of water molecules involved in its homogeneous reaction. Based on the reaction mechanism proposed, it was hypothesized that smaller pores reduce activity but increase selectivity to acrolein, and results of the H-MFI zeolite were consistent with this hypothesis. Our work provides important insight into the overall landscape of the reaction mechanism and can be used to help design reaction systems that have good acrolein selectivity for the liquid phase glycerol dehydration reactions.
Co-reporter:Xufeng Lin, Yanyan Xi, Jian Sun
Computational and Theoretical Chemistry 2012 Volume 999() pp:74-82
Publication Date(Web):1 November 2012
DOI:10.1016/j.comptc.2012.08.016
The yields of amides and aziridines for two cases of dirhodium tetracarboxylate-catalyzed competing amidation and aziridination reactions were successfully interpreted by density functional calculations. For each reaction, four reaction pathways were examined structurally and energetically. The relative reaction rates of different reaction pathways were calculated from the relative free energy of activations for all of the four pathways. Our results may help synthetic chemists better design the substrates and scaffolds of catalyst ligands, for transition metal catalyzed amidation and aziridination reactions.Graphical abstractHighlights► Competing amidation and aziridination pathways on an NR substrate. ► Both pathways are studied with singlet and triplet spin multiplicities. ► Singlet and triplet pathways have different mechanisms. ► Relative reaction rates calculated explain the experimental yields.
Co-reporter:Xufeng Lin ; Yanyan Xi ;Jian Sun
The Journal of Physical Chemistry C 2012 Volume 116(Issue 5) pp:3503-3516
Publication Date(Web):January 6, 2012
DOI:10.1021/jp2088274
Understanding the reaction mechanism for oxidative dehydrogenation (ODH) of alkanes, especially the key intermediate(s) that generates alkene is essential for designing good ODH catalysts. To unravel the mechanisms for Ni-based oxide-catalyzed ODH reactions, we investigated the reactions of C2H6 with Ni3Ox (x = 1, 2, 3) clusters by density functional calculations. For Ni3O3, three pathways were examined for the C–H bond activation step, and the one with concerted mechanism undergoing at two sites is the most favorable pathway, producing an ethylnickel species. Then, four reaction pathways, namely, β-H elimination, α-H abstraction, C–C bond cleavage, and isomerization to an ethoxide species, with 11 reaction channels, were examined to understand the behavior of this ethylnickel species. The selectivity of C2H4 (SC2) for this reaction was calculated based on the relative rates of these four pathways. Similar investigations were carried out on the reactions of Ni3O2 and Ni3O1 clusters with C2H6. The calculated SC2 increases from ∼37 to over 99% with decreasing x value in Ni3Ox.
Co-reporter:Xufeng Lin, Jian Sun, Yanyan Xi, Bo Pang
Computational and Theoretical Chemistry 2011 Volume 963(2–3) pp:284-289
Publication Date(Web):February 2011
DOI:10.1016/j.comptc.2010.10.039
A catalytic cycle of a dirhodium tetracarboxylate (diRh) catalyzed intramolecular amidation reaction was investigated with density functional calculations, and the stereoselectivity of the amidation product was successfully interpreted. The product selectivity was calculated from the free energy of activations for four different reaction pathways. The pathway that forms cis-stereomer on the singlet potential energy surface has the lowest free energy of activation in the four pathways examined. The results may provide deeper insight into transition-metal catalyzed CN bond formation reactions, as well as help synthetic chemists better design/select the catalyst ligands and the reactant substrates for this type of reaction.
Co-reporter:Xufeng Lin, Jian Sun, Yanyan Xi, and Delian Lin
Organometallics 2011 Volume 30(Issue 12) pp:3284-3292
Publication Date(Web):June 1, 2011
DOI:10.1021/om1012049
Two mechanisms, namely, the Ni(0)–Ni(II) and Ni(I)–Ni(III) mechanisms, for nickel-bis(oxazolinyl)pyridine complex catalyzed Negishi cross-coupling reaction were investigated with density functional calculations. The Ni(I)–Ni(III) mechanism, containing sequential steps of transmetalation–oxidative addition–reductive elimination, is more favorable than the Ni(0)–Ni(II) mechanism, based on the energetic span model. The enantioselectivity of the coupled product from a racemic secondary alkyl electrophile was calculated by the relative reaction rate (rS/rR) of the reductive elimination step that forms the coupled product in the S-enantiomer over that leading to the R-enantiomer. The rS/rR can be calculated from the relative free energy of the transition states for these two reductive elimination pathways in the Ni(I)–Ni(III) mechanism. The calculated enantioselectivity for the model reaction is consistent with the experimental report. The influence of the asymmetric steric hindrance of the catalyst ligand on the reductive elimination step is also discussed.
Co-reporter:Xufeng Lin, Yuanyuan Qu, Yanhong Lv, Yanyan Xi, David Lee Phillips and Chenguang Liu
Physical Chemistry Chemical Physics 2013 - vol. 15(Issue 8) pp:NaN2982-2982
Publication Date(Web):2013/01/03
DOI:10.1039/C2CP43644B
The dehydration mechanisms for glucose in β-pyranose (BP) and in open-chain (OC) forms, catalyzed by acids homogeneously and heterogeneously, were investigated using density functional and two-layer ONIOM calculations. The first dehydration reaction and competing reaction pathways are the main focus of the present study. The energetics of five dehydration and two isomerization pathways were examined for the protonated form of BP in acidic aqueous solutions and the most favorable pathway of these was found to be the dehydration at the anomeric site. No dehydration pathway of OC glucose is favored over its isomerization to BP or to fructose. The relative ease of dehydration over isomerization depends on the selection of the reaction media for the protonated form of BP. These two reaction pathways catalyzed by a surface Brönsted acid site were then examined and the isomerization pathway was found to be more favorable than dehydration at the anomeric site on a surface acid site. These mechanistic insights provide an important guide for the catalyst design/selection of the reaction media for glucose dehydration.
Co-reporter:Xufeng Lin, Yanhong Lv, Yuanyuan Qu, Guodong Zhang, Yanyan Xi, David L. Phillips and Chenguang Liu
Physical Chemistry Chemical Physics 2013 - vol. 15(Issue 46) pp:NaN20133-20133
Publication Date(Web):2013/10/10
DOI:10.1039/C3CP53915F
The catalytic activity and the acrolein selectivity for liquid phase glycerol dehydration on β zeolites (HNa-β-k) were found to be dependent on the reaction temperature as well as on the amount of acid sites on the zeolites. An increase in the reaction temperature favors the acrolein selectivity. The acrolein selectivity increases with the Na+/H+ ratio and the glycerol conversion decreases with it so that a maximum acrolein yield is obtained when a certain amount of acidic sites are replaced by non-active Na+ sites. The computational results indicate that 3-hydoxylpropanal (HPA) is an important intermediate that determines the final product selectivity. The relative rates of the different reaction pathways for HPA can be affected by the amount of water molecules involved in its homogeneous reaction. Based on the reaction mechanism proposed, it was hypothesized that smaller pores reduce activity but increase selectivity to acrolein, and results of the H-MFI zeolite were consistent with this hypothesis. Our work provides important insight into the overall landscape of the reaction mechanism and can be used to help design reaction systems that have good acrolein selectivity for the liquid phase glycerol dehydration reactions.

![Ruthenium, carbonyl[5,10,15,20-tetrakis(2,3,4,5,6-pentafluorophenyl)-21H,23H-porphinato(2-)-κN21,κN22,κN23,κN24]-, (SP-5-31)-](/data/chemimg/132000/171899-61-9.png)
![Ruthenium, carbonyl[5,10,15,20-tetrakis(2,3,4,5,6-pentafluorophenyl)-21H,23H-porphinato(2-)-κN21,κN22,κN23,κN24]-, (SP-5-31)-](/data/chemimg/132000/171899-61-9_b.png)