Co-reporter:Adela Jing Li, Ziye Sang, Chi-Hang Chow, Japhet Cheuk-Fung Law, Ying Guo, K.S.-Y. Leung
Journal of Hazardous Materials 2017 Volume 337(Volume 337) pp:
Publication Date(Web):5 September 2017
DOI:10.1016/j.jhazmat.2017.04.067
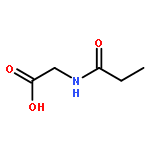
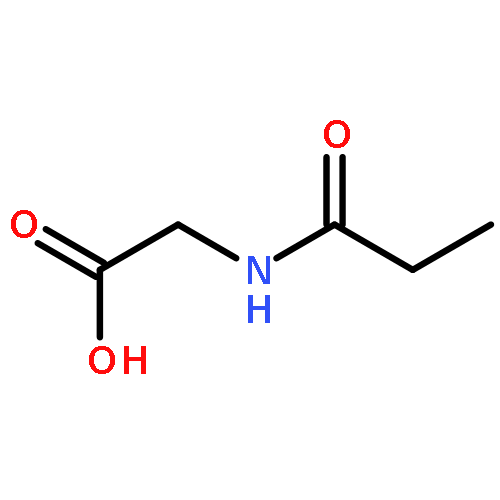
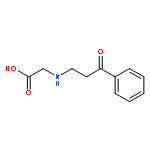
•Et-PABA was not detected in any water sample.•First study of the photocatalysis of Et-PABA.•Eleven transformation products were identified with seven new compounds reported.•Transformation products were less toxic than Et-PABA against Vibrio fischeri.•Photocatalysis may be a valid resolution for Et-PABA removal.Ethyl-4-aminobenzoate (Et-PABA) is currently used as a substitute for 4-aminobenzoate (PABA) in sunscreens and anesthetic ointments. Despite its widespread use and hydrophilicity, Et-PABA has never been found in environmental waters. This study, probed the occurrence of Et-PABA in both seawater and drinking water sources in Hong Kong, and evaluated its transformation products (TPs) and environmental fate via cumulative potency and photocatalytic profile analyses. Another 11 UV filters used in skin-care products were also studied. Et-PABA was not detected in any water sample. Four other UV filters were dominant at ng/L level in both seawater and drinking water sources. UHPLC-QTOF-MS was used to elucidate the structure of TPs. With high resolution accurate mass data and fragment rationalization, 11 Et-PABA TPs were characterized, including seven intermediates firstly proposed as TPs; two compounds were reported for the first time. It is proposed that photocatalysis induces transformation pathways of (de)hydroxylation, demethylation and molecular rearrangement. Luminescent bacteria tests showed decreasing toxicity with increasing irradiation of Et-PABA, suggesting that irradiation TPs are less toxic than the parent compound. Transformation of Et-PABA appears to explain why Et-PABA has not been detected in the natural environment.Download high-res image (75KB)Download full-size image
Co-reporter:Ammaiyappan Selvam;Katrina Kwok;Yumei Chen
Environmental Science and Pollution Research 2017 Volume 24( Issue 10) pp:9058-9066
Publication Date(Web):2017 April
DOI:10.1007/s11356-016-6338-5
Occurrence of 10 antibiotics in the Yuen Long (YLR), Kam Tin (KTR), and Shing Mun (SMR) rivers of Hong Kong and possible influence of livestock activities on the concentrations of antibiotics were investigated. Tetracycline (30–497 ng/L), sulfadiazine (2–80 ng/L), sulfamethoxazole (2–152 ng/L), ofloxacin (5–227 ng/L), and erythromycin (1–315 ng/L) were detected in all the three rivers; chlortetracycline (23–227 ng/L), oxytetracycline (7–104 ng/L), ciprofloxacin (12–68 ng/L), and roxithromycin (1–105 ng/L) were detected in YLR and KTR, whereas norfloxacin (3–34 ng/L) was detected in KTR only. Significant correlation between livestock population and antibiotic contamination was observed in YLR only, indicating the influences of other sources in KTR and SMR. Among the antibiotics, significant correlation was observed between tetracyclines and sulfonamides indicating the major influence of livestock farms, whereas tetracyclines/sulfonamides were negatively correlated with fluoroquinolones/macrolides implying the differential origin of the latter class of antibiotics. Water quality of KTR and YLR were highly influenced by the non-point source pollutions, while of SMR was relatively good. Particularly, Escherichia coli populations of the YLR and KTR were 3–4 logs higher than those of the SMR indicating the involvement of livestock farms and sewerages. Good correlation between tetracyclines (TCs)/sulfonamides (SAs) and number of livestock farms and a negative correlation between TCs/SAs and fluoroquinolones (FQs)/macrolides (MLs) could be used as an indicator to trace the possible source of pollution.
Co-reporter:Tsz-Shan Lum and Kelvin Sze-Yin Leung
Journal of Analytical Atomic Spectrometry 2016 vol. 31(Issue 5) pp:1078-1088
Publication Date(Web):09 Mar 2016
DOI:10.1039/C5JA00497G
ICP-MS serves as a powerful elemental detection method for accurate and precise analysis, especially for quantification purposes. However, interference from spectral overlap with isobaric and polyatomic species has long hindered accurate analysis. The source of interference can come from the sample matrix, solvent medium or plasma gas which makes the problem more complicated. Simple sample dilution may help to lower the background interference on some occasions, but in some cases, additional sample preparation or even advanced instrumentation is required to alleviate the problem. Although there are various solutions to the problem, spectral interference is mainly matrix dependent and analysis specific. Special attention should be paid when performing the analysis to avoid the formation of new interfering species, potential loss or contamination of target analytes. With the help of the developed approaches, analysis can be done with high accuracy and good precision for elemental determination and isotopic analysis. This paper reviews current and emerging strategies for reducing spectral interference in ICP-MS analysis. The strategies to be reviewed are: non-instrumental approaches, alternative methods of sample introduction, instrumental modification, high resolution ICP-MS and emerging methods (interference standard method and tandem ICP-MS). Most of the cited papers were published during or after 2005.
Co-reporter:Tsz-Shan Lum, Yeuk-Ki Tsoi, Patrick Ying-Kit Yue, Kelvin Sze-Yin Leung
Microchemical Journal 2016 Volume 127() pp:94-101
Publication Date(Web):July 2016
DOI:10.1016/j.microc.2016.02.013
•Deposition of Gd and V in mice whisker after drug administration was recorded by LA-ICP-MS.•Deposition behaviors of different complexes and administered dosages were observed.•The potential of LA-ICP-MS for hair (whisker) profiling in therapeutic drug monitoring was reported.As modern drugs become increasingly potent and specific, therapeutic drug monitoring (TDM) becomes ever more important. Theoretically, hair is the best tissue to sample for TDM because it can be sampled noninvasively, any time, and records body chemistry over time. In practice, it is not being used for lack of a good analytical technique. In this study we demonstrate that LA-ICP-MS profiling can be used to trace both deposition and elimination of three metallodrugs in mouse whiskers. Two gadolinium-based and one vanadium-based complexes were administered to ICR mice. After optimization of ablation modes and laser conditions, LA-ICP-MS profiling was performed by line scan with laser spot size of 100 μm along the whiskers of the mice. For gadolinium complexes, gadodiamide and gadoteric acid, different deposition behaviors were recorded, including degree of deposition as a function of dosage levels. Furthermore, the deposition of Gd for high dose (0.1 mmol/kg) was found to be three times that for low dose (0.01 mmol/kg). For the vanadium complex, vanadyl acetylacetonate, asymmetric peaks were observed in the whiskers' profiles at around 1400 and 1900 μm from the follicle, giving valuable information about the absorption/elimination of the drug, and by extension, its metabolism. To the best of our knowledge, this is the first example of LA-ICP-MS bioimaging being used to record the deposition of metallodrugs administered in different dosages. Our results suggest LA-ICP-MS has enormous potential for application in the field of therapeutic drug biomonitoring, particularly using hair for sampling purposes.
Co-reporter:Tsz-Shan Lum, Cheuk-Lam Ho, Yeuk-Ki Tsoi, Chi-Ho Siu, Patrick Ying-Kit Yue, Wai-Yeung Wong, Kelvin Sze-Yin Leung
International Journal of Mass Spectrometry 2016 Volume 404() pp:40-47
Publication Date(Web):20 June 2016
DOI:10.1016/j.ijms.2016.05.005
•Biodistribution and relative bioavailability of platinum complexes were studied.•LA-ICP-MS imaging of mice tissue sections of platinum complexes was performed.•Differential deposition behaviors of platinum complexes were studied.•The use of LA-ICP-MS for molecular design in drug development was reported.In the last decade, the number of applications of laser ablation-inductively coupled plasma-mass spectrometry (LA-ICP-MS) in bioimaging has been increasing. To further extend its capability in drug development, in this study, we used this bioimaging tool to visualize deposition behavior of chemically different metallo-complexes, including two that differed only subtly in structure. A systematic approach with in vitro study and ICP-MS elemental analysis were included to supplement the findings.Two chemically distinct platinum complexes (Pt-1 and Pt-2) were synthesized; their potencies were investigated first on different healthy and cancer cell lines and then on mice. The commercialized anti-cancer drug, cis-platin was used as a reference. In animal studies, the mice were given 4 mg/kg of the complex via intraperitoneal injection and sacrificed 24 h post-injection. ICP-MS analysis was performed on six organs to study the bioavailability of the complexes. Pt accumulated in the organs, from greatest to least, from liver > kidney > lung > testis > heart > brain. Among the complexes, the bioavailability showed a general trend of Pt–2 > cis-platin > Pt-1.In LA-ICP-MS bioimaging analysis of paraffin-embedded mouse liver and kidney sections, a spatial resolution of 50 μm was adopted. Deposition trends matched the findings obtained in elemental analysis. In addition, differential deposition of Pt was observed in the kidney sections of mice treated with different complexes. The LA biomaps successfully distinguished the differential distribution of two structurally similar platinum complexes (Pt-1 and Pt-2) in mice liver and kidney. This information is of particular interest because these two Pt-based complexes can potentially be developed into anti-cancer drugs. This work demonstrates that LA-ICP-MS imaging is a valuable tool for therapeutic drug development, especially in assisting molecular modification of metal-containing complexes.
Co-reporter:Tsz-Shan Lum, Yeuk-Ki Tsoi and Kelvin Sze-Yin Leung
Journal of Analytical Atomic Spectrometry 2014 vol. 29(Issue 2) pp:234-241
Publication Date(Web):28 Nov 2013
DOI:10.1039/C3JA50316J
Sample preconcentration is important for metal analysis in complex clinical matrices due to the low concentration of analytes and high salt content. The limited amount of sample available also increases the challenges of extraction and determination of the analytes. In spite of the increase in sensitivity of instruments, such ultra-trace levels of the metals are still difficult to determine accurately. Researchers have adopted various strategies to improve the efficiency and selectivity of the extraction. The use of chemicals and time for analysis can also be reduced by scaling down the analysis to the micro-level. This review discusses four aspects of the recent development in metal preconcentration in clinical samples, namely the use of ionic liquids (ILs) in extraction, sorption by nanomaterials, preconcentration using surfactants, and automation. The publications reviewed cover the period between 2008 to late 2013.
Co-reporter:Kelvin Sze-Yin Leung;Bonnie Mei-Wah Fong
Analytical and Bioanalytical Chemistry 2014 Volume 406( Issue 9-10) pp:2289-2301
Publication Date(Web):2014 April
DOI:10.1007/s00216-013-7542-5
Liquid chromatography–tandem mass spectrometry (LC–MS/MS) has been increasingly used in routine clinical laboratories during the last two decades. The high specificity, sensitivity, and multi-analyte potential make it an ideal alternative to immunoassays or conventional high-performance liquid chromatography (HPLC). It also provides higher throughput than gas chromatography–mass spectrometry (GC–MS). LC–MS/MS also offers higher flexibility than immunoassays because LC–MS/MS assays are typically developed in-house. In addition, abundant information can be obtained from a single LC–MS/MS run which can produce a large amount of quantitative or qualitative data. In this review, typical LC–MS/MS clinical applications are presented, personal experiences are shared, and strengths and weakness are discussed. It is foreseeable that LC–MS/MS will become a key instrument in routine clinical laboratories.
Co-reporter:Yee-Man Ho, Yeuk-Ki Tsoi, Kelvin Sze-Yin Leung
Analytica Chimica Acta 2013 Volume 775() pp:58-66
Publication Date(Web):2 May 2013
DOI:10.1016/j.aca.2013.02.043
•A rapid, high throughput single DLLME analysis for 10 organophosphorus pesticides in foods or herbs.•Optimization dealt with unique chemistry in 3 sample types: fruit, vegetable, dried herb.•Rapid, micro-scale sample clean-up completes <9 min using about 1 mL solvent.•100-fold enrichment achieved sub-ppt LOD for trace residues based on 0.1 g sample.•High throughput method effectively deals with a wide array of routine samples.The article describes a simple sample pretreatment procedure for the analysis of ten organophosphorus pesticides using dispersive liquid–liquid microextraction (DLLME) followed by gas chromatography–mass spectrometry (GC–MS) in three distinctively different types of matrices: fresh fruits, fresh vegetables and dried herbs. The method was carefully developed, focusing on the chemistry of various dispersive solvents, to achieve simultaneous, comprehensive extraction and preconcentration in a great span of selected matrices. According to matrix-matched validation study, the set of optimized DLLME conditions has been proven robust to determine target OPPs within a wide linear range from 0.1 to 1000 μg L−1. With limited usage of organic extractants, remarkable enrichment factors up to 100-fold were obtained, enabling ultra-trace pesticide quantification down to sub-ppt levels at 0.12–4.92 ng kg−1. Practical application of the method was illustrated by quantitative recovery (70–119%) and good precision (2.6–10% R.S.D.) in a representative range of three fruits and four vegetable commodities featured by the CODEX Alimentarius classification as well as their unique matrix compositions. A careful selection of dried herbs was further classified based on their morphological structures to validate analytical ruggedness of the method. Compared with existing methods for food analysis vis-à-vis OPPs, the present method is superior in terms of high sample throughput, minimal solvent consumption, and small sample size requirement. An additional, significant aspect of this universal DLLME method is that it models sample pretreatment methods with wide coverage of analytical matrices that are more effective, more comprehensive, and more flexible than those currently being used.Graphical abstract
Co-reporter:Yee-Man Ho;Yeuk-Ki Tsoi
Journal of Separation Science 2013 Volume 36( Issue 23) pp:3791-3798
Publication Date(Web):
DOI:10.1002/jssc.201300807
This paper describes an innovation of dispersive liquid–liquid microextraction enabling multiple-component analysis of eight high-priority food contaminants in two chemically distinctive families: Sudan dyes and phthalate plasticizers. To provide convenient sample handling for solid and solid-containing matrices, a modified dispersive liquid–liquid microextraction procedure used an extractant precoated frit to perform simultaneous filtration, solvent mixing, and phase dispersion in one simple step. A binary ionic liquid extractant system was carefully tuned to deliver high quality analysis based only on affordable LC with diode array detector instrumentation. The method is comprehensively validated for robust quantification with good precision (6.9–9.8% RSD) in a linear 2–1000 μg/L range. Having accomplished enrichment factors up to 451, the treatment enables sensitive detection at 0.09–1.01 μg/L levels. Analysis of six high-risk solid condiments and sauces further verified its practical applicability within a 70–120% recovery range. Compared to other approaches, the current dispersive liquid–liquid microextraction treatment offers major advantages in terms of minimal solvent (1.5 mL) and sample (0.1 g) consumption, ultra-high analytical throughput (6 min), and the ability to handle complex solid matrices. The idea of performing simultaneous analysis for multiple contaminants presented here fosters a more effective mode of operation in food control routines.
Co-reporter:PatrickYing-Kit Yue;Yi-Yi Wong;KayYuen-Ki Wong;Yeuk-Ki Tsoi
Chinese Medicine 2013 Volume 8( Issue 1) pp:
Publication Date(Web):2013 December
DOI:10.1186/1749-8546-8-21
Antrodia cinnamomea (AC) is an endemic mushroom species of Taiwan, and has been demonstrated to possess diverse biological and pharmacological activities, such as anti-hypertension, anti-hyperlipidemia, anti-inflammation, anti-oxidation, anti-tumor, and immunomodulation. This review focuses on the inhibitory effects of AC on hepatitis, hepatocarcinoma, and alcohol-induced liver diseases (e.g., fatty liver, fibrosis). The relevant biochemical and molecular mechanisms are addressed. Overall, this review summarizes the hepatoprotective activities in vitro and in vivo. However, there is no doubt that human and clinical trials are still limited, and further studies are required for the development of AC-related products.
Co-reporter:Bonnie Mei-Wah Fong, Sidney Tam, Kelvin Sze-Yin Leung
Talanta 2013 Volume 116() pp:115-121
Publication Date(Web):15 November 2013
DOI:10.1016/j.talanta.2013.04.075
•APCI as the ionization source for quantification of cholesterol sulfate in plasma.•MS/MS detection enables a wider dynamic range analysis.•We establish the first pediatric reference interval in local Chinese.•No significant difference between autistic and non-autistic children.Cholesterol sulfate (CS) has various biological functions. Previously, plasma CS was measured primarily as a means to diagnose X-linked ichthyosis; however, a recent hypothesis suggests that CS deficiency might be related to autism. As such, an assay capable of measuring both very high (in the case of X-linked ichthyosis) and very low (in the case of autism) plasma CS levels is required. Here we describe a novel LC–APCI-MS/MS method for the determination of CS in human plasma, and we propose normal CS ranges for children, based on studies of a local population of normal Chinese children between the ages of 2 and 10. In addition, we have used this method to measure plasma CS in autistic children. CS was isolated by solid-phase extraction, and quantified by isotope-dilution LC–APCI-MS/MS in negative ion mode monitoring 465.3>97.1 m/z (CS) and 472.3>97.1 m/z (CS-d7). Mean recovery of the assay ranged from 88.1 to 112.7%; within- and between-run imprecisions have CVs less than 7.2 and 8.1%, respectively. The assay was linear up to at least 100 µmol L−1. The reference interval of plasma CS in males (range: 1.16–4.23 µmol L−1) was found to be higher than in females (range: 0.86–3.20 µmol L−1). Comparison of normal and autistic children showed no statistically significant difference in the plasma CS level. In conclusion, a robust LC–APCI-MS/MS method for plasma CS was developed, and a pediatric reference interval was derived from applying the method to normal and autistic children.
Co-reporter:Karen Ka-Wing Chung, Sammy Pak-Lam Chen, Sau-Wah Ng, Tony Wing-Lai Mak, Kelvin Sze-Yin Leung
Talanta 2012 Volume 97() pp:491-498
Publication Date(Web):15 August 2012
DOI:10.1016/j.talanta.2012.05.004
Aconite poisoning is one of the most serious types of herb-related medical emergencies. In Hong Kong, many if not most of these poisoning cases are due to confusion in herbal species; that is, the wrong herbs are used in prescriptions. Such human errors, while inevitable perhaps, can be serious, and sometimes fatal. The chemical components responsible for aconite poisoning are yunaconitine and crassicauline A. In the present study, a rapid and sensitive method for the screening and quantification of yunaconitine and crassicauline A in human serum, using LC–MS/MS, was developed and validated. Methyllycaconitine was chosen as the internal standard. The limit of detection (LOD) of yunaconitine and crassicauline A were found to be 0.022 and 0.021 ng/mL, respectively. The limit of quantification (LOQ) was 0.1 ng/mL for both yunaconitine and crassicauline A. The recovery of yunaconitine and crassicauline A ranged from 78.6% to 84.9% and 78.3% to 87.2%, respectively. The matrix effect of yunaconitine and crassicauline A ranged from 110.0% to 130.4% and 121.2 to 130.0%, respectively. Both yunaconitine and crassicauline A were stable in serum for at least 3 months at −20 °C, and the extracts were stable for at least 7 days. For clinical applications, serum samples of two patients confirmed to have had aconite herbs poisoning in 2008 were quantified using the developed method. The result showed that this method can be utilized in clinical routine applications. This screening method expedites the diagnosis in cases of suspected aconite poisoning, thus enabling doctors to treat the condition more quickly and effectively.Highlights► New LC–MS/MS method for yunaconitine and crassicauline A detection in blood matrix. ► High method specificity can reduce endogenous and exogenous interference. ► Results showed that this method can be utilized in clinical routine applications.
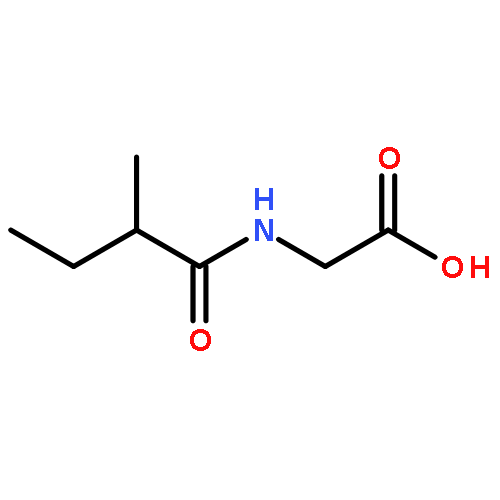
Co-reporter:Bonnie Mei-Wah Fong, Sidney Tam, Kelvin Sze-Yin Leung
Talanta 2012 Volume 88() pp:193-200
Publication Date(Web):15 January 2012
DOI:10.1016/j.talanta.2011.10.031
Urinary organic acids, plasma amino acids and acylcarnitine profile analyses are the main tools used to diagnose inborn errors of metabolisms (IEMs). However, without metabolic decompensation, these parameters are often not helpful. On the other hand, in cases of IEM, acylglycines are consistently raised even when patients appear to be in remission. This study aims to set-up a simple liquid chromatography–electrospray ionization-tandem mass spectrometry (LC–ESI-MS/MS) method for the determination of urine acylglycines, complementary to organic acid and acylcarnitine profiles, for the diagnosis of IEM. In addition, local reference intervals for various acylglycines are established by using this method. Acylglycines were isolated by solid-phase extraction, derivatized with n-butanol, separated by HPLC, and detected by ESI-MS/MS. Acylglycines were quantified with deuterated internal standards. Mean recoveries of acylglycines ranged from 90.2 to 109.3%. Within- and between-run imprecisions for all acylglycines have CVs less than 10%. Linear regression coefficients were greater than 0.99. Reference intervals were established according to CLSI guidelines by analyzing 204 samples from apparently healthy individuals less than 18 years of age. The distributions of AG in the “normal” urine were skewed towards the right. After log transformation, all the results were normally distributed. Partitioning into age group reference intervals was not indicated, according to the Harris and Boyd approach. In this context, a single reference interval for each acylglycine could be used. This method of urine acylglycines analysis is a powerful diagnostic tool, complementary to urine organic acids and plasma acylcarnitine profiling, for detecting certain inborn errors of metabolism.Highlights► Acylglycines (AG) are consistently raised in IEM patients even in remission. ► The first LC–MS/MS method for AG determination, with AG isomers, was developed. ► Reference intervals for acylglycines in local pediatric Chinese were established. ► It is the first data set of AG reference interval in Chinese pediatric population.
Co-reporter:Yeuk-Ki Tsoi, Yee-Man Ho, Kelvin Sze-Yin Leung
Talanta 2012 Volume 89() pp:162-168
Publication Date(Web):30 January 2012
DOI:10.1016/j.talanta.2011.12.007
A novel arsenic-ion imprinted polymer (As-IIP) was firstly synthesized for the separation and recovery of trace elemental As from environmental water samples. Polymers prepared from bifunctional monomers with intrinsic metal-binding capability are a platform for tailoring ion-selectivity via imprinting moiety–template interaction, without complex formation and ligand immobilization. In the present study, As-IIPs based on 1-vinylimidazole, 4-vinylpyridine and styrene were designed to investigate the imprinting mechanism in relation to their structural and functional properties. In terms of selectivity as well as imprinting effects compared with the non-imprinted polymer (NIP), 1-vinylimidazole-based As-IIP exhibited superior analyte recognition for As ion among 23 competing elements, with a 25-fold enhancement in the practical dynamic and static adsorption capacity range (0.048–4.925 μmol g−1). The robust As-IIP sorbent features good reusability up to 20 cycles and a wide working pH 5–7 for a firstly reported solid-phase extraction (SPE) application. As a result of selective sample clean-up, As-IIP-SPE offered limits of detection (LOD) and quantification (LOQ) down to 0.025 and 0.083 μmol L−1, respectively, for environmental sample analysis using inductively coupled plasma-mass spectrometry.Highlights► As-ion imprinted polymer for solid-phase extraction. ► Imprinting mechanism and recognition ability with three bifunctional monomers. ► Superior recognition and selectivity of As-IIP among competing elements.
Co-reporter:Yeuk-Ki Tsoi, Kelvin Sze-Yin Leung
Journal of Chromatography A 2011 Volume 1218(Issue 16) pp:2160-2164
Publication Date(Web):22 April 2011
DOI:10.1016/j.chroma.2011.02.016
This paper describes a novel application of tetrabutylammonium hydroxide-modified activated carbon (AC-TBAH) to the speciation of ultra-trace Se(IV) and Se(VI) using LC-ICP-DRC-MS. The anion exchange functionality was immobilized onto the AC surface enables selective preconcentration of inorganic Se anions in a wide range of working pHs. Simultaneous retention and elution of both analytes, followed by subsequent analysis with LC-ICP-DRC-MS, allows to accomplish speciation analysis in natural samples without complicated redox pre-treatment. The laboratory-made column of immobilized AC (0.4 g of sorbent packed in a 6 mL syringe barrel) has achieved analyte enrichment factors of 76 and 93, respectively, for Se(IV) and Se(VI), thus proving its superior preconcentration efficiency and selectivity over common AC. The considerable enhancement in sensitivity achieved by using the preconcentration column has improved the method's detection limits to 1.9–2.2 ng L−1, which is a 100-fold improvement compared with direct injection. The analyte recoveries from heavily polluted river matrix were between 95.3 and 107.7% with less than 5.0% RSD. The robustness of the preconcentration and speciation method was validated by analysis of natural waters collected from rivers and reservoirs in Hong Kong. The modified AC material is hence presented as a low-cost yet robust substitute for conventional anion exchange resins for routine applications.
Co-reporter:Bonnie Mei-Wah Fong, Sidney Tam, Sik-Hon Tsui, Kelvin Sze-Yin Leung
Talanta 2011 Volume 83(Issue 3) pp:1030-1036
Publication Date(Web):15 January 2011
DOI:10.1016/j.talanta.2010.11.014
A sensitive analytical method for the determination of tetrodotoxin (TTX) in urine and plasma matrices was developed using double solid phase extraction (C18 and hydrophilic interaction liquid chromatography) and subsequent analysis by HPLC coupled with tandem mass spectrometry. The double SPE sample cleanup efficiently reduced matrix and ion suppression effects. Together with the use of ion pair reagent in the mobile phase, isocratic elution became possible which enabled a shorter analysis time of 5.5 min per sample. The assay results were linear up to 500 ng mL−1 for urine and 20 ng mL−1 for plasma. The limit of detection and limit of quantification were 0.13 ng mL−1 and 2.5 ng mL−1, respectively, for both biological matrices. Recoveries were in the range of 75–81%. To eliminate the effect of dehydration and variations in urinary output, urinary creatinine-adjustment was made. TTX was quantified in eight urine samples and seven plasma samples from eight patients suspected of having TTX poisoning. TTX was detected in all urine samples, with concentrations ranging from 17.6 to 460.5 ng mL−1, but was not detected in any of the plasma samples. The creatinine-adjusted TTX concentration in urine (ranging from 7.4 to 41.1 ng μmol−1 creatinine) correlated well with the degree of poisoning as observed from clinical symptoms.
Co-reporter:Yeuk-Ki Tsoi, Sidney Tam and Kelvin Sze-Yin Leung
Journal of Analytical Atomic Spectrometry 2010 vol. 25(Issue 11) pp:1758-1762
Publication Date(Web):03 Aug 2010
DOI:10.1039/C0JA00024H
Solid-phase microextraction coupled to liquid chromatography-inductively coupled plasma-mass spectrometry (SPME-LC-ICP-MS) is presented as a new approach for the extraction and speciation of methylmercury (MeHg) and ethylmercury (EtHg) in a complex urine matrix. Despite the fact that urine is one of the most easily accessible sources of biological specimens for clinical testing, reports of mercury speciation methods are limited. Without any tedious sample pretreatments or derivatization procedures, headspace SPME offers a clean and convenient one-step extraction followed by direct desorption into HPLC. In this work, the fibre was first exposed to the sample headspace for a 45 min extraction, followed by a 5 min desorption. Experimental parameters including extraction time and temperature, sodium chloride salt concentration, agitation rate and desorption time were optimized to achieve detection limits down to 0.06 μg L−1 for both organomercury species. Sample recoveries in spiked urine matrix ranged from 81.7–109.5%. The accuracy of the method was further validated using a certified reference material (TORT-2) and applied in the organomercury speciation of urine samples collected from patients with suspected mercury poisoning.
Co-reporter:Yeuk-Ki Tsoi and Kelvin Sze-Yin Leung
Journal of Analytical Atomic Spectrometry 2010 vol. 25(Issue 6) pp:880-885
Publication Date(Web):17 Mar 2010
DOI:10.1039/B923343A
A new multi-elemental speciation method was developed for seven species of three elements namely As(III), As(VI), MMA, Cr(III), Cr(VI), Se(IV) and Se(VI), using liquid chromatography-inductively coupled plasma-mass spectrometry. The use of dynamic reaction cell technique in removing polyatomic interference dramatically extended the detection sensitivity for all seven species with methane as the reaction gas. The developed condition offered low detection limits in ranges of 0.12–0.44 μg L−1, 0.24–0.29 μg L−1 and 0.26 μg L−1 for species of As, Cr and Se, respectively. For concise quantitative speciation, a controlled chelation condition for Cr species was optimized to minimize species interconversion. In the developed method, the seven species were baseline-separated in an 11-minute elution on an anion exchange column using NH4NO3/NH4H2PO4 as the mobile phase at pH 6. This efficient, high-throughput and sensitive method was fully validated and applied in quantitative speciation of river water samples collected from the Shing Mun River of Hong Kong. Most of the species were found as the toxic forms of the elements studied.
Co-reporter:Chung-Hin Chui, Raymond Siu-Ming Wong, Roberto Gambari, Gregory Yin-Ming Cheng, Marcus Chun-Wah Yuen, Kit-Wah Chan, See-Wai Tong, Fung-Yi Lau, Paul Bo-San Lai, Kim-Hung Lam, Cheuk-Lam Ho, Chi-Wai Kan, Kelvin Sze-Yin Leung, Wai-Yeung Wong
Bioorganic & Medicinal Chemistry 2009 Volume 17(Issue 23) pp:7872-7877
Publication Date(Web):1 December 2009
DOI:10.1016/j.bmc.2009.10.034
A list of diethynylfluorenes and their gold(I) derivatives have been studied for their antitumor activity as a function of their structure–activity relationships. End-capping the fluoren-9-one unit with gold(I) moieties could significantly strengthen the cytotoxic activity in vitro on three human cancer cell lines with induction of reactive oxygen species generation on Hep3B hepatocellular carcinoma cells and exhibit attractive antitumor activity from in vivo nude mice Hep3B xenograft model with limited adverse effects on vital organs including liver and kidney.End-capping the diethynylfluorene unit with gold(I) moieties could significantly strengthen its cytotoxic activity on human cancer cells. Further study of the gold(I) derivative demonstrated its in vivo antitumor activity using Hep3B human hepatocellular carcinoma model with limited adverse effects on vital organs including liver and kidney.
Co-reporter:Dan Chen;Sy Shuo Zhao
Journal of Separation Science 2009 Volume 32( Issue 17) pp:2892-2902
Publication Date(Web):
DOI:10.1002/jssc.200900275
Abstract
Our present study constitutes the first successful attempt to employ eight distinctive chemical groups of compounds for the quality evaluation of a complex traditional Chinese herbal medicine (TCHM) material: BaZhen YiMu (BZYM). Due to the complexity of its matrix, which is composed of nine different herbal ingredients, five representative chemical groups encompassing representative bioactive markers were initially chosen as targets for the quality assessment of this preparation. Furthermore, with the aid of LC-ESI-MS, three additional chemical groups were characterized. In summary, a total of nineteen markers belonging to eight different chemical groups were selectively displayed in the chromatographic fingerprint of BZYM preparation. With this fingerprint, the overall quality of any BZYM preparation can be comprehensively authenticated. The chromatographic separation was performed on an HP C18 AQ column with a gradient elution of ACN and aqueous solution containing 0.1% phosphoric acid at the optimal detection wavelength of 230 nm. The established method was rigorously validated with respect to the ICH guidelines and represents the most extensive and facile HPLC quality control technique for this formulation. Compared with the conventional method of using a single or only a few markers of the same chemical group, this technique provides a new dimension for TCHM quality control.
Co-reporter:Kelvin Chan;Sandy Shuo Zhao
Chinese Medicine 2009 Volume 4( Issue 1) pp:
Publication Date(Web):2009 December
DOI:10.1186/1749-8546-4-18
This article provides an overview on the regulations of Chinese medicinal materials (CMMs) in various countries and regions. Harmonization of CMM monographs would provide standards for the quality control of CMM products and play an important role in the modernization and globalization of Chinese medicine. A harmonized regulatory system would improve the quality of CMMs thereby ensuring the safety of the products and assisting Chinese medicine practitioners in their practice. The fast growing demand worldwide for traditional medicines calls for harmonized monographic standards to safeguard the safety and quality of CMM products.
Co-reporter:Kelvin Sze-Yin Leung;Kelvin Chan;Alan Bensoussan;Mary J. Munroe
Phytochemical Analysis 2007 Volume 18(Issue 2) pp:146-150
Publication Date(Web):5 JAN 2007
DOI:10.1002/pca.962
An HPLC-MS method using an atmospheric pressure chemical ionisation (APCI) source has been developed to assist in the differentiation of three ginseng species: Panax quinquefolium (American ginseng), P. ginseng (Chinese ginseng) and P. notoginseng (sanqi) species. The differentiation method relies on the identification of ginsenosides Rf and F11, and notoginsenoside R1. R1 is observed in both P. notoginseng and Chinese ginseng, whilst F11 is found exclusively in the American species. The presence of these compounds permits the definitive identification of the species to be made. The APCI ionisation source has been employed to tackle the matrix interference in analysing Chinese medicinal materials and to minimise the associated matrix effects that are commonly encountered with other ionisation modes. Moreover, the method allows direct interface to conventional HPLC systems. More importantly, chemical reference standards of ginsenosides are not required in this method. This technique provides an alternative approach to analysing high molecular weight polar compounds that typically encountered in complex matrices of Chinese medicinal materials. Copyright © 2006 John Wiley & Sons, Ltd.
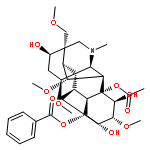
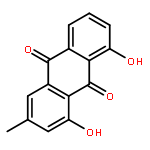
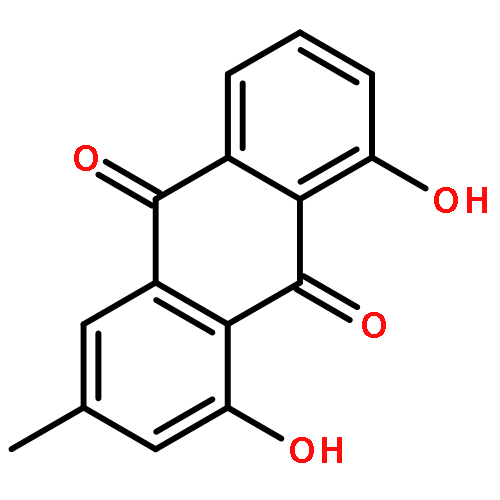
Co-reporter:Tao Yi;Kelvin S. Y. Leung;Guang-Hua Lu;Kelvin Chan;Hao Zhang
Phytochemical Analysis 2007 Volume 18(Issue 3) pp:181-187
Publication Date(Web):29 MAR 2007
DOI:10.1002/pca.963
An HPLC–PAD–MS method has been developed for the qualitative and quantitative analysis of the major chemical constituents in Polygonum multiflorum Thunb. Chromatographic separation was conducted on an Alltima C18 column using water:acetonitrile:acetic acid as the mobile phase. Altogether nine compounds of various classes, including stilbene glucosides, anthraquinone glucosides and anthraquinone derivates, were identified by online ESI/MS. Their identities were ascertained by comparison with data derived from the literature and/or standard compounds. Five components were quantified by HPLC–PAD and the method was fully validated. All the linear regressions were acquired with R2 > 0.99 and the quantification limits (with a signal-to-noise ratio of 3) ranged from 0.63 and 1.57 ng. Repeatability was evaluated by intra- and inter-day assays and RSD value was within 2.47%. Recovery studies for the quantified compounds were found to be within the range 96.32–102.53% with RSD less than 2.35%. The overall procedure is rapid and reproducible and is considered suitable for qualitative and quantitative analysis of a large number of samples. Copyright © 2007 John Wiley & Sons, Ltd.
Co-reporter:Lu-Lu Jia, Xiang-Ying Lou, Ying Guo, Kelvin Sze-Yin Leung, Eddy Y. Zeng
Environment International (January 2017) Volume 98() pp:137-142
Publication Date(Web):1 January 2017
DOI:10.1016/j.envint.2016.10.025
•None over-the-counter medicine in the present study was free of phthalates.•Medicines are not the main source for Chinese population exposure to phthalates.•Health risk of phthalates is low by taking over-the-counter medicine.•Phthalates in patient will decrease to same levels as those before taking medicines shortly.Food, air, personal care products and indoor dust have been recognized as the main routes of exposure to phthalates in Chinese population, but other sources may have been overlooked, e.g., medicines. To fill the knowledge gap, phthalate esters were measured in 96 over-the-counter medicines made in China, including selected 71 Chinese patented medicines and 25 western medicines. It was found that none of the medicines was free of phthalates. The mean concentrations of individual phthalates ranged from 0.001 μg/g (dicyclohexyl phthalate) to 5.85 μg/g (diethyl phthalate). Among 9 targeted phthalates, di-n-butyl phthalate was the dominant congener, accounting for > 65% of the total phthalates in all medicine samples, followed by di-(2-ethylhexyl) phthalate and diethyl phthalate. Phthalates in medicines appeared to derive from gastroresistant film coatings, plastic packing materials or phthalate contaminated rural herbal plants (especially for Chinese patented medicines). Daily human exposure to phthalates was estimated for local patients for one treatment cycle (e.g., one week) based on suggested consumption dosage and phthalate concentrations. Almost all exposure levels were below the guidelines suggested by the United States Environmental Protection Agency or European Food Safety Authority, indicating low health risk with phthalates from consumption of the medicines. In addition, concentration levels of phthalates in patients would increase upon administration but are expected to decrease to the same values as those in patients before they took medicines in several days. Because the number of medicine samples was limited and the concentrations of phthalates varied in a large range, further investigations are needed to acquire more data for better assessment of human health effects for Chinese population.Capsule: Distribution of phthalate esters in over-the-counter medicines and related exposure for Chinese population are examined
Co-reporter:Adela Jing Li, Pengran Wu, Japhet Cheuk-Fung Law, Chi-Hang Chow, Cristina Postigo, Ying Guo, Kelvin Sze-Yin Leung
Water Research (15 June 2017) Volume 117() pp:157-166
Publication Date(Web):15 June 2017
DOI:10.1016/j.watres.2017.03.053
•First study of the chlorination of food additive acesulfame.•Reaction between chlorine and acesulfame is pH-dependent.•Two new transformation products were identified, and then studied in real waters.•Acesulfame is the precursor of several regulated DBPs and N-DBPs.•The byproducts of the chlorination of acesulfame are 1.8-fold more toxic than acesulfame against Vibrio fischeri.Acesulfame (ACE) is one of the most commonly used artificial sweeteners. Because it is not metabolized in the human gut, it reaches the aquatic environment unchanged. In the present study, the reactivity of ACE in free chlorine-containing water was investigated for the first time. The degradation of ACE was found to follow pseudo-first-order kinetics. The first-order rate increased with decreasing pH from 9.4 to 4.8 with estimated half-lives from 693 min to 2 min. Structural elucidation of the detected transformation products (TPs) was performed by ultra-high performance liquid chromatography coupled with quadrupole time-of-flight mass spectrometry. Integration of MS/MS fragments, isotopic pattern and exact mass allowed the characterization of up to 5 different TPs in the ultrapure water extracts analyzed, including two proposed new chlorinated compounds reported for the first time. Unexpectedly, several known and regulated disinfection by-products (DBPs) were present in the ACE chlorinated solution. In addition, two of the six DBPs are proposed as N-DBPs. Time-course profiles of ACE and the identified by-products in tap water and wastewater samples were followed in order to simulate the actual disinfection process. Tap water did not significantly affect degradation, but wastewater did; it reacted with the ACE to produce several brominated-DBPs. A preliminary assessment of chlorinated mixtures by luminescence inhibition of Vibrio fischeri showed that these by-products were up to 1.8-fold more toxic than the parent compound. The generation of these DBPs, both regulated and not, representing enhanced toxicity, make chlorine disinfection a controversial treatment for ACE. Further efforts are urgently needed to both assess the consequences of current water treatment processes on ACE and to develop new processes that will safely treat ACE. Human health and the health of our aquatic ecosystems are at stake.Download high-res image (207KB)Download full-size image
Co-reporter:Yeuk-Ki Tsoi and Kelvin Sze-Yin Leung
Journal of Analytical Atomic Spectrometry 2010 - vol. 25(Issue 6) pp:NaN885-885
Publication Date(Web):2010/03/17
DOI:10.1039/B923343A
A new multi-elemental speciation method was developed for seven species of three elements namely As(III), As(VI), MMA, Cr(III), Cr(VI), Se(IV) and Se(VI), using liquid chromatography-inductively coupled plasma-mass spectrometry. The use of dynamic reaction cell technique in removing polyatomic interference dramatically extended the detection sensitivity for all seven species with methane as the reaction gas. The developed condition offered low detection limits in ranges of 0.12–0.44 μg L−1, 0.24–0.29 μg L−1 and 0.26 μg L−1 for species of As, Cr and Se, respectively. For concise quantitative speciation, a controlled chelation condition for Cr species was optimized to minimize species interconversion. In the developed method, the seven species were baseline-separated in an 11-minute elution on an anion exchange column using NH4NO3/NH4H2PO4 as the mobile phase at pH 6. This efficient, high-throughput and sensitive method was fully validated and applied in quantitative speciation of river water samples collected from the Shing Mun River of Hong Kong. Most of the species were found as the toxic forms of the elements studied.
Co-reporter:Yeuk-Ki Tsoi, Sidney Tam and Kelvin Sze-Yin Leung
Journal of Analytical Atomic Spectrometry 2010 - vol. 25(Issue 11) pp:NaN1762-1762
Publication Date(Web):2010/08/03
DOI:10.1039/C0JA00024H
Solid-phase microextraction coupled to liquid chromatography-inductively coupled plasma-mass spectrometry (SPME-LC-ICP-MS) is presented as a new approach for the extraction and speciation of methylmercury (MeHg) and ethylmercury (EtHg) in a complex urine matrix. Despite the fact that urine is one of the most easily accessible sources of biological specimens for clinical testing, reports of mercury speciation methods are limited. Without any tedious sample pretreatments or derivatization procedures, headspace SPME offers a clean and convenient one-step extraction followed by direct desorption into HPLC. In this work, the fibre was first exposed to the sample headspace for a 45 min extraction, followed by a 5 min desorption. Experimental parameters including extraction time and temperature, sodium chloride salt concentration, agitation rate and desorption time were optimized to achieve detection limits down to 0.06 μg L−1 for both organomercury species. Sample recoveries in spiked urine matrix ranged from 81.7–109.5%. The accuracy of the method was further validated using a certified reference material (TORT-2) and applied in the organomercury speciation of urine samples collected from patients with suspected mercury poisoning.
Co-reporter:Tsz-Shan Lum and Kelvin Sze-Yin Leung
Journal of Analytical Atomic Spectrometry 2016 - vol. 31(Issue 5) pp:
Publication Date(Web):
DOI:10.1039/C5JA00497G
Co-reporter:Tsz-Shan Lum, Yeuk-Ki Tsoi and Kelvin Sze-Yin Leung
Journal of Analytical Atomic Spectrometry 2014 - vol. 29(Issue 2) pp:NaN241-241
Publication Date(Web):2013/11/28
DOI:10.1039/C3JA50316J
Sample preconcentration is important for metal analysis in complex clinical matrices due to the low concentration of analytes and high salt content. The limited amount of sample available also increases the challenges of extraction and determination of the analytes. In spite of the increase in sensitivity of instruments, such ultra-trace levels of the metals are still difficult to determine accurately. Researchers have adopted various strategies to improve the efficiency and selectivity of the extraction. The use of chemicals and time for analysis can also be reduced by scaling down the analysis to the micro-level. This review discusses four aspects of the recent development in metal preconcentration in clinical samples, namely the use of ionic liquids (ILs) in extraction, sorption by nanomaterials, preconcentration using surfactants, and automation. The publications reviewed cover the period between 2008 to late 2013.

![(e)-1-(2,4-dihydroxyphenyl)-3-[4-[(2s,3r,4s,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxyphenyl]prop-2-en-1-one](http://img.cochemist.com/ccimg/5100/5041-81-6.png)
![(e)-1-(2,4-dihydroxyphenyl)-3-[4-[(2s,3r,4s,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxyphenyl]prop-2-en-1-one](http://img.cochemist.com/ccimg/5100/5041-81-6_b.png)




![Dimethyl 3-oxo-3H-pyrrolo[2,1,5-de]quinolizine-1,2-dicarboxylate](http://img.cochemist.com/ccimg/94600/94596-27-7.png)
![Dimethyl 3-oxo-3H-pyrrolo[2,1,5-de]quinolizine-1,2-dicarboxylate](http://img.cochemist.com/ccimg/94600/94596-27-7_b.png)









![1H-Pyrrole-2,5-dione,1-hydroxy-3-[4-[(3-methyl-2-butenyl)oxy]phenyl]-4-(2-methylpropyl)-](http://img.cochemist.com/ccimg/656900/656830-26-1.png)
![1H-Pyrrole-2,5-dione,1-hydroxy-3-[4-[(3-methyl-2-butenyl)oxy]phenyl]-4-(2-methylpropyl)-](http://img.cochemist.com/ccimg/656900/656830-26-1_b.png)
![Gadolinium 2,2',2''-[10-(carboxymethyl)-1,4,7,10-tetraazacyclodod Ecane-1,4,7-triyl]triacetate - 1-deoxy-1-(methylamino)-d-glucitol (1:1:1)](http://img.cochemist.com/ccimg/93000/92943-93-6.png)
![Gadolinium 2,2',2''-[10-(carboxymethyl)-1,4,7,10-tetraazacyclodod Ecane-1,4,7-triyl]triacetate - 1-deoxy-1-(methylamino)-d-glucitol (1:1:1)](http://img.cochemist.com/ccimg/93000/92943-93-6_b.png)








![1,7,7-TRIMETHYLBICYCLO[2.2.1]HEPTAN-2-OL ACETATE](http://img.cochemist.com/ccimg/100/76-49-3.png)
![1,7,7-TRIMETHYLBICYCLO[2.2.1]HEPTAN-2-OL ACETATE](http://img.cochemist.com/ccimg/100/76-49-3_b.png)








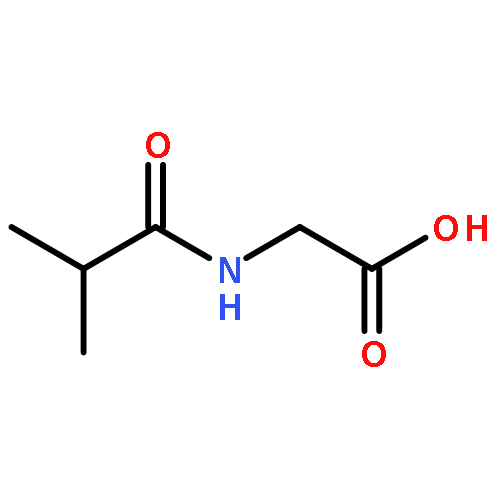
![8-[(carboxymethyl)amino]-8-oxooctanoic acid](http://img.cochemist.com/ccimg/60400/60317-54-6.png)
![8-[(carboxymethyl)amino]-8-oxooctanoic acid](http://img.cochemist.com/ccimg/60400/60317-54-6_b.png)




![(E)-4-((5-Hydroxy-3,7-dimethylocta-2,6-dien-1-yl)oxy)-7H-furo[3,2-g]chromen-7-one](http://img.cochemist.com/ccimg/88300/88206-46-6.png)
![(E)-4-((5-Hydroxy-3,7-dimethylocta-2,6-dien-1-yl)oxy)-7H-furo[3,2-g]chromen-7-one](http://img.cochemist.com/ccimg/88300/88206-46-6_b.png)






![Bicyclo[2.2.1]heptan-2-one,1,7,7-trimethyl-3-(phenylmethylene)-](http://img.cochemist.com/ccimg/15100/15087-24-8.png)
![Bicyclo[2.2.1]heptan-2-one,1,7,7-trimethyl-3-(phenylmethylene)-](http://img.cochemist.com/ccimg/15100/15087-24-8_b.png)

![[(1r)-4-[(1e,3e,5e,7e,9e,11e,13e,15e,17e)-18-[(4r)-4-hexadecanoyloxy-2,6,6-trimethylcyclohexen-1-yl]-3,7,12,16-tetramethyloctadeca-1,3,5,7,9,11,13,15,17-nonaenyl]-3,5,5-trimethylcyclohex-3-en-1-yl] Hexadecanoate](http://img.cochemist.com/ccimg/200/144-67-2.png)
![[(1r)-4-[(1e,3e,5e,7e,9e,11e,13e,15e,17e)-18-[(4r)-4-hexadecanoyloxy-2,6,6-trimethylcyclohexen-1-yl]-3,7,12,16-tetramethyloctadeca-1,3,5,7,9,11,13,15,17-nonaenyl]-3,5,5-trimethylcyclohex-3-en-1-yl] Hexadecanoate](http://img.cochemist.com/ccimg/200/144-67-2_b.png)





















![Dibenz[cd,f]indol-4(5H)-one,2-hydroxy-1-methoxy-](http://img.cochemist.com/ccimg/54000/53948-07-5.png)
![Dibenz[cd,f]indol-4(5H)-one,2-hydroxy-1-methoxy-](http://img.cochemist.com/ccimg/54000/53948-07-5_b.png)
![Benzo[f]-1,3-benzodioxolo[6,5,4-cd]indol-5(6H)-one,8-methoxy-](http://img.cochemist.com/ccimg/13400/13395-02-3.png)
![Benzo[f]-1,3-benzodioxolo[6,5,4-cd]indol-5(6H)-one,8-methoxy-](http://img.cochemist.com/ccimg/13400/13395-02-3_b.png)





![[2,3'-Biindolinylidene]-2',3-dione](http://img.cochemist.com/ccimg/500/479-41-4.png)
![[2,3'-Biindolinylidene]-2',3-dione](http://img.cochemist.com/ccimg/500/479-41-4_b.png)