Co-reporter:Kaijie Xiao, Fan Yu, Zhixin Tian
Journal of Proteomics 2017 Volume 152() pp:41-47
Publication Date(Web):30 January 2017
DOI:10.1016/j.jprot.2016.10.010
•Interpreting protein mass spectra directly using isotopic envelopes instead of “deisotoped” masses•FDR control with decoy database search•Comprehensive handling of amino acid variation, dynamic PTMs and static chemical labeling•Comprehensive leverage of collision-, electron-, and photon-based dissociation as well as various types of product ions in each dissociation method•User friendly GUIs, search results output & visualizationFor top-down protein database search and identification from tandem mass spectra, our isotopic envelope fingerprinting search algorithm and ProteinGoggle search engine have demonstrated their strength of efficiently resolving heavily overlapping data as well separating non-ideal data with non-ideal isotopic envelopes from ideal ones with ideal isotopic envelopes. Here we report our updated ProteinGoggle 2.0 for intact protein database search with full-capacity. The indispensable updates include users' optional definition of dynamic post-translational modifications and static chemical labeling during database creation, comprehensive dissociation methods and ion series, as well as a Proteoform Score for each proteoform. ProteinGoggle has previously been benchmarked with both collision-based dissociation (CID, HCD) and electron-based dissociation (ETD) data of either intact proteins or intact proteomes. Here we report our further benchmarking of the new version of ProteinGoggle with publically available photon-based dissociation (UVPD) data (http://hdl.handle.net/2022/17316) of intact E. coli ribosomal proteins.Biological significanceProtein species (aka proteoforms) function at their molecular level, and diverse structures and biological roles of every proteoform come from often co-occurring proteolysis, amino acid variation and post-translational modifications. Complete and high-throughput capture of this combinatorial information of proteoforms has become possible in evolving top-down proteomics; yet, various methods and technologies, especially database search and bioinformatics identification tools, in the top-down pipeline are still in their infancy stages and demand intensive research and development.
Co-reporter:Kaijie Xiao, Fan Yu, Houqin Fang, Bingbing Xue, Yan Liu, Yunhui Li, Zhixin Tian
Journal of Proteomics 2017 Volume 160(Volume 160) pp:
Publication Date(Web):8 May 2017
DOI:10.1016/j.jprot.2017.03.011
•The composition of all types of fragment ions of representative intact proteins were statistically investigated;•Proteome-level random match probability and utility of neutral loss fragment ions for proteoform identification was studied;•Protein-level random match probability and utility of internal fragment ions for proteoform identification was evaluated.Neutral loss and internal product ions have been found to be significant in both peptide and protein tandem mass spectra and they have been proposed to be included in database search and for protein identification. In addition to common canonical b/y ions in collision-based dissociation or c/z ions in electron-based dissociation, inclusion of neutral loss and internal product ions would certainly make better use of tandem mass spectra data; however, their ultimate utility for protein identification with false discovery rate control remains unclear. Here we report our proteome-level utility benchmarking of neutral loss and internal product ions with tandem mass spectra of intact E. coli proteome. Utility of internal product ions was further evaluated at the protein level using selected tandem mass spectra of individual E. coli proteins. We found that both neutral loss and internal products ions do not have direct utility for protein identification when they were used for scoring of P Score; but they do have indirect utility for provision of more canonical b/y ions when they are included in the database search and overlapping ions between different ion types are resolved.Biological significanceTandem mass spectrometry has evolved to be a state-of-the-art method for characterization of protein primary structures (including amino acid sequence, post-translational modifications (PTMs) as well as their site location), where full study and utilization tandem mass spectra and product ions are indispensable. This primary structure information is essential for higher order structure and eventual function study of proteins.Download high-res image (115KB)Download full-size image
Co-reporter:Kaijie Xiao, Yun Shen, Shasha Li, Zhixin Tian
Analytica Chimica Acta 2017 Volume 996(Volume 996) pp:
Publication Date(Web):15 December 2017
DOI:10.1016/j.aca.2017.09.043
•P-bracket was developed for phosphosite localization using paired phospho-containing site-determining product ions.•In phosphosite localization of 101,520 synthetic phosphopeptides, a false localization rate (FLR) of 0.9% was obtained.•1,601 and 1,393 HeLa phosphopeptides (identified by Mascot and Sequest, respectively) were accurately localized.•P-bracket, as a stand-alone software withuser-friendly GUIs, is available through http://proteingoggle.tongji.edu.cn.Phosphorylation is one of the most important and widely studied protein post-translational modifications. Tandem mass spectrometry using higher-energy collisional dissociation has evolved into a state-of-the-art analytical platform for both phosphorylation identification and site localization. Tens of thousands of phosphopeptides can now be routinely identified from a single shotgun proteomics study; site localization, however, is much more complicated and many challenges still exist. Here, we report our development of P-bracket using direct experimental evidence of phospho-containing site-determining product ions for accurate site localization without the need for additional FLR control. A P-bracket is defined as a complementary product ion pair that forms a bracket to confine a phosphorylation event to a unique site. P-bracket has been successfully benchmarked with a set of six synthetic phosphopeptides with a single phosphorylation event, a set of 96 synthetic peptides and phosphopeptide reference libraries, and two HeLa phosphopeptide LC-MS/MS (HCD) datasets; Accurate phosphosite localization by P-bracket will greatly enhance identification confidence of phosphopeptides and contribute to structural and functional studies of phosphoproteins.Download high-res image (157KB)Download full-size image
Co-reporter:Houqin Fang, Kaijie Xiao, Yunhui Li, Fan Yu, Yan Liu, Bingbing Xue, and Zhixin Tian
Analytical Chemistry 2016 Volume 88(Issue 14) pp:7198
Publication Date(Web):June 30, 2016
DOI:10.1021/acs.analchem.6b01388
Protein structural and functional studies rely on complete qualitative and quantitative information on protein species (proteoforms); thus, it is important to quantify differentially expressed proteins at their molecular level. Here we report our development of universal pseudoisobaric dimethyl labeling (pIDL) of amino groups at both the N-terminal and lysine residues for relative quantitation of intact proteins. Initial proof-of-principle study was conducted on standard protein myoglobin and hepatocellular proteomes (HepG2 vs LO2). The amino groups from both the N-terminal and lysine were dimethylated with HXHO (X = 13C or C) and NaBY3CN (Y = H or D). At the standard protein level, labeling efficiency, effect of product ion size, and mass resolution on quantitation accuracy were explored; and a good linear quantitation dynamic range up to 50-fold was obtained. For the hepatocellular proteome samples, 33 proteins were quantified with RSD ≤ 10% from one-dimensional reversed phase liquid chromatography–tandem mass spectrometry (RPLC–MS/MS) analysis of the 1:1 mixed samples. The method in this study can be extended to quantitation of other intact proteome systems. The universal “one-pot” dimethyl labeling of all the amino groups in a protein without the need of preblocking of those on the lysine residues is made possible by protein identification and quantitation analysis using ProteinGoggle 2.0 with customized databases of both precursor and product ions containing heavy isotopes.
Co-reporter:Zhixin Tian, Dana R. Reed, Steven R. Kass
International Journal of Mass Spectrometry 2015 Volume 377() pp:130-138
Publication Date(Web):1 February 2015
DOI:10.1016/j.ijms.2014.04.020
•H/D exchange mechanisms of 16 small rigid anions were examined.•Four deuterated reagents were used to probe relay and flip-flop processes.•Distortion energies were found to account for relay mechanism barriers.Hydrogen–deuterium exchange reactions of 16 small rigid anions were examined using D2O, CH3OD, CH3CO2D, and (CF3)2C(CH3)OD. Bimolecular rate constants and extensive computations are reported. These deuterated reagents were used because D2O and CH3OD are weak acids that cannot react via sequential deuteron/proton transfers (i.e., the conventional H/D exchange mechanism of DePuy et al.) with the selected anions, and they provide a means to probe alternative pathways. The relative acidity of CH3CO2D and (CF3)2C(CH3)OD was measured (ΔΔHacid° = 1.1 ± 0.8 kcal mol−1) and their thermodynamic similarity but structural differences (i.e., the presence of a CO in the carboxylic acid but not the alcohol) were exploited to systematically investigate flip-flop and relay mechanisms. This has led to a number of generalizations relating to the occurrence of these pathways.
Co-reporter:Zhixin Tian, Lev Lis, and Steven R. Kass
The Journal of Organic Chemistry 2013 Volume 78(Issue 24) pp:12650-12653
Publication Date(Web):November 18, 2013
DOI:10.1021/jo402263v
The ionization energy (IE) of the 3-cyclopropenyl radical (6.00 ± 0.17 eV) was measured in the gas phase by reacting 3-cyclopropenium cation (c-C3H3+) with a series of reference reagents of known IEs. This result was combined in a thermodynamic cycle to obtain the heat of formation of c–C3H3• (118.9 ± 4.0 kcal mol–1) and the allylic C–H bond dissociation energy (BDE) of cyclopropene (104.4 ± 4.0 kcal mol–1). These experimental values are well reproduced by high level G3 and W1 computations and reveal that the BDE is similar to that for cyclopropane and the vinyl position of cyclopropene. This is unprecedented and is a reflection of the unusual nature of cyclopropene.



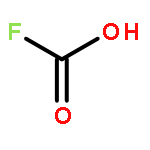
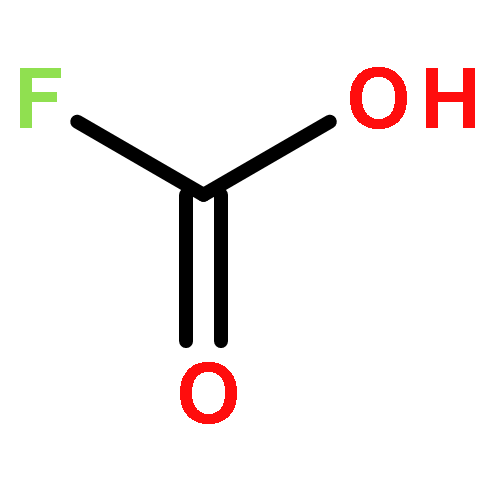

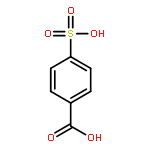



![4-[(E)-2-(4-fluorophenyl)ethenyl]-N,N-dimethylaniline](http://img.cochemist.com/ccimg/38700/38695-34-0.png)
![4-[(E)-2-(4-fluorophenyl)ethenyl]-N,N-dimethylaniline](http://img.cochemist.com/ccimg/38700/38695-34-0_b.png)




![Benzenamine,4-[(1E)-2-(4-methoxyphenyl)ethenyl]-N,N-dimethyl-](http://img.cochemist.com/ccimg/2900/2844-24-8.png)
![Benzenamine,4-[(1E)-2-(4-methoxyphenyl)ethenyl]-N,N-dimethyl-](http://img.cochemist.com/ccimg/2900/2844-24-8_b.png)



