Co-reporter:Takahiro Suto, Yuta Yanagita, Yoshiyuki Nagashima, Shinsaku Takikawa, Yasuhiro Kurosu, Naoya Matsuo, Takaaki Sato, and Noritaka Chida
Journal of the American Chemical Society March 1, 2017 Volume 139(Issue 8) pp:2952-2952
Publication Date(Web):February 11, 2017
DOI:10.1021/jacs.7b00807
A stereodivergent strategy for the synthesis of skipped dienes is developed. The method consists of hydroboration of allenes and Migita–Kosugi–Stille coupling, which allows for access to all four possible stereoisomers of the skipped dienes. The hydroboration is especially useful for providing both E-allylic and Z-allylic alcohols from the same allene by simply changing the organoborane reagent. The strategy was successfully applied to a unified total synthesis of the madangamine alkaloids via a common ABCE-tetracyclic intermediate with a (Z,Z)-skipped diene. The late-stage variation of the D-ring enabled the supply of synthetic madangamines A, C, and E for the first time.
Co-reporter:Seiya Katahara; Shoichiro Kobayashi; Kanami Fujita; Tsutomu Matsumoto; Takaaki Sato
Journal of the American Chemical Society 2016 Volume 138(Issue 16) pp:5246-5249
Publication Date(Web):April 13, 2016
DOI:10.1021/jacs.6b02324
An Ir-catalyzed reductive formation of functionalized nitrones from N-hydroxyamides was reported. The reaction took place through two types of iridium-catalyzed reactions including dehydrosilylation and hydrosilylation. The method showed high chemoselectivity in the presence of sensitive functional groups, such as methyl esters, and was successfully applied to the synthesis of cyclic and macrocyclic nitrones, which are known to be challenging compounds to access by conventional methods. 1H NMR studies strongly supported generation of an N-siloxyamide and an N,O-acetal as the actual intermediates.
Co-reporter:Keisuke Fukaya, Yu Yamaguchi, Ami Watanabe, Hiroaki Yamamoto, Tomoya Sugai, Takeshi Sugai, Takaaki Sato and Noritaka Chida
The Journal of Antibiotics 2016 69(4) pp:273-279
Publication Date(Web):February 10, 2016
DOI:10.1038/ja.2016.6
The practical synthesis of the C-ring precursor of paclitaxel starting from 3-methoxytoluene is described. Lipase-catalyzed kinetic resolution of a substituted cyclohexane-1,2-diol, derived from 3-methoxytoluene in three steps, successfully afforded a desired enantiomer with >99% ee, which was transformed to a cyclohexenone. 1,4-Addition of a vinyl metal species, followed by Mukaiyama aldol reaction with formalin in the presence of a Lewis acid provided the known C-ring precursor of paclitaxel in a 10 g scale.
Co-reporter:Shun Tsuzaki, Shunme Usui, Hiroki Oishi, Daichi Yasushima, Takahiro Fukuyasu, Takeshi Oishi, Takaaki Sato, and Noritaka Chida
Organic Letters 2015 Volume 17(Issue 7) pp:1704-1707
Publication Date(Web):March 13, 2015
DOI:10.1021/acs.orglett.5b00475
The total synthesis of sphingofungin F through the Overman rearrangement of an unsaturated ester, which is known to be an unsuitable substrate under standard conditions due to the competitive aza-Michael reaction, is described. The developed conditions enabled the ester to be compatible with the original Overman rearrangement, providing quick access to α,α-disubstituted amino acids by minimizing extra protecting group manipulations and redox reactions.
Co-reporter:Keisuke Fukaya, Keisuke Kodama, Yuta Tanaka, Hirohisa Yamazaki, Tomoya Sugai, Yu Yamaguchi, Ami Watanabe, Takeshi Oishi, Takaaki Sato, and Noritaka Chida
Organic Letters 2015 Volume 17(Issue 11) pp:2574-2577
Publication Date(Web):May 26, 2015
DOI:10.1021/acs.orglett.5b01174
A formal synthesis of the antitumor diterpenoid paclitaxel (Taxol) is described. The ABC ring of paclitaxel, synthesized starting from 1,3-cyclohexanedione and tri-O-acetyl-d-glucal by SmI2-mediated cyclization as the key transformation, was successfully converted to Takahashi’s tetracyclic oxetane intermediate. A double Chugaev reaction was employed for introduction of the strained bridgehead olefin, and stereoselective formation of the oxetane ring afforded the known synthetic intermediate, completing the formal synthesis of paclitaxel.
Co-reporter:Keisuke Fukaya, Yuta Tanaka, Ayako C. Sato, Keisuke Kodama, Hirohisa Yamazaki, Takeru Ishimoto, Yasuyoshi Nozaki, Yuki M. Iwaki, Yohei Yuki, Kentaro Umei, Tomoya Sugai, Yu Yamaguchi, Ami Watanabe, Takeshi Oishi, Takaaki Sato, and Noritaka Chida
Organic Letters 2015 Volume 17(Issue 11) pp:2570-2573
Publication Date(Web):May 26, 2015
DOI:10.1021/acs.orglett.5b01173
A convergent synthesis of the ABC ring of antitumor natural product paclitaxel (Taxol) is described. SmI2-mediated reductive cyclization of an allylic benzoate possessing an aldehyde function, synthesized from tri-O-acetyl-d-glucal and 1,3-cyclohexanedione, smoothly afforded the highly strained 6–8–6 tricarbocyclic structure in 66% yield.
Co-reporter:Yuta Yanagita, Takahiro Suto, Naoya Matsuo, Yasuhiro Kurosu, Takaaki Sato, and Noritaka Chida
Organic Letters 2015 Volume 17(Issue 8) pp:1946-1949
Publication Date(Web):March 27, 2015
DOI:10.1021/acs.orglett.5b00661
A general synthetic route toward a diazatricyclic core common to the madangamine family is described. Ring-closing metathesis and palladium-catalyzed cycloisomerization provided the cis-fused diazadecalin structure, accompanied by formation of the N-Boc-enamine, which was utilized as an N-acyliminium ion equivalent. Direct cyclization from the N-Boc-enamine was achieved through the in situ formation of an N,O-acetal.
Co-reporter:Minami Nakajima, Takaaki Sato, and Noritaka Chida
Organic Letters 2015 Volume 17(Issue 7) pp:1696-1699
Publication Date(Web):March 27, 2015
DOI:10.1021/acs.orglett.5b00664
Iridium-catalyzed reductive nucleophilic addition to N-methoxyamides is reported. The reaction took place in high yields in the presence of a variety of sensitive functional groups such as esters and nitro groups. Mechanistic studies revealed that the reaction of N-methoxyamides proceeded without equilibrium to an enamine intermediate in contrast to that with tert-amides.
Co-reporter:Noritaka Chida;Takaaki Sato
The Chemical Record 2014 Volume 14( Issue 4) pp:592-605
Publication Date(Web):
DOI:10.1002/tcr.201402024
Abstract
The Ferrier carbocyclization reaction is one of the most powerful transformations of carbohydrates. This reaction provides enantiomerically pure cyclohexanone derivatives from aldohexoses, and is particularly useful in the chiral pool synthesis of cyclohexane-containing natural products from carbohydrates. We have investigated the synthesis of natural products utilizing the Ferrier carbocyclization reaction. This account provides a brief overview of the Ferrier carbocyclization and its application to natural product synthesis. The utility and versatility of the Ferrier carbocyclization reaction are showcased with the syntheses of hygromycin A, lycoricidine, actinobolin, galanthamine, and morphine starting from carbohydrates.
Co-reporter:Minami Nakajima;Yukiko Oda;Takamasa Wada;Ryo Minamikawa;Dr. Kenji Shirokane;Dr. Takaaki Sato
Chemistry - A European Journal 2014 Volume 20( Issue 52) pp:17565-17571
Publication Date(Web):
DOI:10.1002/chem.201404648
Abstract
As the complexity of targeted molecules increases in modern organic synthesis, chemoselectivity is recognized as an important factor in the development of new methodologies. Chemoselective nucleophilic addition to amide carbonyl centers is a challenge because classical methods require harsh reaction conditions to overcome the poor electrophilicity of the amide carbonyl group. We have successfully developed a reductive nucleophilic addition of mild nucleophiles to tertiary amides, secondary amides, and N-methoxyamides that uses the Schwartz reagent [Cp2ZrHCl]. The reaction took place in a highly chemoselective fashion in the presence of a variety of sensitive functional groups, such as methyl esters, which conventionally require protection prior to nucleophilic addition. The reaction will be applicable to the concise synthesis of complex natural alkaloids from readily available amide groups.
Co-reporter:Makoto Yoritate;Tatsuhiko Meguro;Naoya Matsuo;Kenji Shirokane;Dr. Takaaki Sato; Noritaka Chida
Chemistry - A European Journal 2014 Volume 20( Issue 26) pp:
Publication Date(Web):
DOI:10.1002/chem.201403607
Abstract
Invited for the cover of this issue is the group of Prof. Noritaka Chida and Dr. Takaaki Sato at Keio University, Japan. The cover illustrates the N-methoxy group as the reactivity control element to allow for beautiful chemical transformations to blossom in organic synthesis. Read the full text of the article at 10.1002/chem.201402231.
Co-reporter:Makoto Yoritate;Tatsuhiko Meguro;Naoya Matsuo;Kenji Shirokane;Dr. Takaaki Sato; Noritaka Chida
Chemistry - A European Journal 2014 Volume 20( Issue 26) pp:
Publication Date(Web):
DOI:10.1002/chem.201490103
Co-reporter:Makoto Yoritate;Tatsuhiko Meguro;Naoya Matsuo;Kenji Shirokane;Dr. Takaaki Sato; Noritaka Chida
Chemistry - A European Journal 2014 Volume 20( Issue 26) pp:8210-8216
Publication Date(Web):
DOI:10.1002/chem.201402231
Abstract
The development of a two-step synthesis of multi-substituted N-methoxyamines from N-methoxyamides is reported. Utilization of the N-methoxy group as a reactivity control element was the key to success in this two-step synthesis. The first reaction involves a N-methoxyamide/aldehyde coupling reaction. Whereas ordinary amides cannot condense with aldehydes intermolecularly due to the poor nucleophilicity of the amide nitrogen, the N-methoxy group enhances the nucleophilicity of the nitrogen, enabling the direct coupling reaction. The second reaction in the two-step process was nucleophilic addition to the N-methoxyamides. Incorporation of the N-methoxy group into the amides increased the electrophilicity of the amide carbonyls and promoted the chelation effect. This nucleophilic addition enabled quick diversification of the products derived from the first step. The developed strategy was applicable to a variety of substrates, resulting in the elaboration of multi-substituted piperidines and acyclic amines, as well as a substructure of a complex natural alkaloid.
Co-reporter:Kenji Shirokane;Takamasa Wada;Makoto Yoritate;Ryo Minamikawa;Nobuaki Takayama;Dr. Takaaki Sato;Dr. Noritaka Chida
Angewandte Chemie International Edition 2014 Volume 53( Issue 2) pp:512-516
Publication Date(Web):
DOI:10.1002/anie.201308905
Abstract
A chemoselective approach for the total synthesis of (±)-gephyrotoxin has been developed. The key to success was the utilization of N-methoxyamides, which enabled the direct coupling of the amide with an aldehyde and selective reductive nucleophilic addition to the amide in the presence of a variety of sensitive and electrophilic functional groups, such as a methyl ester. This chemoselective approach minimized the use of protecting-group manipulations and redox reactions, which resulted in the most concise and efficient total synthesis of (±)-gephyrotoxin described to date.
Co-reporter:Yasuaki Nakayama;Ruriko Sekiya;Hiroki Oishi;Naoto Hama;Miki Yamazaki;Dr. Takaaki Sato;Dr. Noritaka Chida
Chemistry - A European Journal 2013 Volume 19( Issue 36) pp:12052-12058
Publication Date(Web):
DOI:10.1002/chem.201301216
Abstract
This article describes the details of two new types of Overman rearrangement from allylic vicinal diols. Starting from identical diols, both bis(imidate)s and cyclic orthoamides were selectively synthesized by simply changing the reaction conditions. Whilst exposure of the bis(imidate)s to thermal conditions initiated the double Overman rearrangement to introduce two identical nitrogen groups in a single operation (the cascade-type Overman rearrangement), the reaction of cyclic orthoamides resulted in a single rearrangement (the orthoamide-type Overman rearrangement). The newly generated allylic alcohols from the orthoamide-type reaction can potentially undergo a variety of further transformations. For instance, we demonstrated an Overman/Claisen sequence in one pot. The most conspicuous feature of this method is that it offers precise control over the number of Overman rearrangements from the same allylic vicinal diols. This method also excludes the tedious protecting-group manipulations of the homoallylic alcohols, which are necessary in conventional Overman rearrangements. All of the performed rearrangements proceeded in a completely diastereoselective fashion through a chair-like transition state.
Co-reporter:Yuta Yanagita;Hugh Nakamura;Kenji Shirokane;Yusuke Kurosaki;Dr. Takaaki Sato; , Dr. Noritaka Chida
Chemistry - A European Journal 2013 Volume 19( Issue 2) pp:678-684
Publication Date(Web):
DOI:10.1002/chem.201202639
Abstract
While the synthesis of amide bonds is now one of the most reliable organic reactions, functionalization of amide carbonyl groups has been a long-standing issue due to their high stability. As an ongoing program aimed at practical transformation of amides, we developed a direct nucleophilic addition to N-alkoxyamides to access multisubstituted amines. The reaction enabled installation of two different functional groups to amide carbonyl groups in one pot. The N-alkoxy group played important roles in this reaction. First, it removed the requirement for an extra preactivation step prior to nucleophilic addition to activate inert amide carbonyl groups. Second, the N-alkoxy group formed a five-membered chelated complex after the first nucleophilic addition, resulting in suppression of an extra addition of the first nucleophile. While diisobutylaluminum hydride (DIBAL-H) and organolithium reagents were suitable as the first nucleophile, allylation, cyanation, and vinylation were possible in the second addition including inter- and intramolecular reactions. The yields were generally high, even in the synthesis of sterically hindered α-trisubstituted amines. The reaction exhibited wide substrate scope, including acyclic amides, five- and six-membered lactams, and macrolactams.
Co-reporter:Masato Ichiki;Dr. Hiroki Tanimoto;Shohei Miwa;Ryosuke Saito;Dr. Takaaki Sato ; Noritaka Chida
Chemistry - A European Journal 2013 Volume 19( Issue 1) pp:264-269
Publication Date(Web):
DOI:10.1002/chem.201203284
Abstract
A detailed exploration of the synthesis of (−)-morphine based on sequential [3,3]-sigmatropic rearrangements is described. The sequential Claisen/Claisen rearrangements of an allylic vicinal diol resulted in the stereoselective formation of the two contiguous carbon centers, including a sterically encumbered quaternary carbon, in a single operation. The two ethyl esters generated in this reaction were successfully differentiated during a subsequent Friedel–Crafts-type cyclization. The (−)-morphine double bond was introduced at a late stage in our first-generation synthesis, but was formed at an earlier stage in the second-generation synthesis, resulting in a more efficient route to the end product.
Co-reporter:Yusuke Kurosaki, Kenji Shirokane, Takeshi Oishi, Takaaki Sato, and Noritaka Chida
Organic Letters 2012 Volume 14(Issue 8) pp:2098-2101
Publication Date(Web):April 4, 2012
DOI:10.1021/ol300622r
Three-component allylation and cyanation utilizing a ketone and an N-methoxyamine are reported. The high nucleophilicity of the N-methoxyamine and high electrophilicity of the corresponding iminium ion enable the concise synthesis of α-trisubstituted amines in a single step.
Co-reporter:Yukiko Oda, Takaaki Sato, and Noritaka Chida
Organic Letters 2012 Volume 14(Issue 3) pp:950-953
Publication Date(Web):January 19, 2012
DOI:10.1021/ol3000316
Direct allylation of inert amide carbonyls utilizing the Schwartz reagent afforded either substituted tertiary or secondary amines. A preactivation step was successfully avoided, which is generally a requisite to increase the electrophilicity of amides. The reaction exhibited remarkable functional group tolerance and proceeded even in the presence of methyl esters and nitro groups.
Co-reporter:Katsunori Kitamoto;Yasuaki Nakayama;Mana Sampei;Masato Ichiki;Naoya Furuya;Takaaki Sato
European Journal of Organic Chemistry 2012 Volume 2012( Issue 22) pp:4217-4231
Publication Date(Web):
DOI:10.1002/ejoc.201200523
Abstract
A detailed description is presented of two sequential sigmatropic rearrangements starting from enantiopure allylic vicinal diols. Starting from the same allylic diol, the sequential Claisen/Claisen rearrangement can install two identical functional groups in a one-pot reaction, whereas, the sequential Claisen/Overman rearrangement can introduce two different functional groups, both occurring without protecting group manipulation. Both sequential reactions proceeded with complete stereoselectivity, which was easily predictable by the judicious choice of two factors regarding the allylic diols: (1) the stereochemistry of the hydroxy groups and (2) the geometry of the olefin. To demonstrate this sequential rearrangement methodology, we accomplished the total synthesis of (–)-kainic acid, whose three contiguous stereocenters were completely established by three chirality transfer reactions (SN2′ and sequential Claisen/Overman reactions) of flexible acyclic intermediates derived from D-arabinose.
Co-reporter:Naoto Hama, Toshihiro Aoki, Shohei Miwa, Miki Yamazaki, Takaaki Sato, and Noritaka Chida
Organic Letters 2011 Volume 13(Issue 4) pp:616-619
Publication Date(Web):December 31, 2010
DOI:10.1021/ol102856j
A first total synthesis of broussonetine F from diethyl l-tartrate was achieved. The cornerstone of our synthesis was an orthoamide Overman rearrangement, which provided an allylic amino alcohol with complete diastereoselectivity.
Co-reporter:Katsunori Kitamoto, Mana Sampei, Yasuaki Nakayama, Takaaki Sato, and Noritaka Chida
Organic Letters 2010 Volume 12(Issue 24) pp:5756-5759
Publication Date(Web):November 24, 2010
DOI:10.1021/ol1026602
Sequential sigmatropic rearrangements (Claisen/Claisen and Claisen/Overman) of enantiopure allylic diols are described. The reactions proceeded in complete diastereoselectivity without protecting group manipulations. The sequential Claisen/Overman rearrangement was successfully applied to the total synthesis of (−)-kainic acid.
Co-reporter:Kenji Shirokane;Yusuke Kurosaki;Takaaki Sato Dr. Dr.
Angewandte Chemie International Edition 2010 Volume 49( Issue 36) pp:6369-6372
Publication Date(Web):
DOI:10.1002/anie.201001127
Co-reporter:Naoto Hama, Tomoki Matsuda, Takaaki Sato and Noritaka Chida
Organic Letters 2009 Volume 11(Issue 12) pp:2687-2690
Publication Date(Web):May 18, 2009
DOI:10.1021/ol900799e
An enantioselective total synthesis of (−)-agelastatin A from (−)-2,3-O-isopropylidene-d-threitol is described. The sequential Overman/Mislow−Evans rearrangement of the allylic bistrichloroimidate is the key step, which efficiently installed a diaminohydroxy group.
Co-reporter:Hiroyoshi Yamanaka, Kazuya Sato, Hideyuki Sato, Masatoshi Iida, Takeshi Oishi, Noritaka Chida
Tetrahedron 2009 65(45) pp: 9188-9201
Publication Date(Web):
DOI:10.1016/j.tet.2009.09.012
Co-reporter:Hideyuki Sato, Takaki Maeba, Ryota Yanase, Akiko Yamaji-Hasegawa, Toshihide Kobayashi and Noritaka Chida
The Journal of Antibiotics 2005 58(1) pp:37-49
Publication Date(Web):
DOI:10.1038/ja.2005.4
The first total synthesis of (+)-sulfamisterin (AB5366), a naturally occurring -substituted -amino acid derivative possessing a sulfonated hydroxy function, is described. Overman rearrangement of an allylic trichloroacetimidate derived from D-tartrate effectively generated the tetrasubstituted carbon containing a nitrogen substituent. Construction of the amino acid moiety and sulfonation of the hydroxy group, followed by deprotection completed the total synthesis, which fully confirmed the proposed absolute structure of the natural product. The possible stereoisomers of (+)-sulfamisterin and their desulfonated derivatives were also synthesized. Biological assessment of all synthetic compounds revealed that natural (+)-sulfamisterin and its 3-epimer as well as their desulfonated derivatives possessing 2S-configuration strongly inhibit the serine palmitoyl transferase both in vitro and in vivo, whereas compounds with 2R-configuration were found to show much weaker inhibitory activity.
Co-reporter:Masahiro Bohno, Hidetomo Imase and Noritaka Chida
Chemical Communications 2004 (Issue 9) pp:1086-1087
Publication Date(Web):30 Mar 2004
DOI:10.1039/B402762K
The stereoselective and chiral synthesis of the Amaryllidaceae alkaloid, (+)-vittatine 1, is described; the quaternary carbon in 1 was generated by Claisen rearrangement of a cyclohexenol derived from D-glucose by way of a Ferrier's carbocyclisation reaction and a hexahydroindole skeleton was effectively constructed by intramolecular aminomercuration–demercuration, followed by Chugaev reaction.
Co-reporter:Kazuo Kurosawa, Toshihiko Nagase and Noritaka Chida
Chemical Communications 2002 (Issue 12) pp:1280-1281
Publication Date(Web):16 May 2002
DOI:10.1039/B202298B
The total synthesis and an unambiguous structure confirmation of stevastelin B 1, a novel 15-membered cyclic depsipeptide, are described; the fatty acid moiety in 1, prepared stereoselectively from L-quebrachitol was converted into the amino carboxylic acid, whose macrolactamization by Shioiri’s procedure effectively constructed the cyclic structure of 1.
Co-reporter:Takeshi Oishi, Koji Ando and Noritaka Chida
Chemical Communications 2001 (Issue 19) pp:1932-1933
Publication Date(Web):04 Sep 2001
DOI:10.1039/B104864N
The stereoselective total synthesis of myriocin 1 from D-mannose is described; the carbon framework with three contiguous chiral centers including a tetra-substituted carbon with nitrogen was effectively constructed using Overman rearrangement as the key reaction.
Co-reporter:Takayuki Momose, Masaki Setoguchi, Toshiki Fujita, Hitoshi Tamura and Noritaka Chida
Chemical Communications 2000 (Issue 22) pp:2237-2238
Publication Date(Web):31 Oct 2000
DOI:10.1039/B006170K
The chiral synthesis of the fully functionalized CD ring unit
of paclitaxel 3 is described; the three component coupling reaction of a
cyclohexenone derived from D-glucal by way of Ferrier’s
carbocyclization with vinyl cuprate and formaldehyde effectively
constructed the carbon framework of 3.

![(3AR,4R,5R,6AS)-4-[(1E)-4-(3-CHLOROPHENOXY)-3-OXO-1-BUTEN-1-YL]-2-OXOHEXAHYDRO-2H-CYCLOPENTA[B]FURAN-5-YL 4-BIPHENYLCARBOXYLATE](http://img.cochemist.com/ccimg/478000/477953-07-4.png)
![(3AR,4R,5R,6AS)-4-[(1E)-4-(3-CHLOROPHENOXY)-3-OXO-1-BUTEN-1-YL]-2-OXOHEXAHYDRO-2H-CYCLOPENTA[B]FURAN-5-YL 4-BIPHENYLCARBOXYLATE](http://img.cochemist.com/ccimg/478000/477953-07-4_b.png)
![Benzonitrile, 4-[1-[(phenylmethyl)amino]-3-buten-1-yl]-](http://img.cochemist.com/ccimg/927500/927437-54-5.png)
![Benzonitrile, 4-[1-[(phenylmethyl)amino]-3-buten-1-yl]-](http://img.cochemist.com/ccimg/927500/927437-54-5_b.png)








![BENZONITRILE, 4-[(METHOXYAMINO)METHYL]-](http://img.cochemist.com/ccimg/543800/543731-34-6.png)
![BENZONITRILE, 4-[(METHOXYAMINO)METHYL]-](http://img.cochemist.com/ccimg/543800/543731-34-6_b.png)










![Benzenemethanamine, N-[1-[(1E)-2-phenylethenyl]-3-butenyl]-](http://img.cochemist.com/ccimg/173500/173416-02-9.png)
![Benzenemethanamine, N-[1-[(1E)-2-phenylethenyl]-3-butenyl]-](http://img.cochemist.com/ccimg/173500/173416-02-9_b.png)






![Butanal, 4-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-](http://img.cochemist.com/ccimg/127800/127793-62-8.png)
![Butanal, 4-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-](http://img.cochemist.com/ccimg/127800/127793-62-8_b.png)





![1,3-Dioxolane-4-carboxaldehyde, 5-[[[(1,1-dimethylethyl)dimethylsilyl]oxy]methyl]-2,2-dimethyl-, (4R,5S)-](http://img.cochemist.com/ccimg/108900/108817-97-6.png)
![1,3-Dioxolane-4-carboxaldehyde, 5-[[[(1,1-dimethylethyl)dimethylsilyl]oxy]methyl]-2,2-dimethyl-, (4R,5S)-](http://img.cochemist.com/ccimg/108900/108817-97-6_b.png)
![1,3-Dioxolane-4-methanol,5-[[[(1,1-dimethylethyl)dimethylsilyl]oxy]methyl]-2,2-dimethyl-, (4S,5S)-](http://img.cochemist.com/ccimg/108900/108817-96-5.png)
![1,3-Dioxolane-4-methanol,5-[[[(1,1-dimethylethyl)dimethylsilyl]oxy]methyl]-2,2-dimethyl-, (4S,5S)-](http://img.cochemist.com/ccimg/108900/108817-96-5_b.png)










![(2s)-2-[tert-butyl(dimethyl)silyl]oxypropanal](http://img.cochemist.com/ccimg/87800/87727-28-4.png)
![(2s)-2-[tert-butyl(dimethyl)silyl]oxypropanal](http://img.cochemist.com/ccimg/87800/87727-28-4_b.png)




![1,4-Dioxaspiro[4.5]dec-7-ene, 7-methyl-](http://img.cochemist.com/ccimg/83400/83313-55-7.png)
![1,4-Dioxaspiro[4.5]dec-7-ene, 7-methyl-](http://img.cochemist.com/ccimg/83400/83313-55-7_b.png)
![1,5-DIOXASPIRO[5.5]UNDECAN-8-ONE, 3,3,7,7-TETRAMETHYL-](http://img.cochemist.com/ccimg/75100/75072-38-7.png)
![1,5-DIOXASPIRO[5.5]UNDECAN-8-ONE, 3,3,7,7-TETRAMETHYL-](http://img.cochemist.com/ccimg/75100/75072-38-7_b.png)


![BENZOIC ACID, 4-[(METHYLAMINO)METHYL]-, METHYL ESTER](http://img.cochemist.com/ccimg/70800/70785-70-5.png)
![BENZOIC ACID, 4-[(METHYLAMINO)METHYL]-, METHYL ESTER](http://img.cochemist.com/ccimg/70800/70785-70-5_b.png)





![Benzoic acid, 4-[(hydroxyamino)methyl]-, methyl ester](http://img.cochemist.com/ccimg/57200/57124-42-2.png)
![Benzoic acid, 4-[(hydroxyamino)methyl]-, methyl ester](http://img.cochemist.com/ccimg/57200/57124-42-2_b.png)




















![N-[(4-NITROPHENYL)METHYL]HYDROXYLAMINE](http://img.cochemist.com/ccimg/3000/2912-97-2.png)
![N-[(4-NITROPHENYL)METHYL]HYDROXYLAMINE](http://img.cochemist.com/ccimg/3000/2912-97-2_b.png)




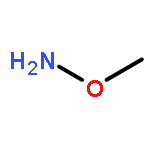
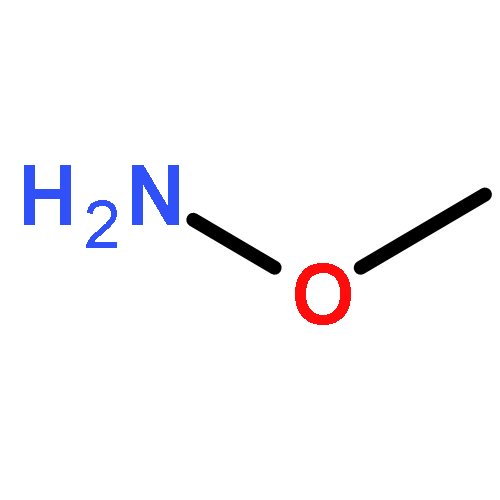




![BENZOIC ACID, 4-[(METHOXYAMINO)METHYL]-, METHYL ESTER](http://img.cochemist.com/ccimg/543800/543731-23-3.png)
![BENZOIC ACID, 4-[(METHOXYAMINO)METHYL]-, METHYL ESTER](http://img.cochemist.com/ccimg/543800/543731-23-3_b.png)


![Furo[3,4-d]-1,3-dioxol-4-ol, tetrahydro-2,2-dimethyl-, (3aS,6aS)-](http://img.cochemist.com/ccimg/213800/213767-09-0.png)
![Furo[3,4-d]-1,3-dioxol-4-ol, tetrahydro-2,2-dimethyl-, (3aS,6aS)-](http://img.cochemist.com/ccimg/213800/213767-09-0_b.png)


![Pyrrolo[1,2-a]quinoline-1-ethanol,dodecahydro-6-(2Z)-2-penten-4-yn-1-yl-, (1R,3aR,5aR,6R,9aS)-rel-](http://img.cochemist.com/ccimg/75700/75685-48-2.png)
![Pyrrolo[1,2-a]quinoline-1-ethanol,dodecahydro-6-(2Z)-2-penten-4-yn-1-yl-, (1R,3aR,5aR,6R,9aS)-rel-](http://img.cochemist.com/ccimg/75700/75685-48-2_b.png)
![Pyrrolo[1,2-a]quinoline-1-ethanol,dodecahydro-6-(2Z)-2-penten-4-ynyl-, (1R,3aR,5aR,6R,9aS)- (9CI)](http://img.cochemist.com/ccimg/55900/55893-12-4.png)
![Pyrrolo[1,2-a]quinoline-1-ethanol,dodecahydro-6-(2Z)-2-penten-4-ynyl-, (1R,3aR,5aR,6R,9aS)- (9CI)](http://img.cochemist.com/ccimg/55900/55893-12-4_b.png)
![Benzoic acid, 4-[(methoxyimino)methyl]-, methyl ester](http://img.cochemist.com/ccimg/33500/33499-35-3.png)
![Benzoic acid, 4-[(methoxyimino)methyl]-, methyl ester](http://img.cochemist.com/ccimg/33500/33499-35-3_b.png)


![Heptanoic acid, 7-[[(phenylmethoxy)carbonyl]amino]-](http://img.cochemist.com/ccimg/23500/23434-37-9.png)
![Heptanoic acid, 7-[[(phenylmethoxy)carbonyl]amino]-](http://img.cochemist.com/ccimg/23500/23434-37-9_b.png)
![Silane, [(1-ethoxyethenyl)oxy]trimethyl-](http://img.cochemist.com/ccimg/18300/18295-66-4.png)
![Silane, [(1-ethoxyethenyl)oxy]trimethyl-](http://img.cochemist.com/ccimg/18300/18295-66-4_b.png)







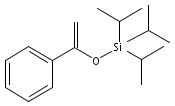
![Octanoic acid, 8-[(tetrahydro-2H-pyran-2-yl)oxy]-](http://img.cochemist.com/ccimg/54700/54699-43-3.png)
![Octanoic acid, 8-[(tetrahydro-2H-pyran-2-yl)oxy]-](http://img.cochemist.com/ccimg/54700/54699-43-3_b.png)