Co-reporter:Hosea Nelson;Qingyu Sun;Tony Ly;Brian M. Stoltz
Journal of Proteome Research February 6, 2009 Volume 8(Issue 2) pp:958-966
Publication Date(Web):2017-2-22
DOI:10.1021/pr800592t
A crown ether based, photolabile radical precursor which forms noncovalent complexes with peptides has been prepared. The peptide/precursor complexes can be electrosprayed, isolated in an ion trap, and then subjected to laser photolysis and collision induced dissociation to generate hydrogen deficient peptide radicals. It is demonstrated that these peptide radicals behave very differently from the hydrogen rich peptide radicals generated by electron capture methods. In fact, it is shown that side chain chemistry dictates both the occurrence and relative abundance of backbone fragments that are observed. Fragmentation at aromatic residues occurs preferentially over most other amino acids. The origin of this selectivity relates to the mechanism by which backbone dissociation is initiated. The first step is abstraction of a β-hydrogen from the side chain, followed by beta-elimination to yield primarily a-type fragment ions. Calculations reveal that those side chains which can easily lose a β-hydrogen correlate well with experimentally favored sites for backbone fragmentation. In addition, radical mediated side chain losses from the parent peptide are frequently observed. Eleven amino acids exhibit unique mass losses from side chains which positively identify that particular amino acid as part of the parent peptide. Therefore, side chain losses allow one to unambiguously narrow the possible sequences for a parent peptide, which when combined with predictable backbone fragmentation should lead to greatly increased confidence in peptide identification.Keywords: 18-crown-6 ether; direct dissociation; iodine; photodissociation; proteomics; ultraviolet;
Co-reporter:Huong T. Pham and Ryan R. Julian
Analyst 2016 vol. 141(Issue 4) pp:1273-1278
Publication Date(Web):15 Jan 2016
DOI:10.1039/C5AN02383A
Glycosphingolipids (GSLs) are important metabolic regulators that control critical cellular functions and are found in all cell membranes. GSL epimers which only differ in the orientation of a single OH group are indicators for different lysosomal storage diseases. In this study, we investigate structural discrimination of GSL isomers using radical-directed dissociation (RDD). Radical fragmentation of different GSL species can be enabled by either noncovalent or covalent modification strategies. The results reveal that RDD can quantitatively distinguish lyso-GSL epimers without the requirement of prior separation by liquid chromatography. In addition, it is demonstrated that covalent labeling with boronic acid enables chemical separation of the two glycolipids isomers due to differential reactiviy of the respective sugar moiteties, offering another avenue for both molecular identification and radical characterization.
Co-reporter:Nathan G. Hendricks and Ryan R. Julian
Analyst 2016 vol. 141(Issue 15) pp:4534-4540
Publication Date(Web):31 May 2016
DOI:10.1039/C6AN01020B
Recent advances in mass spectrometry and lasers have facilitated the development of novel experiments combining the benefits of both technologies. This minireview focuses on the coupling of visible/ultraviolet photons with mass spectrometry for analysis of peptide and protein three-dimensional structure. Practical aspects of instrument design and the relationship between experiment and theory are discussed. Experiments utilizing spectroscopy, action spectroscopy, excitation energy transfer, photodissociation, and photoactivated radical chemistry are described. The strengths and weaknesses of each approach are discussed in relation to the type of information typically obtained. A significant body of data suggests that under appropriate source conditions, kinetically trapped structures are observed in these experiments rather than true gas phase minima, suggesting retention of solution phase structural features is possible. Further refinement and exploration of these methods promises to accelerate protein structure discovery in the near future.
Co-reporter:Yuanqi Tao, Ryan R. Julian
International Journal of Mass Spectrometry 2016 Volume 409() pp:81-86
Publication Date(Web):1 November 2016
DOI:10.1016/j.ijms.2016.10.001
•Radical Directed Dissociation (RDD) is a sensitive technique suitable for probing peptide structure.•18-Crown-6 attachment influences peptide structure in the gas phase.•Long peptides with basic side chains are influenced most by 18-crown-6 complexation.Peptide structure is often correlated with biological function, and recently interest in developing gas-phase based methods for examining peptide structure has grown. The relationship between solution and gas phase structures is unclear, partially due to removal of solvent during the transition. 18-Crown-6 (18C6) is a small molecule that can noncovalently attach to peptides in the gas phase via basic residues, perhaps replacing water and helping retain solution-like structures. Herein, we investigate structural differences between naked peptides and those solvated by 18C6 with radical directed dissociation (RDD), a structurally sensitive fragmentation method. Peptides with and without 18C6 attached often yield disparate RDD spectra, indicating significant structural differences between them. The effects of solvation by 18C6 were explored as a function of peptide size and sequence. Although general trends can be observed with regard to factors that influence solvation, the results suggest that solvation is unique for each peptide and should be examined on a case by case basis.
Co-reporter:James G. Bonner;Nathan G. Hendricks
Journal of The American Society for Mass Spectrometry 2016 Volume 27( Issue 10) pp:1661-1669
Publication Date(Web):2016 October
DOI:10.1007/s13361-016-1456-3
Significant effort is being employed to utilize the inherent speed and sensitivity of mass spectrometry for rapid structural determination of proteins; however, a thorough understanding of factors influencing the transition from solution to gas phase is critical for correct interpretation of the results from such experiments. It was previously shown that combined use of action excitation energy transfer (EET) and simulated annealing can reveal detailed structural information about gaseous peptide ions. Herein, we utilize this method to study microsolvation of charged groups by retention of 18-crown-6 (18C6) in the gas phase. In the case of GTP (CEGNVRVSRE LAGHTGY), solvation of the 2+ charge state leads to reduced EET, whereas the opposite result is obtained for the 3+ ion. For the mini-protein C-Trpcage, solvation by 18C6 leads to dramatic increase in EET for the 3+ ion. Examination of structural details probed by molecular dynamics calculations illustrate that solvation by 18C6 alleviates the tendency of charged side chains to seek intramolecular solvation, potentially preserving native-like structures in the gas phase. These results suggest that microsolvation may be an important tool for facilitating examination of native-like protein structures in gas phase experiments.
Co-reporter:Nathan G. Hendricks and Ryan R. Julian
Chemical Communications 2015 vol. 51(Issue 64) pp:12720-12723
Publication Date(Web):23 Jun 2015
DOI:10.1039/C5CC03779D
Two-step energy transfer is potentially useful for exploring macromolecular structure, but it has not been observed previously in the gas-phase. Single step excitation energy transfer (EET) has been recently documented for tyrosine and tryptophan containing peptides, but not for phenylalanine. Herein, we report sequential energy transfer from phenylalanine to tyrosine to a disulfide, resulting in homolytic cleavage of a sulfur–sulfur bond. Interestingly, energy transfer from phenylalanine is only observed in the presence of tyrosine and only occurs within certain distance constraints. Isolated, electronically excited phenylalanine is known to have an extremely long lifetime in the gas phase, potentially suggesting quicker relaxation occurs via energy transfer to tyrosine. Alternatively, the direct overlap of states between phenylalanine and disulfide bonds is predicted to be poor, in which case tyrosine would serve to bridge the gap. In either case, the distance constraints imposed by this two-step EET are shown to be useful for evaluation and determination of gaseous biomolecular structure.
Co-reporter:Omar M. Hamdy, Arman Alizadeh and Ryan R. Julian
Analyst 2015 vol. 140(Issue 15) pp:5023-5028
Publication Date(Web):05 Jun 2015
DOI:10.1039/C5AN00798D
Maintaining redox homeostasis, or the balance of oxidant and antioxidant forces, is essential for proper cellular functioning in biology. Although the antioxidant nature of many small molecules such as vitamin c and glutathione have been thoroughly investigated, contributions to redox homeostasis from larger biomolecules have received less attention. Evidence has shown that some proteins are antioxidant (in a non-catalytic sense), but large scale examination of this property for a diverse set of proteins has proven difficult. Herein, radical-directed dissociation mass spectrometry (RDD-MS) is used to examine the antioxidant capacity of a series of proteins with diverse biological roles, persistence intervals, and localizations. Digestion of these proteins reveals that all contain antioxidant peptide regions. Examination of the amino acid content of the antioxidant peptides does not reveal significant differences relative to normal peptides, suggesting that sequence may be more important than residue content. Sequence homology analysis across organisms reveals that antioxidant regions are frequently conserved, although many of these regions are also known to have other functions which may have influenced evolutionary pressure. Regardless of the origin, it is clear that many proteins may play secondary roles as sacrificial antioxidants within the cellular milieu.
Co-reporter:Nathan G. Hendricks and Ryan R. Julian
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 39) pp:25822-25827
Publication Date(Web):30 Apr 2015
DOI:10.1039/C5CP01617G
Evaluation of biomolecular structure in the gas phase is challenging, but worthwhile due to advantages in sensitivity and speed relative to traditional condensed phase approaches. Herein, we demonstrate that a recently developed method utilizing energy transfer to establish distance constraints can be combined with molecular dynamics calculations to rapidly and accurately reveal gaseous peptide structures. Three peptides in various charge states are examined. The influence of increasing charge state on peptide structure is easily observed. The presence of multiple conformations can be detected. Furthermore, the method is demonstrated to aid the assignment of charge, which is frequently nontrivial for peptides containing numerous acidic and basic residues that could adopt a variety of conformers of equal charge state. Comparison with ion mobility reveals that many low energy structures that are distinguishable by distance constraints would not be resolvable by collision cross section. Action-EET is demonstrated to be a powerful new tool for structure elucidation.
Co-reporter:Huong T. Pham, Ryan R. Julian
International Journal of Mass Spectrometry 2015 Volume 378() pp:225-231
Publication Date(Web):15 February 2015
DOI:10.1016/j.ijms.2014.08.015
•Non-covalent binding of crown ethers is used to examine specific classes of lipids.•Two efficient methods for generating distonic radical lipid ions are demonstrated.•Radical directed dissociation (RDD) yields more structural information for plasmalogen PE compared to conventional CID.•The approach is used to rapidly separate and characterize the major PE lipid isomers present in soybean.Conventional tandem mass spectrometry relies on even-electron fragmentation that provides limited structural information for glycerophospholipids (GP), which are key constituents of all cell membranes. Different GP classes are chemically very similar and subtle variations in carbon-carbon bonding features and linkages can lead to numerous isomeric structures that are challenging to distinguish with traditional mass spectrometry. In this study, we demonstrate that the primary amine groups in many GP classes can be modified with either noncovalent attachment of crown ether derivatives containing an iodobenzoyl moiety, or by direct covalent attachment of the iodobenzoyl moiety. Radical lipids can be generated using these modifications via photoactivation of labile carbon-iodine bonds, providing rich information about headgroup and fatty acid chain structure. The method is demonstrated for lipid standards containing various carbon chain motifs and linkages, as well as phospholipids extracted from a soybean mixture. Numerous lipids were examined, including plasmalogen-, lyso-, diacyl- types of phospholipids containing mono-/poly- unsaturated fatty acid (FA) substituents, and branched-/nonbranched-FA chains. Interestingly, the presence of double bond and/or vinyl ether linkage leads to the formation of a signature fragment ion that facilitates rapid structural identification.
Co-reporter:Nathan G. Hendricks ; Nichole M. Lareau ; Sarah M. Stow ; John A. McLean
Journal of the American Chemical Society 2014 Volume 136(Issue 38) pp:13363-13370
Publication Date(Web):September 1, 2014
DOI:10.1021/ja507215q
Herein, we report chemistry that enables excitation energy transfer (EET) to be accurately measured via action spectroscopy on gaseous ions in an ion trap. It is demonstrated that EET between tryptophan or tyrosine and a disulfide bond leads to excited state, homolytic fragmentation of the disulfide bond. This phenomenon exhibits a tight distance dependence, which is consistent with Dexter exchange transfer. The extent of fragmentation of the disulfide bond can be used to determine the distance between the chromophore and disulfide bond. The chemistry is well suited for the examination of protein structure in the gas phase because native amino acids can serve as the donor/acceptor moieties. Furthermore, both tyrosine and tryptophan exhibit unique action spectra, meaning that the identity of the donating chromophore can be easily determined in addition to the distance between donor/acceptor. Application of the method to the Trpcage miniprotein reveals distance constraints that are consistent with a native-like fold for the +2 charge state in the gas phase. This structure is stabilized by several salt bridges, which have also been observed to be important previously in proteins that retain native-like structures in the gas phase. The ability of this method to measure specific distance constraints, potentially at numerous positions if combined with site-directed mutagenesis, significantly enhances our ability to examine protein structure in the gas phase.
Co-reporter:Omar M. Hamdy, Steven Lam, and Ryan R. Julian
Analytical Chemistry 2014 Volume 86(Issue 7) pp:3653
Publication Date(Web):March 12, 2014
DOI:10.1021/ac500425f
Antioxidant peptides such as glutathione play critically important roles within cells by opposing the action of oxidative species. Similarly all proteins may, as a secondary function, potentially contribute to the antioxidant capacity of the cellular milieu, though this possibility has not been thoroughly explored previously. Herein it is demonstrated that, in addition to radical quenching solution-phase behavior, antioxidant peptides display an astonishing ability to sequester radicals in the gas phase. Compared to other peptides of similar sequence and size, radical antioxidant peptides exhibit very little radical-directed dissociation when subjected to collisional activation in the gas phase. Importantly, this property can be leveraged in highly sensitive and rapid mass spectrometry based experiments to identify antioxidant peptides. Examination of peptides derived from human serum albumin (HSA), which is a protein known to behave as an antioxidant, revealed three previously unknown peptide regions that exhibit antioxidant capacity. One of these peptides, VAHRFK, shows antioxidant capacity comparable to that of glutathione. It is likely that these peptide regions contribute to the overall antioxidant capacity of HSA. In comparison with previous methods, the present technique is significantly more sensitive and less time-consuming, which should enable more wide-scale examination of antioxidant peptides that are relevant to redox homeostasis, food chemistry, and disease.
Co-reporter:Huong T. Pham and Ryan R. Julian
Analytical Chemistry 2014 Volume 86(Issue 6) pp:3020
Publication Date(Web):February 18, 2014
DOI:10.1021/ac403754j
Phosphatidylethanolamines (PE) and phosphatidylcholines (PC) are important phospholipids frequently present in many types of cells. In some cases, PE has been equated with PC because they are chemically very similar and are often isomeric species. In this study, we demonstrate that noncovalent complexation between PE and 18-crown-6 ether (18C6) can be used to quantitatively mass shift and separate PE from PC phospholipids. Detection of PE is also more sensitive by approximately an order of magnitude with addition of 18C6. This noncovalent complexation approach is used to separate and quantitatively characterize PE in a soy bean asolectin extract. 18C6 (modified with an iodobenzoyl moiety) can also be used to efficiently generate radical PE lipids following photoactivation in the gas phase. Subsequent collisional activation of these lipid radical ions leads to radical directed dissociation (RDD), which generates unique fragment ions relative to dissociation of comparable even electron ions. Interestingly, RDD produces fragment ions that reveal carbon bonding features within the lipid acyl chain substituents, such as double bond location or the presence of branching. Furthermore, several novel and abundant fragments were observed in unsaturated lipids. Mechanisms that can account for the high abundance of some of these product ions are proposed.
Co-reporter:Yuanqi Tao and Ryan R. Julian
Analytical Chemistry 2014 Volume 86(Issue 19) pp:9733
Publication Date(Web):September 4, 2014
DOI:10.1021/ac502296c
Post-translational modifications that do not result in a change in mass are particularly difficult to detect by mass spectrometry. For example, isomerization of aspartic acid or epimerization of any chiral residue within a peptide do not lead to mass shifts but can be identified by examination of independently acquired tandem mass spectra or by combination with another technique. For analysis of a biological sample, this means that liquid chromatography or some other type of separation must be used to first separate the isomers from one another. Furthermore, each specific m/z of interest must be sampled repeatedly to allow for comparison of the tandem mass spectra from each separated isomer, which contrasts with the traditional approach in proteomics where the goal is typically to avoid resampling the same m/z. We illustrate that isomerization and epimerization of peptides can be identified in this fashion by examination of long-lived crystallin proteins extracted from a sheep eye lens. Tandem mass spectrometry relying on a combination of radical directed dissociation (RDD) and collision induced dissociation (CID) following separation by liquid chromatography was used to identify modified peptides. Numerous sites of isomerization and epimerization, including several that have not been previously identified, were determined with peptide specificity. It is demonstrated that the specific sites of amino acid isomerization within each peptide can be identified by comparison with synthetic peptides. For α-crystallin proteins, the sites that undergo the greatest degree of isomerization correspond to disordered regions, which may have important implications on chaperone functionality within the context of aging.
Co-reporter:Xing Zhang, Ryan R. Julian
International Journal of Mass Spectrometry 2014 Volume 372() pp:22-28
Publication Date(Web):1 November 2014
DOI:10.1016/j.ijms.2014.07.045
•Radical directed dissociation of saccharides is developed with photodissociation.•Two methods that generate radical saccharides are described.•Glycosidic and cross-ring bond cleavage products are generated.•Isomeric saccharides are easily distinguished with radical directed dissociation.Radical chemistry continues to play an increasingly important role in tandem mass spectrometry based experiments on biomolecules. Oligosaccharides represent a very important class of target molecules that require structural characterization in terms of both monosaccharide identity and overall connectivity. Herein, two methods that generate radical oligosaccharides for subsequent activation are described. In one approach, a radical precursor is covalently attached to the oligosaccharide by reductive amination. Radicals can then be generated by homolytic bond cleavage of specific carbon–iodine bonds in protonated systems by either collisional activation or photodissociation. Subsequent activation of the radical species generates information rich spectra including numerous cross-ring fragments. Alternatively, noncovalent complexation with iodophthalic acid can be used to generate radical disaccharides by photoactivation. Subsequent radical transfer, loss of the radical precursor adduct, and collisional activation of the radical disaccharide results in characteristic glycosidic bond cleavage and cross-ring cleavage products that can easily distinguish isomeric species. Radical chemistry is demonstrated to have several advantages for the characterization of oligosaccharides relative to other approaches, including the identification of isomeric molecules of various sizes or analysis of various charge states.
Co-reporter:Xing Zhang
Journal of The American Society for Mass Spectrometry 2014 Volume 25( Issue 4) pp:626-635
Publication Date(Web):2014 April
DOI:10.1007/s13361-013-0810-y
Accurate identification of fragments in tandem mass spectrometry experiments is aided by knowledge of relevant fragmentation mechanisms. Herein, novel radical addition reactions that direct unexpected side-chain dissociations at tryptophan and tyrosine residues are reported. Various mechanisms that can account for the observed dissociation channels are investigated by experiment and theory. The propensity for radical addition at a particular site is found to be primarily under kinetic control, which is largely dictated by molecular structure. In certain peptides, intramolecular radical addition reactions are favored, which leads to the observation of numerous unexpected fragments. In one pathway, radical addition leads to migration of an aromatic side chain to another residue. Alternatively, radical addition followed by hydrogen atom loss leads to cyclization of the peptide and increased observation of internal sequence fragments. Radical addition reactions should be considered when assigning fragmentation spectra obtained from activation of hydrogen deficient peptides.
Co-reporter:Ryan R. Julian;Richard W. Vachet
Journal of The American Society for Mass Spectrometry 2014 Volume 25( Issue 8) pp:1307-1309
Publication Date(Web):2014 August
DOI:10.1007/s13361-014-0897-9
Co-reporter:Huong T. Pham, Ryan R. Julian
International Journal of Mass Spectrometry 2014 370() pp: 58-65
Publication Date(Web):
DOI:10.1016/j.ijms.2014.06.022
Co-reporter:Benjamin B. Kirk, Adam J. Trevitt, Stephen J. Blanksby, Yuanqi Tao, Benjamin N. Moore, and Ryan R. Julian
The Journal of Physical Chemistry A 2013 Volume 117(Issue 6) pp:1228-1232
Publication Date(Web):August 20, 2012
DOI:10.1021/jp305470j
Structural investigations of large biomolecules in the gas phase are challenging. Herein, it is reported that action spectroscopy taking advantage of facile carbon–iodine bond dissociation can be used to examine the structures of large molecules, including whole proteins. Iodotyrosine serves as the active chromophore, which yields distinctive spectra depending on the solvation of the side chain by the remainder of the molecule. Isolation of the chromophore yields a double featured peak at ∼290 nm, which becomes a single peak with increasing solvation. Deprotonation of the side chain also leads to reduced apparent intensity and broadening of the action spectrum. The method can be successfully applied to both negatively and positively charged ions in various charge states, although electron detachment becomes a competitive channel for multiply charged anions. In all other cases, loss of iodine is by far the dominant channel which leads to high sensitivity and simple data analysis. The action spectra for iodotyrosine, the iodinated peptides KGYDAKA, DAYLDAG, and the small protein ubiquitin are reported in various charge states.
Co-reporter:Xing Zhang
Journal of The American Society for Mass Spectrometry 2013 Volume 24( Issue 4) pp:524-533
Publication Date(Web):2013 April
DOI:10.1007/s13361-012-0540-6
One of the keys for understanding radical directed dissociation in peptides is a detailed knowledge of the factors that mediate radical migration. Peptide radicals can be created by a variety of means; however, in most circumstances, the originally created radicals must migrate to alternate locations in order to facilitate fragmentation such as backbone cleavage or side chain loss. The kinetics of radical migration are examined herein by comparing results from ortho-, meta-, and para-benzoyl radical positional isomers for several peptides. Isomers of a constrained cyclic peptide generated by several orthogonal radical initiators are also probed as a function of charge state. Cumulatively, the results suggest that small changes in radical position can significantly impact radical migration, and overall structural flexibility of the peptide is also an important controlling factor. A particularly interesting pathway for the peptide RGYALG that is sensitive to ortho versus meta or para substitution was fully mapped out by a suite of deuterium labeled peptides. This data was then used to optimize parameters in molecular dynamics-based simulations, which were subsequently used to obtain further insight into the structural underpinnings that most strongly influence the kinetics of radical migration.
Co-reporter:Yuanqi Tao
Journal of The American Society for Mass Spectrometry 2013 Volume 24( Issue 11) pp:1634-1640
Publication Date(Web):2013 November
DOI:10.1007/s13361-013-0684-z
The ability of 18-crown-6 (18C6) to form noncovalent complexes with cationic groups in the gas phase has been leveraged in numerous, largely orthogonal mass spectrometry-based applications. Although the fundamental interaction between 18C6 and a charged group in the gas phase is quite strong, the strength of attachment of 18C6 to large molecules is more difficult to predict because intramolecular binding of the cation can be competitive. Herein, we demonstrate in experiments with model peptides that 18C6 adducts are not strongly attached to flexible molecules with numerous potential hydrogen bonding sites. 18C6 adduct stability is increased if intramolecular charge complexation is inhibited by sterics or competitive binding. It is demonstrated with molecular mechanics that significant structural changes occur upon loss of 18C6 in model peptides. Examination of the loss of 18C6 adducts from proteins following collisional activation reveals that lower charge states lose the most 18C6. The degree of 18C6 adduct stability may reflect the degree of structural reorganization that occurs following collisional activation, suggesting that lower charge states represent structures that are not similar to gas phase idealized states. In this regard, 18C6 may serve the function of protecting solution phase protein structure. Collisional activation of holomyoglobin with 18C6 adducts attached reveals that heme loss occurs primarily after 18C6 loss, further supporting the notion that 18C6 protects native structure by solvating charged sites.
Co-reporter:Yuanqi Tao, Neil R. Quebbemann, and Ryan R. Julian
Analytical Chemistry 2012 Volume 84(Issue 15) pp:6814
Publication Date(Web):July 18, 2012
DOI:10.1021/ac3013434
The presence of a single d-amino acid in a peptide is very difficult to detect. Mass spectrometry-based approaches rely on differences in fragmentation between all l-amino acid-containing peptides and single d-amino acid-containing peptides (which are epimers) for identification. The success of this approach is dependent on the structural sensitivity of the fragmentation method. Recently, experiments have demonstrated that fragmentation initiated by radical chemistry, or radical-directed dissociation (RDD), is particularly sensitive to the structure of the ion being fragmented. Herein, RDD is used to identify the presence of d-serine, d-alanine, or d-aspartic acid in eight biologically relevant peptides. It is demonstrated that chiral disambiguation by RDD is dependent on both the initial radical site and subsequent radical migration. Fortuitously, RDD can be initiated by a variety of different radical precursors which can be associated with the peptide via covalent or noncovalent means, and RDD can be examined in all observable charge states (both positive and negative). This diversity enables numerous initial radical sites and migration pathways to be explored. For all but one of the peptides that were examined, RDD provides significantly better chiral discrimination than CID. Quantitation of peptide epimers by RDD is also described.
Co-reporter:Benjamin N. Moore and Ryan R. Julian
Physical Chemistry Chemical Physics 2012 vol. 14(Issue 9) pp:3148-3154
Publication Date(Web):30 Jan 2012
DOI:10.1039/C2CP23443B
In biochemistry, free radicals are versatile species which can perform diverse functions including: signaling, synthesis, and destructive modification. It is of interest to understand how radicals behave within all biomolecules and specifically within peptides and proteins. The 20 standard amino acids contain a wide range of chemical structures, which give proteins their complexity and ultimately their functionality. Many factors influence how radicals interact with these complex molecules, including the bond dissociation energies (BDEs) for homolytically cleaving any X–H bonds. The BDEs provide a simple measure for comparing the thermodynamic favorability of abstracting hydrogen atoms from various sites within a protein. BDEs for abstractable hydrogen atoms have been calculated for each amino acid, the peptide backbone, and peptide termini in order to compile a roadmap of the relative thermodynamics which influence protein radical chemistry. With this information it is possible to gain insight into what contributions both kinetics and thermodynamics will make to various radical mediated reaction pathways.
Co-reporter:Xing Zhang and Ryan R. Julian
Physical Chemistry Chemical Physics 2012 vol. 14(Issue 47) pp:16243-16249
Publication Date(Web):31 Oct 2012
DOI:10.1039/C2CP42242E
Polyproline is a fascinating polymer with interesting structural properties that have been studied in both solution and the gas phase. Herein, a method capable of measuring structural dynamics over long timescales is developed and applied to examination of polyproline in the gas phase. This method is based on measuring the probability of two radicals recombining to form a new covalent bond within a single molecule, which provides distance constraint information. To examine polyproline peptides of various lengths, radical precursors were selectively placed at the termini. Photoactivation with 266 nm light can then be used to create a diradical species, and recombination of the two radicals can be used to evaluate end-to-end distances and structural flexibility. The results reveal that interaction of the polyproline termini is more favorable for shorter chain lengths and lower charge states. As charge states increase, Coulombic repulsion favors formation of more extended structures where the termini no longer come in close contact. With increasing chain length, the greater conformational space also appears to decrease the likelihood of the termini being able to recombine. The amount of radical recombination observed for short polyproline peptides in low charge states is not consistent with what would be expected for helical conformations. Rather, molecular mechanics calculations reveal that lower charge state polyproline peptides tends to adopt globular conformations in the gas phase.
Co-reporter:Yuanqi Tao and Ryan R. Julian
Biochemistry 2012 Volume 51(Issue 8) pp:
Publication Date(Web):February 2, 2012
DOI:10.1021/bi2018199
A simple mass spectrometry-based method capable of examining protein structure called SNAPP (selective noncovalent adduct protein probing) is used to evaluate the structural consequences of point mutations in naturally occurring sequence variants from different species. SNAPP monitors changes in the attachment of noncovalent adducts to proteins as a function of structural state. Mutations that lead to perturbations to the electrostatic surface structure of a protein affect noncovalent attachment and are easily observed with SNAPP. Mutations that do not alter the tertiary structure or electrostatic surface structure yield similar results by SNAPP. For example, bovine, porcine, and human insulin all have very similar backbone structures and no basic or acidic residue mutations, and the SNAPP distributions for all three proteins are very similar. In contrast, four variants of cytochrome c (cytc) have varying degrees of sequence homology, which are reflected in the observed SNAPP distributions. Bovine and pigeon cytc have several basic or acidic residue substitutions relative to horse cytc, but the SNAPP distributions for all three proteins are similar. This suggests that these mutations do not significantly influence the protein surface structure. On the other hand, yeast cytc has the least sequence homology and exhibits a unique, though related, SNAPP distribution. Even greater differences are observed for lysozyme. Hen and human lysozyme have identical tertiary structures but significant variations in the locations of numerous basic and acidic residues. The SNAPP distributions are quite distinct for the two forms of lysozyme, suggesting significant differences in the surface structures. In summary, SNAPP experiments are relatively easy to perform, require minimal sample consumption, and provide a facile route for comparison of protein surface structure between highly homologous proteins.
Co-reporter:Benjamin Moore;Qingyu Sun;Julie C. Hsu
Journal of The American Society for Mass Spectrometry 2012 Volume 23( Issue 3) pp:460-468
Publication Date(Web):2012 March
DOI:10.1007/s13361-011-0318-2
The fragmentation chemistry of anionic deprotonated hydrogen-deficient radical peptides is investigated. Homolytic photodissociation of carbon–iodine bonds with 266 nm light is used to generate the radical species, which are subsequently subjected to collisional activation to induce further dissociation. The charges do not play a central role in the fragmentation chemistry; hence deprotonated peptides that fragment via radical directed dissociation do so via mechanisms which have been reported previously for protonated peptides. However, charge polarity does influence the overall fragmentation of the peptide. For example, the absence of mobile protons favors radical directed dissociation for singly deprotonated peptides. Similarly, a favorable dissociation mechanism initiated at the N-terminus is more notable for anionic peptides where the N-terminus is not protonated (which inhibits the mechanism). In addition, collisional activation of the anionic peptides containing carbon–iodine bonds leads to homolytic cleavage and generation of the radical species, which is not observed for protonated peptides presumably due to competition from lower energy dissociation channels. Finally, for multiply deprotonated radical peptides, electron detachment becomes a competitive channel both during the initial photoactivation and following subsequent collisional activation of the radical. Possible mechanisms that might account for this novel collision-induced electron detachment are discussed.
Co-reporter:Benjamin N. Moore, Omar Hamdy, Ryan R. Julian
International Journal of Mass Spectrometry 2012 s 330–332() pp: 220-225
Publication Date(Web):
DOI:10.1016/j.ijms.2012.08.013
Co-reporter:Jolene K. Diedrich
Analytical and Bioanalytical Chemistry 2012 Volume 403( Issue 8) pp:2269-2277
Publication Date(Web):2012 June
DOI:10.1007/s00216-012-5867-0
Described herein is a method which combines bond selective fragmentation by photodissociation with online liquid chromatographic separation and mass spectrometric analysis. Photoexcitation of proteins or peptides with 266-nm light does not normally yield abundant fragmentation; however, incorporation of a suitable carbon–sulfur or carbon–halogen bond that is proximal to a chromophore allows access to direct dissociation pathways, resulting in homolytic cleavage of these bonds. Radicals generated through this process can cause further dissociation of the peptide backbone, which is useful for site specifically identifying the point of modification. Two specific applications of this technique for peptide analysis in model systems are presented: (1) identification of reactive metabolites which covalently modify cysteine residues, and (2) characterization of halogenated tyrosine residues which are biomarkers related to oxidative stress. In both cases, these naturally occurring post translational modifications create photocleavable bonds which can be fragmented by 266-nm light. The selectivity offered by photodissociation allows facile identification of the peptides of interest even in complex mixtures, and subsequent selective radical directed backbone fragmentation pinpoints the site of modification. This combination greatly simplifies data analysis and provides more confident assignments.
Co-reporter:Omar M. Hamdy
Journal of The American Society for Mass Spectrometry 2012 Volume 23( Issue 1) pp:1-6
Publication Date(Web):2012 January
DOI:10.1007/s13361-011-0284-8
The connection between charge state distributions, protein structure, and mechanistic details of electrospray are discussed in relation to the emerging field of gas phase structural biology. Comparisons are drawn with the established area of enzymatic catalysis in organic solvents, which shares many similar challenges. Charge solvation emerges as a dominant force in both systems that must be dealt with to enable kinetic trapping of native structures in foreign environments. Potential methods for mediating unfavorable charge solvation effects are discussed and, ironically, do not include partial solvation by water. The importance of timescale in relation to the evolution of protein structure during the process of electrospray ionization is discussed. Finally several prospects for future endeavors are highlighted.
Co-reporter:Tony Ly, Xing Zhang, Qingyu Sun, Benjamin Moore, Yuanqi Tao and Ryan R. Julian
Chemical Communications 2011 vol. 47(Issue 10) pp:2835-2837
Publication Date(Web):24 Jan 2011
DOI:10.1039/C0CC03363D
Novel p-iodobenzoate-based labelling reagents are shown to be effective photocaged precursors for synthesizing biomolecular radicals site-selectively in the gaseous and condensed phases. In vacuo, a single pulse of UV photons (266 nm) is sufficient to quantitatively photolyse the C–I bond. In aqueous solutions, the photolysis half-life is estimated to be 2.5 minutes when irradiating with a 15 W compact fluorescent lamp (254 nm).
Co-reporter:Arun Agarwal, Jolene K. Diedrich, and Ryan R. Julian
Analytical Chemistry 2011 Volume 83(Issue 17) pp:6455
Publication Date(Web):July 28, 2011
DOI:10.1021/ac201650v
Disulfide bonds stabilize the tertiary and quaternary structure of proteins. Identifying the correct disulfide bond pairs can be extremely useful to understand the nature of a protein. However identifying correct disulfide linkages remains a challenge for many proteins. We report the use of ultraviolet photodissociation (UVPD) at 266 nm to selectively cleave disulfide bonds in the gas phase, while leaving all other bonds intact. This methodology can be used to identify disulfide bonded pairs in complex systems with multiple disulfide bond partners. We have explored UVPD chemistry on pairs of model peptides with one disulfide bond to evaluate the importance of various sequence and structural effects. In addition, online experiments were performed on whole protein digests. Bond selective UVPD was able to correctly identify and characterize all known disulfide bonded pairs. The method also proved sufficiently sensitive to identify and characterize several non-native disulfide-bound peptide pairs which were present in trace amounts. Photodissociation at 266 nm can be a valuable tool for disulfide bond identification and pair assignment in high-throughput proteomics studies.
Co-reporter:Jolene K. Diedrich and Ryan R. Julian
Analytical Chemistry 2011 Volume 83(Issue 17) pp:6818
Publication Date(Web):July 25, 2011
DOI:10.1021/ac201647w
Identification of phosphorylation sites is of interest due to their importance in protein regulation; however, the identification of the exact sites of this modification is not always easily obtained due to the dynamic nature of phosphorylation and the challenges faced during mass spectrometric analysis. Herein we elaborate on our previous communication (Diedrich, J. K.; Julian, R. R. J. Am. Chem. Soc.2008, 130, 12212–12213) describing a novel technique for assignment of phosphorylation in a site-specific and facile manner. Phosphorylation sites are selectively modified through β elimination followed by Michael addition chemistry to install a photolabile group. Photodissociation with 266 nm light yields homolytic cleavage at the modification site, generating a β radical which is poised to fragment the peptide backbone. Dissociation primarily yields d-type ions at the previously phosphorylated residue, allowing facile identification. Radical directed fragmentation also occurs in smaller abundances at neighboring residues. The mechanisms behind this selective radical fragmentation are presented and the utility is discussed. Fragmentation is shown to be independent of charge state allowing analysis of a wide variety of peptide sequences including peptides with multiple phosphorylation sites. A comparison of this technique is made with collision induced dissociation (CID) and electron capture dissociation (ECD) for representative peptides.
Co-reporter:Xing Zhang, Ryan R. Julian
International Journal of Mass Spectrometry 2011 Volume 308(2–3) pp:225-231
Publication Date(Web):1 December 2011
DOI:10.1016/j.ijms.2011.07.010
Evaluating protein structure in the gas phase is useful for understanding the intrinsic forces which influence protein folding and for determining the feasibility of probing condensed phase structure with gas phase interrogation. KIX is a three-helix bundle protein that has been reported previously to preserve the condensed phase structure in the gas phase. Herein, structure dependent radical directed dissociation (RDD) is used to examine the gas phase structure of KIX by establishing residue specific distance constraints which can be used to assess candidate structures obtained from molecular dynamics simulations. The data obtained by RDD is consistent with KIX structures that largely retain condensed phase structure as determined previously by NMR. There are several factors that favor retention of the KIX native fold in the gas phase. The structure is largely comprised of alpha helices, which are known to be stable in the gas phase. This is particularly true if the C-terminus of the helix is capped with a positive charge, which occurs for the two most stable helices in KIX. There are several arginine based salt bridges which link critical portions of KIX together. KIX also has an abundance of basic residues; this multiplicity increases the chance that sites which require little structural reorganization following desolvation can be charge carriers. Thus under appropriate conditions, solution phase structure can be largely retained and meaningfully examined in the gas phase.Graphical abstractHighlights► Radical directed dissociation is used to examine protein structure in the gas phase. ► KIX is a three helix bundle protein that retains its native structure in the gas phase. ► Various electrostatic forces stabilize the native structure in the gas phase.
Co-reporter:Qingyu Sun;Robert C. Tyler
Journal of The American Society for Mass Spectrometry 2011 Volume 22( Issue 3) pp:399-407
Publication Date(Web):2011 March
DOI:10.1007/s13361-010-0042-3
The human chemokine lymphotactin (Ltn) is a remarkable protein that interconverts between two unrelated native state structures in the condensed phase. It is possible to shift the equilibrium toward either conformation with selected sequence substitutions. Previous results have shown that a disulfide-stabilized variant preferentially adopts the canonical chemokine fold (Ltn10), while a single amino acid change (W55D) favors the novel Ltn40 dimeric structure. Selective noncovalent adduct protein probing (SNAPP) is a recently developed method for examining solution phase protein structure. Herein, it is demonstrated that SNAPP can easily recognize and distinguish between the Ltn10 and Ltn40 states of lymphotactin in aqueous solution. The effects of organic denaturants, acid, and disulfide bond reduction and blocking were also examined using SNAPP for the CC3, W55D, and wild type proteins. Only disulfide reduction was shown to significantly perturb the protein, and resulted in considerably decreased adduct formation consistent with loss of tertiary/secondary structure. Cold denaturation experiments demonstrated that wild-type Ltn is the most temperature sensitive of the three proteins. Examination of the higher charge states in all experiments, which are presumed to represent transition state structures between Ltn-10 and Ltn-40, reveals increased 18C6 attachment relative to the more folded structures. This observation is consistent with increased competitive intramolecular hydrogen bonding, which may guide the transition. Experiments examining the gas phase structures revealed that all three proteins can be structurally distinguished in the gas phase. In addition, the gas phase experiments enabled identification of preferred adduct binding sites.
Co-reporter:Tony Ly
Journal of the American Chemical Society 2010 Volume 132(Issue 25) pp:8602-8609
Publication Date(Web):June 4, 2010
DOI:10.1021/ja910665d
A new method for identifying residue specific through space contacts as a function of protein secondary and tertiary structure in the gas phase is presented. Photodissociation of a non-native carbon−iodine bond incorporated into Tyr59 of ubiquitin yields a radical site specifically at that residue. The subsequent radical migration is shown to be highly dependent on the structure of the protein. Radical-directed dissociation (RDD) of low charge states, which adopt compact structures, generates backbone fragmentation that is prominently distributed throughout the protein sequence, including residues that are distant in sequence from Tyr59. Higher charge states of ubiquitin, which adopt elongated, unfolded structures, yield RDD that is primarily nearby in sequence to Tyr59. Regardless of which structure is probed, information at the residue-level is obtained by examining specific radical-donor and radical-acceptor pairs. The relative importance of a particular interaction pair for a specific conformation can be revealed by tracking the charge state dependence of the dissociation. Structurally important contact pairs exhibit strong and concerted changes in relative intensities as a function of charge state and can also be used to reveal structural features which persist among different protein structures. Moreover, incorporation of distance constraint information into molecular mechanics conformational searches can be used to drive the search toward relevant conformational space. Implementation of this approach has revealed highly stable, previously undiscovered structures for the +4 and +6 charge states of ubiquitin, which bear little resemblance to the crystal structure.
Co-reporter:Jolene K. Diedrich and Ryan R. Julian
Analytical Chemistry 2010 Volume 82(Issue 10) pp:4006
Publication Date(Web):April 20, 2010
DOI:10.1021/ac902786q
Described herein are several unique analytical applications utilizing mass spectrometry and the selective modification of the free thiol form of cysteine in both peptides and proteins by various quinones. This simple modification can be used to quantify the number of free or disulfide bound cysteines in a protein. In addition, quinone modification can also be used to easily probe the solvent accessibility of cysteine residues, which provides information about protein structure or folding state. Furthermore, the chromophoric properties of the quinone moiety can be leveraged for site specific photodissociation of the backbone. The photodissociation reveals both the presence and location of modified cysteine residues. For example, cleavage of the protein backbone of α-hemoglobin is observed selectively at a single cysteine out of 140 residues in the whole protein. This selective backbone fragmentation is accompanied by a parent ion mass loss, which is unique to the modifying quinone. When combined, this information can be used to determine both the presence and site of modification generated by naturally occurring molecules, such as dopamine, which can harness quinone chemistry to modify proteins.
Co-reporter:Qingyu Sun, Sheng Yin, Joseph A. Loo and Ryan R. Julian
Analytical Chemistry 2010 Volume 82(Issue 9) pp:3826
Publication Date(Web):March 31, 2010
DOI:10.1021/ac100256v
Iodination of tyrosine residues in proteins has many uses in chemistry, biology, and medicine. Site specific identification of the sites of iodination is important for many of these uses. Reported herein is a facile method employing photodissociation and mass spectrometry to localize sites of iodination in whole proteins. Absorption of ultraviolet photons by iodotyrosine results in loss of iodine via homolytic bond dissociation. The resulting protein radical fragments in the vicinity of the iodotyrosine upon collisional activation. Analysis of the fragments within the vicinity of each tyrosine residue in the protein enables quantitative evaluation of the likelihood for iodination at each site. The results are compared with both traditional bottom up and top down mass spectrometric methods. Radical directed dissociation yields results in agreement with traditional approaches but requires significantly less effort and is inherently more sensitive. One limitation occurs when multiple tyrosine residues are in close proximity, in which case the extent of iodination at each residue may be difficult to determine. This limitation is frequently problematic for traditional approaches as well.
Co-reporter:Eric R. Knudsen, Ryan R. Julian
International Journal of Mass Spectrometry 2010 Volume 294(2–3) pp:83-87
Publication Date(Web):1 July 2010
DOI:10.1016/j.ijms.2010.05.012
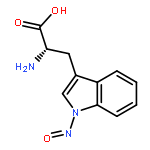
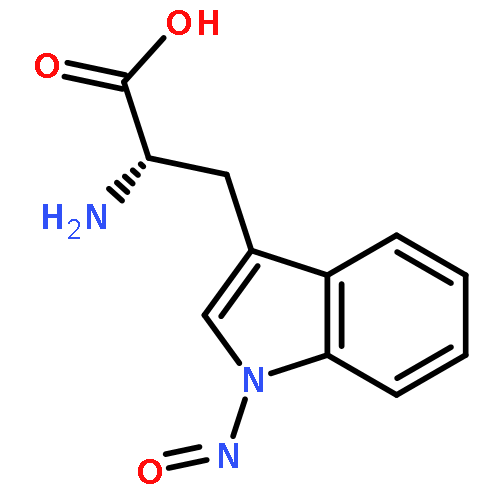
The N-nitrosation reaction has been utilized to add an NO group to the indole nitrogen of tryptophan in several peptides. These peptides can be electrosprayed and then subjected to collision-induced dissociation (CID). The input of CID energy causes the homolytic cleavage of the labile nitrogen–nitrogen single bond resulting in removal of the NO group and generation of a hydrogen deficient peptide radical. This N-nitrosation reaction serves as a simple way to create peptide radicals in the gas phase. Here we examine features of CID for several N-nitrosopeptides and give mechanisms which explain the observed chemistry. In particular, tryptophan side chain loss is frequently an abundant fragmentation channel. Interestingly dissociation of the N–NO bond occurs concomitantly with CO2 loss for peptides with C-terminal tryptophan residues. The location of the nascent radical is an important factor in both of these dissociation pathways. Other fragmentation channels are observed to occur via radical- or proton-catalyzed pathways, depending on the mobility of available protons. Theoretical calculations were also performed to study the energetics of the proposed mechanisms.The dissociation chemistry for peptides containing radical tryptophan is described. One favorable fragmentation channel is the loss of the tryptophan side chain itself.
Co-reporter:Benjamin N. Moore, Stephen J. Blanksby and Ryan R. Julian
Chemical Communications 2009 (Issue 33) pp:5015-5017
Publication Date(Web):13 Jul 2009
DOI:10.1039/B907833A
Ion–molecule reactions between molecular oxygen and peptide radicals in the gas phase demonstrate that radical migration occurs easily within large biomolecules without addition of collisional activation energy.
Co-reporter:Tony Ly, Ryan R. Julian
Journal of the American Society for Mass Spectrometry 2009 Volume 20(Issue 6) pp:1148-1158
Publication Date(Web):June 2009
DOI:10.1016/j.jasms.2009.02.009
Photodissociation of iodo-tyrosine modified peptides yields localized radicals on the tyrosine side chain, which can be further dissociated by collisional activation. We have performed extensive experiments on model peptides, RGYALG, RGYG, and their derivatives, to elucidate the mechanisms underlying backbone fragmentation at tyrosine. Neither acetylation nor deuteration of the tyrosyl phenolic hydrogen significantly affects backbone fragmentation. However, deuterium migration from the tyrosyl β carbon is concomitant with cleavage at tyrosine. Substitution of tyrosine with 4-hydroxyphenylglycine, which does not have β hydrogens, results in almost complete elimination of backbone fragmentation at tyrosine. These results suggest that a radical situated on the β carbon is required for a-type fragmentation in hydrogen-deficient radical peptides. Replacement of the αH of the residue adjacent to tyrosine with methyl groups results in significant diminution of backbone fragmentation. The initial radical abstracts an αH from the adjacent amino acid, which is poised to “rebound” and abstract the βH of tyrosine through a six-membered transition-state. Subsequent β-scission leads to the observed a-type backbone fragment. These results from deuterated peptides clearly reveal that radical migration in peptides can occur and that multiple migrations are not infrequent. Counterintuitively, close examination of all experimental results reveals that the probability for fragmentation at a particular residue is well correlated with thermodynamic radical stability. A-type fragmentation therefore appears to be most likely when favorable thermodynamics are combined with the relevant kinetic control. These results are consistent with ab initio calculations, which demonstrate that barriers to migration are significantly smaller in magnitude than probable dissociation thresholds.Radical migration is shown to occur in large peptides and accounts for selective fragmentation at tyrosine residues and other sites.Figure optionsDownload full-size imageDownload high-quality image (28 K)Download as PowerPoint slide
Co-reporter:Geoffrey K. Yeh;Qingyu Sun;Claudia Meneses
Journal of The American Society for Mass Spectrometry 2009 Volume 20( Issue 3) pp:385-393
Publication Date(Web):2009 March
DOI:10.1016/j.jasms.2008.10.019
Ultraviolet photodissociation of peptides followed by mass analysis has several desirable advantages relative to other methods, yet it has not found widespread use due to several limitations. One shortcoming is the inefficiency with which peptides absorb in the ultraviolet. This issue has a simple solution and can be circumvented by the attachment of noncovalent adducts that contain appropriate chromophores. Subsequent photoactivation of the chromophore leads to vibrational excitation of the complex and eventually to fragmentation of the peptide. Herein, the energetics that control the efficiency of this process are examined as a function of the characteristics of both the peptide and the noncovalently attached chromophore. Fragmentation efficiency decreases with increasing peptide size and is also constrained by the binding energy of the noncovalent adduct. The optimum chromophore should have excellent absorption at the excitation wavelength and a low luminescence quantum yield. It is demonstrated that a naphthyl based 18-crown-6 adduct is ideally suited for attaching to a variety peptides and fragmenting them following absorption of 266 nm light. Potential applications and limitations of this methodology are discussed.
Co-reporter:Zhenjiu Liu
Journal of The American Society for Mass Spectrometry 2009 Volume 20( Issue 6) pp:965-971
Publication Date(Web):2009 June
DOI:10.1016/j.jasms.2008.12.014
Iodination of tyrosine was recently discovered as a useful method for generating radical peptides via photodissociation of carbon-iodine bonds by an ultraviolet photon in the gas phase. The subsequent fragmentation behavior of the resulting odd-electron peptides is largely controlled by the radical. Although previous experiments have focused on mono-iodination of tyrosine, peptides and proteins can also be multiply iodinated. Tyrosine and, to a lesser extent, histidine can both be iodinated or doubly iodinated. The behavior of doubly iodinated residues is explored under conditions where the sites of iodination are carefully controlled. It is found that radical peptides generated by the loss of a single iodine from doubly iodinated tyrosine behave effectively identically to singly iodinated peptides. This suggests that the remaining iodine does not interfere with radical directed dissociation pathways. In contrast, the concerted loss of two iodines from doubly iodinated peptides yields substantially different results that suggest that radical recombination can occur. However, sequential activation can be used to generate multiple usable radicals in different steps of an MSn experiment. Furthermore, it is demonstrated that in actual peptides, the rate of iodination for tyrosine versus mono-iodotyrosine cannot be predicted easily a priori. In other words, previous assumptions that mono-iodination of tyrosine is the rate-limiting step to the formation of doubly iodinated tyrosine are incorrect.
Co-reporter:Tony Ly ;RyanR. Julian
Angewandte Chemie 2009 Volume 121( Issue 39) pp:7266-7273
Publication Date(Web):
DOI:10.1002/ange.200900613
Abstract
Die Auswertung aller Informationen in einem Proteom ist eine Herausforderung für die chemische Analyse, doch winkt als Lohn ein enormer Wissenszuwachs. Die Massenspektrometrie bietet eine einmalige Auswahl an Verfahren für verschiedene Ansätze der Proteomanalyse. Vor kurzem wurde gezeigt, dass man mit UV-Bestrahlung eine eindeutige Fragmentierung von Peptiden und Proteinen induzieren kann. Dabei ist eine unspezifische Dissoziation ebenso möglich wie eine hochspezifische Dissoziation an künstlich eingeführten Chromophoren, nachdem diese ein Photon absorbiert haben. Die Spezifität und Kontrollierbarkeit der Fragmentierung in solchen Experimenten sind besser als bei anderen Fragmentierungsverfahren. Von neuen Methoden, die auf dieser Technik beruhen, erwartet man wichtige Beiträge zur Proteomforschung.
Co-reporter:Tony Ly ;RyanR. Julian
Angewandte Chemie International Edition 2009 Volume 48( Issue 39) pp:7130-7137
Publication Date(Web):
DOI:10.1002/anie.200900613
Abstract
Unraveling of all of the information contained in proteomes poses a tremendous chemical challenge, which is balanced by the promise of potentially transformational knowledge. Mass spectrometry offers an unprecedented arsenal of tools for diverse proteomic investigations. Recently, it was demonstrated that ultraviolet light can be utilized to initiate unique and potentially useful fragmentations in peptides and proteins. Either nonspecific dissociation or highly specific dissociation at engineered chromophoric sites is possible following photon absorption. The level of specificity and control over fragmentation in these experiments is greater than with other fragmentation methods. Novel techniques made possible by this technology are poised to make substantial contributions to the field of proteomics.
Co-reporter:Zhenjiu Liu, Shijun Cheng, Daniel R. Gallie and Ryan R. Julian
Analytical Chemistry 2008 Volume 80(Issue 10) pp:3846
Publication Date(Web):April 12, 2008
DOI:10.1021/ac800176u
Mass spectrometry (MS) is emerging as an additional tool for examining protein structure by way of experiments where structurally related mass changes induced in solution are subsequently detected in the gas phase. Selective noncovalent adduct protein probing (SNAPP) is a recent addition to this type of experiment. SNAPP utilizes noncovalent recognition of lysine residues with 18-crown-6 (18C6) to monitor changes in protein structure. It has been observed that the number of 18C6 adducts that attach to a protein is a function of the structure of the protein. The present work seeks to examine the underlying chemistry which controls the differential attachment of 18C6 to lysine by using ubiquitin as a model system. Ubiquitin is a small protein with a structure that has been well characterized by multiple techniques. Site-directed mutagenesis was used to create a series of ubiquitin mutants where the lysine residues were exchanged for asparagine one at a time. These mutants were then evaluated by SNAPP-MS to determine the relative contribution of each lysine as a binding site for 18C6. It was found that attachment of 18C6 is largely controlled by the strength of intramolecular interactions involving lysine residues. Salt bridges provide the greatest interference, followed by hydrogen bonds. In addition to determining the mechanism for SNAPP, insights are provided about the structure of ubiquitin including confirmation of the existence of two dynamic states for the native structure. These results are discussed in relation to the biological functions of ubiquitin.
Co-reporter:Tony Ly, Zhenjiu Liu, Brian G. Pujanauski, Richmond Sarpong and Ryan R. Julian
Analytical Chemistry 2008 Volume 80(Issue 13) pp:5059
Publication Date(Web):May 23, 2008
DOI:10.1021/ac800177s
Selective noncovalent adduct protein probing (SNAPP) mass spectrometry was recently developed to study solution-phase conformations of proteins by exploiting the specific affinity between 18-crown-6 ether (18C6) and lysine side chains. To obtain more detailed information about protein tertiary structure, a novel noncovalent cross-linking reagent with two 18C6 molecules bridged by a covalent phenyl linker (called PBC for phenyl bis-crown) was synthesized. PBC introduces a distance constraint into SNAPP experiments where pairs of lysine side chains that are held in proximity by tertiary structure should be the most favored binding sites. Application of this method to ubiquitin reveals that PBC can bind to one lysine in a monodentate fashion or bind to two lysines via a bidentate interaction. Comparison with 18C6 can be used to reveal the mode of binding. For the native state of ubiquitin, bidentate binding of PBC is not observed. The partially denatured A-state, however, contains a single pair of lysines that are both chemically available and spaced by less than ∼19 Å (the maximum distance spanning the crown ether binding sites in PBC). Collision-induced dissociation and site-directed mutagenesis reveal that the bidentate PBC attaches to K29 and K33, which is in agreement with previous structural data on the A-state of ubiquitin. PBC is shown to be an effective probe of protein structure in SNAPP experiments, although assigning the specific residues to which PBC is attached can be experimentally challenging.
Co-reporter:Tony Ly
Journal of The American Society for Mass Spectrometry 2008 Volume 19( Issue 11) pp:1663-1672
Publication Date(Web):2008 November
DOI:10.1016/j.jasms.2008.07.006
The metal binding properties of proteins are biologically significant, particularly in relationship to the molecular origins of disease and the discovery of therapeutic pharmaceutical treatments. Herein, we demonstrate that selective noncovalent adduct protein probing mass spectrometry (SNAPP-MS) is a sensitive technique to investigate the structural effects of protein-metal interactions. We utilize specific, noncovalent interactions between 18-crown-6 ether (18C6) and lysine to probe protein structure in the presence and absence of metal ions. Application of SNAPP-MS to the calmodulin-Ca2+ system demonstrates that changes in protein structure are reflected in a substantial change in the number and intensity of 18C6s, which bind to the protein as observed by MS. In this manner, SNAPP is demonstrated to be a sensitive technique for monitoring ligand-induced conformational rearrangements in proteins. In addition, SNAPP is well-suited to examine the properties of natively unfolded proteins, where structural changes are more difficult to detect by other methods. For example, α-synuclein is a protein associated in the pathology of Parkinson’s disease, which is known to aggregate more rapidly in the presence of Al3+ and Cu2+. The 18C6 SNAPP distributions for α-synuclein change dramatically in the presence of 3 µM Al3+, revealing that Al3+ binding causes a significant change in the conformational dynamics of the monomeric form of this disordered protein. In contrast, binding of Cu2+ does not induce a significant shift in 18C6 binding, suggesting that noteworthy structural reorganizations at the monomeric level are minimal. These results are consistent with the idea that the metal-induced aggregation caused by Al3+ and Cu2+ proceed by independent pathways.
Co-reporter:Tony Ly, Ryan R. Julian
Journal of the American Society for Mass Spectrometry 2006 Volume 17(Issue 9) pp:1209-1215
Publication Date(Web):September 2006
DOI:10.1016/j.jasms.2006.05.007
A new method for probing the equilibrium structures and folding states of proteins utilizing electrospray ionization mass spectrometry is described. Protein structure is explored as a function of side-chain availability as determined by a specific interaction between lysine and 18-crown-6 ether (18C6). Various intramolecular interactions are competitive with the lysine/18C6 interaction and can prevent noncovalent attachment of 18C6. Changes to protein structure modify these inhibiting intramolecular interactions, which leads to a change in the number of 18C6s that attach to the protein. Experiments conducted with cytochrome c, ubiquitin, and melittin reveal that the method is sensitive to changes in both tertiary and secondary structure. In addition, the structure of each charge state can be examined independently. Experiments can be performed under conditions where the pH and amount of organic cosolvent are varied. Control experiments conducted with pentalysine, which lacks structural organization, are also presented.
Co-reporter:Tony Ly, Ryan R. Julian
Journal of the American Society for Mass Spectrometry (November 2008) Volume 19(Issue 11) pp:1663-1672
Publication Date(Web):1 November 2008
DOI:10.1016/j.jasms.2008.07.006
The metal binding properties of proteins are biologically significant, particularly in relationship to the molecular origins of disease and the discovery of therapeutic pharmaceutical treatments. Herein, we demonstrate that selective noncovalent adduct protein probing mass spectrometry (SNAPP-MS) is a sensitive technique to investigate the structural effects of protein–metal interactions. We utilize specific, noncovalent interactions between 18-crown-6 ether (18C6) and lysine to probe protein structure in the presence and absence of metal ions. Application of SNAPP-MS to the calmodulin-Ca2+ system demonstrates that changes in protein structure are reflected in a substantial change in the number and intensity of 18C6s, which bind to the protein as observed by MS. In this manner, SNAPP is demonstrated to be a sensitive technique for monitoring ligand-induced conformational rearrangements in proteins. In addition, SNAPP is well-suited to examine the properties of natively unfolded proteins, where structural changes are more difficult to detect by other methods. For example, α-synuclein is a protein associated in the pathology of Parkinson's disease, which is known to aggregate more rapidly in the presence of Al3+ and Cu2+. The 18C6 SNAPP distributions for α-synuclein change dramatically in the presence of 3 μM Al3+, revealing that Al3+ binding causes a significant change in the conformational dynamics of the monomeric form of this disordered protein. In contrast, binding of Cu2+ does not induce a significant shift in 18C6 binding, suggesting that noteworthy structural reorganizations at the monomeric level are minimal. These results are consistent with the idea that the metal-induced aggregation caused by Al3+ and Cu2+ proceed by independent pathways.Protein-metal binding is detected by a change in the noncovalent attachment of 18-crown-6 ether to protein, reflecting a reorganization of the electrostatic interactions involving lysine.Download high-res image (139KB)Download full-size image
Co-reporter:Emily A.C. Spencer, Tony Ly, Ryan R. Julian
International Journal of Mass Spectrometry (1 March 2008) Volume 270(Issue 3) pp:166-172
Publication Date(Web):1 March 2008
DOI:10.1016/j.ijms.2007.12.011
Co-reporter:Benjamin N. Moore, Stephen J. Blanksby and Ryan R. Julian
Chemical Communications 2009(Issue 33) pp:
Publication Date(Web):
DOI:10.1039/B907833A
Co-reporter:Nathan G. Hendricks and Ryan R. Julian
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 39) pp:NaN25827-25827
Publication Date(Web):2015/04/30
DOI:10.1039/C5CP01617G
Evaluation of biomolecular structure in the gas phase is challenging, but worthwhile due to advantages in sensitivity and speed relative to traditional condensed phase approaches. Herein, we demonstrate that a recently developed method utilizing energy transfer to establish distance constraints can be combined with molecular dynamics calculations to rapidly and accurately reveal gaseous peptide structures. Three peptides in various charge states are examined. The influence of increasing charge state on peptide structure is easily observed. The presence of multiple conformations can be detected. Furthermore, the method is demonstrated to aid the assignment of charge, which is frequently nontrivial for peptides containing numerous acidic and basic residues that could adopt a variety of conformers of equal charge state. Comparison with ion mobility reveals that many low energy structures that are distinguishable by distance constraints would not be resolvable by collision cross section. Action-EET is demonstrated to be a powerful new tool for structure elucidation.
Co-reporter:Xing Zhang and Ryan R. Julian
Physical Chemistry Chemical Physics 2012 - vol. 14(Issue 47) pp:NaN16249-16249
Publication Date(Web):2012/10/31
DOI:10.1039/C2CP42242E
Polyproline is a fascinating polymer with interesting structural properties that have been studied in both solution and the gas phase. Herein, a method capable of measuring structural dynamics over long timescales is developed and applied to examination of polyproline in the gas phase. This method is based on measuring the probability of two radicals recombining to form a new covalent bond within a single molecule, which provides distance constraint information. To examine polyproline peptides of various lengths, radical precursors were selectively placed at the termini. Photoactivation with 266 nm light can then be used to create a diradical species, and recombination of the two radicals can be used to evaluate end-to-end distances and structural flexibility. The results reveal that interaction of the polyproline termini is more favorable for shorter chain lengths and lower charge states. As charge states increase, Coulombic repulsion favors formation of more extended structures where the termini no longer come in close contact. With increasing chain length, the greater conformational space also appears to decrease the likelihood of the termini being able to recombine. The amount of radical recombination observed for short polyproline peptides in low charge states is not consistent with what would be expected for helical conformations. Rather, molecular mechanics calculations reveal that lower charge state polyproline peptides tends to adopt globular conformations in the gas phase.
Co-reporter:Nathan G. Hendricks and Ryan R. Julian
Chemical Communications 2015 - vol. 51(Issue 64) pp:NaN12723-12723
Publication Date(Web):2015/06/23
DOI:10.1039/C5CC03779D
Two-step energy transfer is potentially useful for exploring macromolecular structure, but it has not been observed previously in the gas-phase. Single step excitation energy transfer (EET) has been recently documented for tyrosine and tryptophan containing peptides, but not for phenylalanine. Herein, we report sequential energy transfer from phenylalanine to tyrosine to a disulfide, resulting in homolytic cleavage of a sulfur–sulfur bond. Interestingly, energy transfer from phenylalanine is only observed in the presence of tyrosine and only occurs within certain distance constraints. Isolated, electronically excited phenylalanine is known to have an extremely long lifetime in the gas phase, potentially suggesting quicker relaxation occurs via energy transfer to tyrosine. Alternatively, the direct overlap of states between phenylalanine and disulfide bonds is predicted to be poor, in which case tyrosine would serve to bridge the gap. In either case, the distance constraints imposed by this two-step EET are shown to be useful for evaluation and determination of gaseous biomolecular structure.
Co-reporter:Tony Ly, Xing Zhang, Qingyu Sun, Benjamin Moore, Yuanqi Tao and Ryan R. Julian
Chemical Communications 2011 - vol. 47(Issue 10) pp:NaN2837-2837
Publication Date(Web):2011/01/24
DOI:10.1039/C0CC03363D
Novel p-iodobenzoate-based labelling reagents are shown to be effective photocaged precursors for synthesizing biomolecular radicals site-selectively in the gaseous and condensed phases. In vacuo, a single pulse of UV photons (266 nm) is sufficient to quantitatively photolyse the C–I bond. In aqueous solutions, the photolysis half-life is estimated to be 2.5 minutes when irradiating with a 15 W compact fluorescent lamp (254 nm).
Co-reporter:Benjamin N. Moore and Ryan R. Julian
Physical Chemistry Chemical Physics 2012 - vol. 14(Issue 9) pp:NaN3154-3154
Publication Date(Web):2012/01/30
DOI:10.1039/C2CP23443B
In biochemistry, free radicals are versatile species which can perform diverse functions including: signaling, synthesis, and destructive modification. It is of interest to understand how radicals behave within all biomolecules and specifically within peptides and proteins. The 20 standard amino acids contain a wide range of chemical structures, which give proteins their complexity and ultimately their functionality. Many factors influence how radicals interact with these complex molecules, including the bond dissociation energies (BDEs) for homolytically cleaving any X–H bonds. The BDEs provide a simple measure for comparing the thermodynamic favorability of abstracting hydrogen atoms from various sites within a protein. BDEs for abstractable hydrogen atoms have been calculated for each amino acid, the peptide backbone, and peptide termini in order to compile a roadmap of the relative thermodynamics which influence protein radical chemistry. With this information it is possible to gain insight into what contributions both kinetics and thermodynamics will make to various radical mediated reaction pathways.


![5H-Dibenzo[a,d]cyclohepten-5-one, 3,7-diamino-](http://img.cochemist.com/ccimg/97100/97049-28-0.png)
![5H-Dibenzo[a,d]cyclohepten-5-one, 3,7-diamino-](http://img.cochemist.com/ccimg/97100/97049-28-0_b.png)
























![9,12-Octadecadienoic acid (9Z,12Z)-,
(1R)-1-[[[(2-aminoethoxy)hydroxyphosphinyl]oxy]methyl]-2-[(1-oxohexad
ecyl)oxy]ethyl ester](http://img.cochemist.com/ccimg/26700/26662-95-3.png)
![9,12-Octadecadienoic acid (9Z,12Z)-,
(1R)-1-[[[(2-aminoethoxy)hydroxyphosphinyl]oxy]methyl]-2-[(1-oxohexad
ecyl)oxy]ethyl ester](http://img.cochemist.com/ccimg/26700/26662-95-3_b.png)
![9-Octadecenoic acid(9Z)-,(1R)-1-[[[(2-aminoethoxy)hydroxyphosphinyl]oxy]methyl]-2-[(1-oxohexadecyl)oxy]ethylester](http://img.cochemist.com/ccimg/26700/26662-94-2.png)
![9-Octadecenoic acid(9Z)-,(1R)-1-[[[(2-aminoethoxy)hydroxyphosphinyl]oxy]methyl]-2-[(1-oxohexadecyl)oxy]ethylester](http://img.cochemist.com/ccimg/26700/26662-94-2_b.png)












![9-Octadecenoic acid(9Z)-,1,1'-[(1R)-1-[[[(2-aminoethoxy)hydroxyphosphinyl]oxy]methyl]-1,2-ethanediyl]ester](http://img.cochemist.com/ccimg/4100/4004-05-1.png)
![9-Octadecenoic acid(9Z)-,1,1'-[(1R)-1-[[[(2-aminoethoxy)hydroxyphosphinyl]oxy]methyl]-1,2-ethanediyl]ester](http://img.cochemist.com/ccimg/4100/4004-05-1_b.png)