Co-reporter:Peter Larson, Tamara A. Kucaba, Zhengming Xiong, Michael Olin, Thomas S. Griffith, and David M. Ferguson
ACS Medicinal Chemistry Letters November 9, 2017 Volume 8(Issue 11) pp:1148-1148
Publication Date(Web):October 16, 2017
DOI:10.1021/acsmedchemlett.7b00256
A series of N1-modified imidazoquinolines were synthesized and screened for Toll-like receptors (TLR) 7 and 8 activities to identify recognition elements that confer high affinity binding and selectivity. These receptors are key targets in the development of immunomodulatory agents that signal the NF-κB mediated transcription of pro-inflammatory chemokines and cytokines. Results are presented showing both TLR7/8 activations are highly correlated to N1-substitution, with TLR8 selectivity achieved through inclusion of an ethyl-, propyl-, or butylamino group at this position. While the structure–activity relationship analysis indicates TLR7 activity is less sensitive to N1-modification, extension of the aminoalkyl chain length to pentyl and p-methylbenzyl elicited high affinity TLR7 binding. Cytokine profiles are also reported that show the pure TLR8 agonist [4-amino-2-butyl-1-(2-aminoethyl)-7-methoxycarbonyl-1H-imidazo[4,5-c]quinoline] induces higher levels of IL-1β, IL-12, and IFNγ when compared with TLR7 selective or mixed TLR7/8 agonists. The results are consistent with previous work suggesting TLR8 agonists are Th1 polarizing and may help promote cell-mediated immunity.Keywords: cytokine; imidazoquinoline; SAR; TLR7; TLR8; Toll-like receptor;
Co-reporter:Charles E. Schiaffo ; Ce Shi ; Zhengming Xiong ; Michael Olin ; John R. Ohlfest ; Courtney C. Aldrich ;David M. Ferguson
Journal of Medicinal Chemistry 2014 Volume 57(Issue 2) pp:339-347
Publication Date(Web):January 2, 2014
DOI:10.1021/jm4004957
Toll-like receptors 7 and 8 (TLRs) have emerged as key targets in the design of small molecule adjuvants and stimulants for use in immunotherapies. This study examines the structure–activity relationship of a series of C2- and N1-substituted C7-methoxycarbonylimidazoquinolines to gain insight to the structural basis to TLR-7 and -8 selective activity. The analysis is further applied to evaluate the induction of multiple cytokines, including IL-10, IL-12, IL-1β, TNF-α, IFN-α, and IFN-γ, using murine BMDCs and human PBMCs. The results show TLR-7/8 activity is correlated to the C2-alkyl chain length, with peak activity occurring for the butyl (TLR-7) and pentyl (TLR-8) derivatives. A similar SAR is identified in the production of IL-1β, IL-12, and IFN-γ, which are shown to depend on both the C2-alkyl chain length and substitution to the N1-position. The compounds were also potent stimulators of IFN-α and IL-10 production but with less pronounced structure-based correlations.
Co-reporter:Adam R. Benoit, Charles Schiaffo, Christine E. Salomon, John R. Goodell, Hiroshi Hiasa, David M. Ferguson
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 14) pp:3014-3017
Publication Date(Web):15 July 2014
DOI:10.1016/j.bmcl.2014.05.037
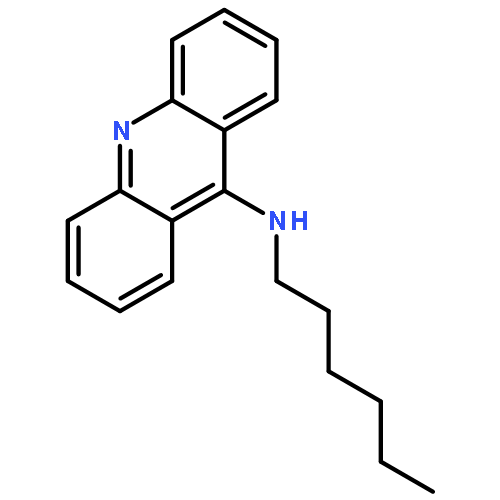
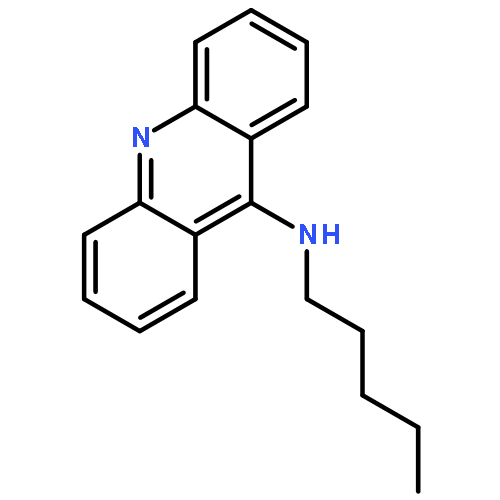
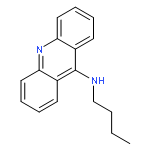
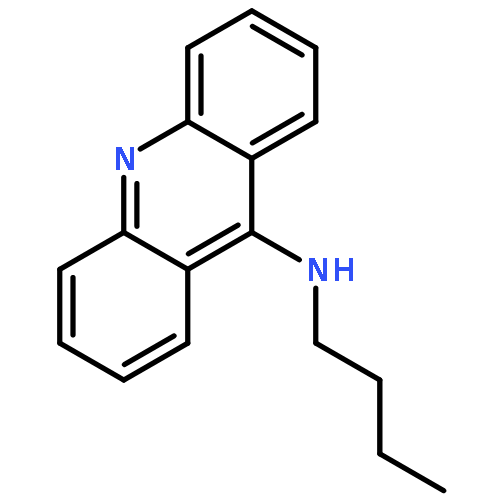
A series of 9-alkylaminoacridines were synthesized and evaluated for activity against two strains of methicillin-resistant and one strain of methicillin-sensitive Staphylococcus aureus. Results are presented that show a clear structure activity relationship between the N-alkyl chain length and antibacterial activity with peak MIC99 values of 2–3 μM for alkyl chains ranging from 10 to 14 carbons in length. Although prior work has linked the function of acridine-based compounds to intercalation and topoisomerase inhibition, the present results show that 9-alkylaminoacridines likely function as amphiphilic membrane-active disruptors potentially in a similar manner as quaternary ammonium antimicrobials.
Co-reporter:Ce Shi, Zhengming Xiong, Padmaja Chittepu, Courtney C. Aldrich, John R. Ohlfest, and David M. Ferguson
ACS Medicinal Chemistry Letters 2012 Volume 3(Issue 6) pp:501
Publication Date(Web):May 12, 2012
DOI:10.1021/ml300079e
Toll-like receptors (TLRs) are key targets in the design of immunomodulating agents for use as vaccine adjuvants and anticancer treatments. The imidazoquinolines, imiquimod and resiquimod, have been shown to activate TLR-7 and -8, which in turn induce cytokine production as part of the innate immune response. Herein, we report the synthesis and discovery of a C7-methoxycarbonyl derivative of imiquimod that stimulates cytokine production but is devoid of TLR-7/8 activity. Data are presented that shows that this analogue not only induces IL-12p40 and TNF production, similar to that of imiquimod and resiquimod, but greatly enhances the production of IL-1β, a key cytokine involved in the activation of CD4 T cells. It is further demonstrated that TLR-7/8 activation can be recovered by the addition of a C2-alkyl substituent to this newly discovered analogue. The results support the existence of an alternative mechanism of action by which imidazoquinolines can stimulate cytokine production.Keywords: cytokine induction; imidazoquinolines; toll-like receptor 7/8
Co-reporter:Ahmad Raza;Blake A. Jacobson;Adam Benoit;Manish R. Patel
Investigational New Drugs 2012 Volume 30( Issue 4) pp:1443-1448
Publication Date(Web):2012 August
DOI:10.1007/s10637-011-9720-7
Human topoisomerase II (hTopoII) inhibitors are important chemotherapeutic agents in many different settings including treatment of malignant mesothelioma. Topoisomerase poisons, such as etoposide and doxorubicin, function by trapping the DNA-enzyme covalent complex producing DNA strand breaks which can ultimately lead to cancer cell death, as well as development of secondary malignancies. While these compounds have been used successfully in treating a wide variety of cancers, their use against mesothelioma has been limited. This study evaluates the anti-proliferative activity of series of acridine-based catalytic inhibitors of hTopoII using four mesothelioma cell lines (H513, H2372, H2461, and H2596). The results indicate these compounds inhibit malignant cell proliferation with EC50 values ranging from 6.9 to 32 μM. Experiments are also performed that show that combination therapies may be used to increase potency. Based on the results of PARP cleavage and Guava Nexin assay, it is concluded that the primary mode of cell death is by apoptosis. The results are consistent with prior work involving pancreatic cancer and hTopoII catalytic inhibitors and suggest substituted acridines may hold promise in treating malignant mesothelioma.
Co-reporter:Aaron M. Teitelbaum;Jose L. Gallardo;Jessica Bedi;Rajan Giri;Adam R. Benoit;Michael R. Olin;Kate M. Morizio;John R. Ohlfest;Rory P. Remmel;David M. Ferguson
Cancer Chemotherapy and Pharmacology 2012 Volume 69( Issue 6) pp:1519-1527
Publication Date(Web):2012/06/01
DOI:10.1007/s00280-012-1855-5
The delivery of drugs to the brain is a major obstacle in the design and development of useful treatments for malignant glioma. Previous studies by our laboratory have identified a series of 9-amino acridine compounds that block the catalytic cycle of topoisomerase II resulting in apoptosis and cell death in a variety of cancer cell lines.This study reports the in vitro and in vivo activity of two promising lead compounds, [{9-[2-(1H-Indol-3-yl)-ethylamino]-acridin-4-yl}-(4-methyl-piperazin-1-yl)-methanone (1) and [9-(1-Benzyl-piperidin-4-ylamino)-acridin-3-yl]-(4-methyl-piperazin-1-yl)-methanone] (2), using an orthotopic glioblastoma mouse model. In addition, the absorption, distribution, and metabolism properties are characterized by determining metabolic stability, MDCK accumulation, Pgp efflux transport, plasma protein binding, and brain distribution in mouse pharmacokinetic studies.The efficacy results indicate low micromolar ED50 values against glioma cells and a significant increase in the survival of glioma-bearing mice dosed with (2) (p < 0.05). Pharmacokinetic data collected at time intervals following a 60 mg/kg oral dose of acridine 1 and 2 showed both compounds penetrate the blood–brain barrier yielding peak concentrations of 0.25 μM and 0.6 μM, respectively. Peak plasma concentrations were determined to be 2.25 μM (1) and 20.38 μM (2). The results were further compared with data collected using a 15 mg/kg intravenous dose of 2 which yielded a peak concentration in the brain of 1.7 μM at 2.0 h relative to a 2.04 μM peak plasma concentration. The bioavailability was calculated to be 83.8%.Taken overall, the results suggest compounds in this series may offer new strategies for the design of chemotherapeutics for treating brain cancers with high oral bioavailability and improved efficacy.
Co-reporter:Rajan Giri, John R. Goodell, Chenguo Xing, Adam Benoit, Harneet Kaur, Hiroshi Hiasa, David M. Ferguson
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 4) pp:1456-1463
Publication Date(Web):15 February 2010
DOI:10.1016/j.bmc.2010.01.018
A series of substituted xanthenes was synthesized and screened for activity using DU-145, MCF-7, and HeLa cancer cell growth inhibition assays. The most potent compound, 9g ([N,N-diethyl]-9-hydroxy-9-(3-methoxyphenyl)-9H-xanthene-3-carboxamide), was found to inhibit cancer cell growth with IC50 values ranging from 36 to 50 μM across all three cancer cell lines. Structure–activity relationship (SAR) data is presented that indicates additional gains in potency may be realized through further derivatization of the compounds (e.g., the incorporation of a 7-fluoro substituent to 9g). Results are also presented that suggest the compounds function through a unique mechanism of action as compared to that of related acridine and xanthone anticancer agents (which have been shown to intercalate into DNA and inhibit topoisomerase II activity). A structural comparison of these compounds suggests the differences in function may be due to the structure of the xanthene heterocycle which adopts a nonplanar conformation about the pyran ring.
Co-reporter:John R. Goodell ; Andrei V. Ougolkov ; Hiroshi Hiasa ; Harneet Kaur ; Rory Remmel ; Daniel D. Billadeau ;David M. Ferguson
Journal of Medicinal Chemistry 2008 Volume 51(Issue 2) pp:179-182
Publication Date(Web):December 29, 2007
DOI:10.1021/jm701228e
A series of substituted 9-aminoacridines is evaluated for antiproliferative activity toward pancreatic cancer cells. The results indicate that the compounds inhibit cell proliferation by inducing a G1-S phase arrest. A model is also developed that explains the molecular basis to inhibition through a DNA “threading” mechanism. We conclude that the drug–DNA complex formed blocks topoisomerase II binding and activity leading to catalytic inhibition of the enzyme and the induction of apoptosis and programmed cell death.
Co-reporter:Brian E. Kane ; Christopher R. McCurdy ;David M. Ferguson
Journal of Medicinal Chemistry 2008 Volume 51(Issue 6) pp:1824-1830
Publication Date(Web):February 23, 2008
DOI:10.1021/jm701040v
The structural basis to salvinorin A recognition of the κ-opioid receptor is evaluated using a combination of site-directed mutagenesis and molecular-modeling techniques. The results show that salvinorin A recognizes a collection of residues in transmembrane II and VII, including Q115, Y119, Y313, I316, and Y320. The mutation of one hydrophobic residue in particular, I316, was found to completely abolish salvinorin A binding. As expected, none of the residues in transmembrane III or VI commonly associated with opiate recognition (such as D138 or E297) appear to be required for ligand binding. On the basis of the results presented here and elsewhere, a binding site model is proposed that aligns salvinorin A vertically within a pocket spanning transmembrane II and VII, with the 2′ substituent directed toward the extracellular domains. The model explains the role that hydrophobic contacts play in binding this lipophilic ligand and gives insight into the structural basis to the μ-opioid receptor selectivity of 2′-benzoyl salvinorin (herkinorin).
Co-reporter:Nicholas P. Labello ; Eric M. Bennett ; David M. Ferguson ;Courtney C. Aldrich
Journal of Medicinal Chemistry 2008 Volume 51(Issue 22) pp:7154-7160
Publication Date(Web):October 30, 2008
DOI:10.1021/jm800668u
MbtA (a salicyl AMP ligase) is a key target for the design of new antitubercular agents. On the basis of structure−activity relationship (SAR) data generated in our laboratory, a structure-based model is developed to predict the binding affinities of aryl acid−AMP bisubstrate inhibitors of MbtA. The approach described takes advantage of the linear interaction energy (LIE) technique to derive linear equations relating ligand structure to function. With only two parameters derived from molecular dynamics simulations, good correlation (R2 = 0.70) was achieved for a set of 31 inhibitors with binding affinities spanning 6 orders of magnitude. The results were applied to understand the effect of steric and heteroatom substitutions on bisubstrate ligand binding and to predict second generation inhibitors of MbtA. The resulting model was further validated by chemical synthesis of a novel inhibitor with a predicted LIE binding affinity of 1.6 nM and a subsequently determined experimental Kiapp of 0.7 nM.
Co-reporter:Brian E. Kane, Marianne K.O. Grant, Esam E. El-Fakahany, David M. Ferguson
Bioorganic & Medicinal Chemistry 2008 Volume 16(Issue 3) pp:1376-1392
Publication Date(Web):1 February 2008
DOI:10.1016/j.bmc.2007.10.058
A series of xanomeline analogs were synthesized and evaluated for binding at the M1 muscarinic acetylcholine receptor (M1 receptor). Specifically, compounds that substitute the O-hexyl chain of xanomeline with polar, ionizable, or conformationally restricted moieties were assessed for their ability to bind to the M1 receptor in a wash-resistant manner (persistent binding). From our screen, several novel ligands that persistently bind to the M1 receptor with greater affinity than xanomeline were discovered. Results indicate that persistent binding may arise not only from hydrophobic interactions but also from ionic interactions with a secondary M1 receptor binding site. Herein, a qualitative model that accounts for both binding scenarios is proposed and applied to understand the structural basis to wash-resistant binding and long-acting effects of xanomeline-based compounds.

![1-[4-AMINO-2-(ETHYLAMINOMETHYL)IMIDAZO[4,5-C]QUINOLIN-1-YL]-2-METHYLPROPAN-2-OL](http://img.cochemist.com/ccimg/1020500/1020412-43-4.png)
![1-[4-AMINO-2-(ETHYLAMINOMETHYL)IMIDAZO[4,5-C]QUINOLIN-1-YL]-2-METHYLPROPAN-2-OL](http://img.cochemist.com/ccimg/1020500/1020412-43-4_b.png)
![Piperazine, 1-methyl-4-[(9-phenoxy-3-acridinyl)carbonyl]-](http://img.cochemist.com/ccimg/877700/877629-44-2.png)
![Piperazine, 1-methyl-4-[(9-phenoxy-3-acridinyl)carbonyl]-](http://img.cochemist.com/ccimg/877700/877629-44-2_b.png)



![Ethanamine, 2-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-](http://img.cochemist.com/ccimg/91600/91578-89-1.png)
![Ethanamine, 2-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-](http://img.cochemist.com/ccimg/91600/91578-89-1_b.png)




![4-Thia-1-azabicyclo[3.2.0]heptane-2-carboxylicacid, 6-[(2,6-dimethoxybenzoyl)amino]-3,3-dimethyl-7-oxo-, (2S,5R,6R)-](http://img.cochemist.com/ccimg/100/61-32-5.png)
![4-Thia-1-azabicyclo[3.2.0]heptane-2-carboxylicacid, 6-[(2,6-dimethoxybenzoyl)amino]-3,3-dimethyl-7-oxo-, (2S,5R,6R)-](http://img.cochemist.com/ccimg/100/61-32-5_b.png)