Co-reporter:Lea Thomas, Zhigang Rao, Jana Gerstmeier, Martin Raasch, ... Oliver Werz
Biochemical Pharmacology 2017 Volume 130(Volume 130) pp:
Publication Date(Web):15 April 2017
DOI:10.1016/j.bcp.2017.02.004
Pharmacological interference with vacuolar-type H(+)-ATPase (V-ATPase), a proton-translocating enzyme involved in protein transport and pH regulation of cell organelles, is considered a potential strategy for cancer therapy. Macrophages are critically involved in tumor progression and may occur as pro-tumoral M2 phenotype, whereas classically-activated M1 can inhibit tumor development for example by releasing tumor-suppressing molecules, including tumor necrosis factor (TNF)α. Here, we show that targeting V-ATPase by selective inhibitors such as archazolid upregulates the expression and secretion of TNFα in lipopolysaccharide (LPS)- or LPS/interferon (INF)γ-activated M1-like macrophages derived from human blood monocytes. In contrast, archazolid failed to elevate TNFα production from uncommitted (M0) or interleukin (IL)-4-treated M2-like macrophages. Secretion of other relevant cytokines (i.e., IL-1β, IL-6, IL-10) or chemokines (i.e. IL-8 and monocyte chemotactic protein-1) from M1 was not affected by archazolid. Though V-ATPase inhibitors elevated the lysosomal pH in M1 comparable to chloroquine or ammonium chloride, the latter agents suppressed TNFα secretion. Archazolid selectively increased TNFα mRNA levels, which was abolished by dexamethasone. Interestingly, archazolid enhanced the phosphorylation and nuclear translocation of the p65 subunit of NFκB and stimulated phosphorylation of SAPK/JNK. In a microfluidically-supported human tumor biochip model, archazolid-treated M1 significantly reduced tumor cell viability. Together, our data show that V-ATPase inhibition selectively upregulates TNFα production in classically-activated macrophages along with NFκB and SAPK/JNK activation. Such increased TNFα release caused by V-ATPase inhibitors may contribute to tumor suppression in addition to direct targeting cancer cells.Download high-res image (145KB)Download full-size image
Co-reporter:Sebastian Thiede, Paul R. Wosniok, Daniel Herkommer, Anna-Christina Schulz-Fincke, Michael Gütschow, and Dirk Menche
Organic Letters 2016 Volume 18(Issue 16) pp:3964-3967
Publication Date(Web):August 3, 2016
DOI:10.1021/acs.orglett.6b01724
The total synthesis of leupyrrin B1 was accomplished by an expedient strategy that involves an optimized HATU-mediated amide coupling protocol of elaborate substrates. The generally useful procedure was also successfully applied in an improved total synthesis of leupyrrin A1. Finally, leupyrrins A1 and B1 were evaluated toward a panel of proteases, and human leukocyte elastase was discovered as a molecular target of the leupyrrins.
Co-reporter:Sebastian Essig and Dirk Menche
The Journal of Organic Chemistry 2016 Volume 81(Issue 5) pp:1943-1966
Publication Date(Web):January 29, 2016
DOI:10.1021/acs.joc.5b02781
Full details on the design, development, and application of a highly stereoselective strategy for the synthesis of isochromanones are reported. The method is based on an asymmetric ortho lithiation with aldehyde electrophiles and utilizes the chiral memory of a preoriented atropisomeric amide axis for stereocontrol. For direct transformation of sterically hindered amides to isochromanones, efficient and mild one-pot protocols involving either O-alkylation or acidic microwave activation were developed. The procedures may be applied also to highly functionalized as well as stereochemically complex and sensitive substrates and demonstrate a high protective group tolerance. Furthermore, asymmetric crotylborations of axially chiral amides were studied in detail. These methodologies enable a general access to all possible stereoisomers of hydroxyl-isochromanones with up to three contiguous stereocenters. The true applicability of our approach was finally demonstrated by synthesis of the authentic anti,anti-configured isochromanone core of the ajudazols, highly potent inhibitors of the mitochondrial respiratory chain from myxobacteria.
Co-reporter:Sebastian Essig, Björn Schmalzbauer, Sebastian Bretzke, Olga Scherer, Andreas Koeberle, Oliver Werz, Rolf Müller, and Dirk Menche
The Journal of Organic Chemistry 2016 Volume 81(Issue 4) pp:1333-1357
Publication Date(Web):January 21, 2016
DOI:10.1021/acs.joc.5b02844
Full details on the evaluation and application of an easily feasible and generally useful method for configurational assignments of isolated methyl-bearing stereocenters are reported. The analytical tool relies on a bioinformatic gene cluster analysis and utilizes a predictive enoylreductase alignment, and its feasibility was demonstrated by the full stereochemical determination of the ajudazols, highly potent inhibitors of the mitochondrial respiratory chain. Furthermore, a full account of our strategies and tactics that culminated in the total synthesis of ajudazol B, the most potent and least abundant of these structurally unique class of myxobacterial natural products, is presented. Key features include an application of an asymmetric ortholithiation strategy for synthesis of the characteristic anti-configured hydroxyisochromanone core bearing three contiguous stereocenters, a modular oxazole formation, a flexible cross-metathesis approach for terminal allyl amide synthesis, and a late-stage Z,Z-selective Suzuki coupling. This total synthesis unambiguously proves the correct stereochemistry, which was further corroborated by comparison with reisolated natural material. Finally, 5-lipoxygenase was discovered as an additional molecular target of ajudazol B. Activities against this clinically validated key enzyme of the biosynthesis of proinflammatory leukotrienes were in the range of the approved drug zileuton, which further underlines the biological importance of this unique natural product.
Co-reporter:Jan Philipp Gölz;Dr. Yaser NejatyJahromy;Mirko Bauer;Ashraf Muhammad;Dr. Gregor Schnakenburg;Dr. Stefan Grimme;Dr. Olav Schiemann;Dr. Dirk Menche
Chemistry - A European Journal 2016 Volume 22( Issue 28) pp:9591-9598
Publication Date(Web):
DOI:10.1002/chem.201600528
Abstract
Novel types of spin-labeled N,N′-dicyclohexylcarbodiimides (DCC) are reported that bear a 2,2,6,6-tetramethylpiperidinyloxyl (TEMPO) residue on one side and different aromatic and aliphatic cyclohexyl analogues on the other side of the diimide core. These readily available novel reagents add efficiently to aliphatic and aromatic carboxylic acids, forming two possible spin-labeled amide derivatives with different radical distances of the resulting amide. The addition of aromatic DCC analogues proceeds with excellent selectivity, giving amides where the carboxylic acid is exclusively connected to the aromatic residue, while little or no selectivity was observed for the aliphatic congeners. The usefulness of these adducts in structural studies was demonstrated by EPR (electron paramagnetic resonance) measurements of biradical adducts of biphenyl-4,4′-dicarboxylic acids. These analyses also reveal high degrees of conformational bias for aromatic DCC derivatives, which further underlines the powerfulness of these novel reagents. This observation was further corroborated by quantum chemical calculations, giving a detailed understanding of the structural dynamics, while detailed information on the solid state structure of all novel reagents was obtained by X-ray structure analyses.
Co-reporter:Daniel Herkommer; Sebastian Thiede; Paul R. Wosniok; Sandra Dreisigacker; Maoqun Tian; Thomas Debnar; Herbert Irschik
Journal of the American Chemical Society 2015 Volume 137(Issue 12) pp:4086-4089
Publication Date(Web):March 13, 2015
DOI:10.1021/jacs.5b01894
The stereochemical determination of the potent antifungal agents leupyrrin A1 and B1 and the total synthesis of leupyrrin A1 are reported. The relative and absolute configuration was determined by a combination of high field NMR studies, molecular modeling, and chemical derivatization. The expedient total synthesis involves a one-pot sequential Zr-mediated oxidative diyne-cyclization/regioselective opening sequence for preparation of the unique dihydrofuran ring, a highly stereoselective one-pot approach to the butyrolactone, a challenging sp2–sp3 Suzuki coupling and a high-yielding Shiina macrolactonization.
Co-reporter:Björn Schmalzbauer and Dirk Menche
Organic Letters 2015 Volume 17(Issue 12) pp:2956-2959
Publication Date(Web):May 28, 2015
DOI:10.1021/acs.orglett.5b01231
A concise synthesis of the tricyclic core 2 of the structurally unique marine myxobacterial natural product salimabromide has been developed. Compound 2 contains the tetraline subunit including the two quaternary centers and the eight-membered ring of salimabromide. Major features for its synthesis include a Lewis base catalyzed Denmark-crotylation for stereoselective construction of the highly hindered quaternary stereocenter, an innovative iodine/selectfluor induced endo-carbocylization, and a unique chemoselective carbonylative lactonization of the eight-membered ring.
Co-reporter:Michael Schrempp;Sebastian Thiede;Daniel Herkommer;Dr. Andreas Gansäuer ;Dr. Dirk Menche
Chemistry - A European Journal 2015 Volume 21( Issue 45) pp:16266-16271
Publication Date(Web):
DOI:10.1002/chem.201502263
Abstract
Inspired by the bioactive natural metabolites leupyrrin A1 and B1, two novel stereoselective methods for the highly concise synthesis of densely substituted α-chiral butyrolactones are reported. The first approach relies on an innovative three-step TiIII-catalyzed radical reaction that proceeds with excellent chemo-, regio-, and stereoselectivity. The alternative route utilizes sequential asymmetric alkylations and enables asymmetric synthesis of the authentic α-tetrasubstituted butyrolactone motif of the leupyrrins in only four steps from commercially available substrates.
Co-reporter:Daniel Herkommer;Dr. Sra Dreisigacker;Dr. Galina Sergeev;Dr. Florenz Sasse;Dr. Holger Gohlke;Dr. Dirk Menche
ChemMedChem 2015 Volume 10( Issue 3) pp:470-489
Publication Date(Web):
DOI:10.1002/cmdc.201402508
Abstract
The natural products rhizopodin and bistramide belong to an elite class of highly potent actin binding agents. They show powerful antiproliferative activities against a range of tumor cell lines, with IC50 values in the low-nanomolar range. At the molecular level they disrupt the actin cytoskeleton by binding specifically to a few critical sites of G-actin, resulting in actin filament stabilization. The important biological properties of rhizopodin and bistramide, coupled with their unique and intriguing molecular architectures, render them attractive compounds for further development. However, this is severely hampered by the structural complexity of these metabolites. We initiated an interdisciplinary approach at the interface between molecular modeling, organic synthesis, and chemical biology to support further biological applications. We also wanted to expand structure–activity relationship studies with the goal of accessing simplified analogues with potent biological properties. We report computational analyses of actin–inhibitor interactions involving molecular docking, validated on known actin binding ligands, that show a close match between the crystal and modeled structures. Based on these results, the ligand shape was simplified, and more readily accessible rhizopodin–bistramide mimetics were designed. A flexible and modular strategy was applied for the synthesis of these compounds, enabling diverse access to dramatically simplified rhizopodin–bistramide hybrids. This novel analogue class was analyzed for its antiproliferative and actin binding properties.
Co-reporter:Daniel Herkommer, Björn Schmalzbauer and Dirk Menche
Natural Product Reports 2014 vol. 31(Issue 4) pp:456-467
Publication Date(Web):23 Dec 2013
DOI:10.1039/C3NP70093C
Covering: the literature up to 2012
This review presents recent advances in sequential catalytic methods developed in our group for the rapid and stereoselective synthesis of key structural features of complex polyketides. These include a novel domino reaction, based on a combination of a nucleophilic addition and a Tsuji–Trost reaction, an innovative sequence relying on an oxidative diyne cyclization and regioselective opening, as well as sequential cross coupling strategies. The design and scope of these methods are discussed and applications in complex target syntheses presented.
Co-reporter:Thomas Debnar, Sandra Dreisigacker and Dirk Menche
Chemical Communications 2013 vol. 49(Issue 7) pp:725-727
Publication Date(Web):30 Nov 2012
DOI:10.1039/C2CC37678D
An efficient protocol for the highly regioselective opening of aliphatic zirconacyclopentadienes is reported. The one-pot process involves a zirconocene-mediated cyclization of 1,6-diynes and highly selective cleavage of the metallacycles with NBS and enables a concise synthesis of the tetrahydrofuran-core of the leupyrrins.
Co-reporter:Thomas Debnar, Tongtong Wang, and Dirk Menche
Organic Letters 2013 Volume 15(Issue 11) pp:2774-2777
Publication Date(Web):May 17, 2013
DOI:10.1021/ol401110x
Stereoselective syntheses of the Northern and the Southern fragments 2 and 3 of leupyrrin A1 are reported. The convergent preparation of 2 is highlighted by a zirconocene-mediated one-pot cyclization–regioselective opening of an advanced diyne while the route to 3 involves a Krische allylation and a one-pot Sharpless dihydroxylation–cyclization. Comparison of the spectroscopic data with those reported for the natural product supports a relative stereochemical assignment within these heterocycles.
Co-reporter:Michael Dieckmann and Dirk Menche
Organic Letters 2013 Volume 15(Issue 1) pp:228-231
Publication Date(Web):December 21, 2012
DOI:10.1021/ol3033303
A novel domino process for 1,3-anti diol synthesis by the union of a methyl ketone with an aldehyde is described. The operationally simple procedure is based on an Ipc-boron-aldol coupling and subsequent Ipc-mediated reduction of the intermediate β-hydroxy-ketone. The sequence proceeds with excellent anti-selectivities and enables the rapid construction of complex polyketide fragments.
Co-reporter:Björn Schmalzbauer, Jennifer Herrmann, Rolf Müller, and Dirk Menche
Organic Letters 2013 Volume 15(Issue 4) pp:964-967
Publication Date(Web):February 7, 2013
DOI:10.1021/ol400156u
A concise total synthesis of dysidavarone A possessing the new “dysidavarane” carbon skeleton has been accomplished by a convergent strategy, involving a stereoselective reductive alkylation of a Wieland-Miescher type ketone under Birch conditions and an advantageous intramolecular palladium-catalyzed α-arylation of a sterically hindered ketone. Dysidavarone A showed potent antimicrobial and antiproliferative activities based on characteristic morphological changes of treated cells.
Co-reporter:Mario Altendorfer, Aruna Raja, Florenz Sasse, Herbert Irschik and Dirk Menche
Organic & Biomolecular Chemistry 2013 vol. 11(Issue 13) pp:2116-2139
Publication Date(Web):19 Nov 2012
DOI:10.1039/C2OB26906F
An efficient procedure for the concise synthesis of hetero-bis-metallated alkenes as useful building blocks for the modular access to highly elaborate polyenes and stabilized analogues is reported. By applying these bifunctional olefins in convergent Stille/Suzuki–Miyaura couplings, novel, carefully selected side chain analogues of the potent RNA polymerase inhibitor etnangien were synthesized by a modular late stage coupling strategy and evaluated for antibacterial and antiproliferative activities.
Co-reporter:Dr. Manuel Kretschmer;Dr. Michael Dieckmann ;Dr. Pengfei Li ;Dr. Sven Rudolph ;Dipl.-Chem. Daniel Herkommer;Dr. Johannes Troendlin;Dr. Dirk Menche
Chemistry - A European Journal 2013 Volume 19( Issue 47) pp:15993-16018
Publication Date(Web):
DOI:10.1002/chem.201302197
Abstract
A highly convergent total synthesis of the potent polyketide macrolide rhizopodin has been achieved in 29 steps by employing a concise strategy that exploits the molecule′s C2 symmetry. Notable features of this convergent approach include a rapid assembly of the macrocycle through a site-directed sequential cross-coupling strategy and the bidirectional attachment of the side chains by means of Horner–Wadsworth–Emmons (HWE) coupling reactions. During the course of this endeavor, scalable routes for synthesis of three main building blocks of similar complexity were developed that allowed for their stereocontrolled construction. This modular route will be amenable to the development of syntheses of other analogues of rhizopodin.
Co-reporter:Sebastian Essig ; Sebastian Bretzke ; Rolf Müller
Journal of the American Chemical Society 2012 Volume 134(Issue 47) pp:19362-19365
Publication Date(Web):November 5, 2012
DOI:10.1021/ja309685n
The stereochemical determination of the potent respiratory chain inhibitors ajudazols A and B and the total synthesis of ajudazol B are reported. Configurational assignment was exclusively based on biosynthetic gene cluster analysis of both ketoreductase domains for hydroxyl-bearing stereocenters and one of the first predictive enoylreductase alignments for methyl-bearing stereocenters. The expedient total synthesis resulting in unambiguous proof of the predicted stereochemistry involves a short stereoselective approach to the challenging isochromanone stereotriad by an innovative asymmetric ortholithiation strategy, a modular oxazole formation, and a late-stage Z,Z-selective Suzuki coupling.
Co-reporter:Mario Altendorfer and Dirk Menche
Chemical Communications 2012 vol. 48(Issue 66) pp:8267-8269
Publication Date(Web):02 Jul 2012
DOI:10.1039/C2CC34052F
An efficient synthesis of diverse hetero-bis-metallated alkenes with conjugated and isolated olefin subunits is reported. Relying on those useful tin/boron reagents a convergent, palladium-catalyzed fragment coupling strategy has been developed as an elegant methodology to construct highly conjugated polyenes and stabilized olefinic analogues thereof. The utility of these reagents was further demonstrated in a concise and modular construction of extended polyene side chains of the potent polyketide antibiotic etnangien.
Co-reporter:Manuel Kretschmer and Dirk Menche
Organic Letters 2012 Volume 14(Issue 1) pp:382-385
Publication Date(Web):December 13, 2011
DOI:10.1021/ol203130b
A convergent synthesis of the central C8–C22 core of the potent macrolide antibiotic rhizopodin is reported. Notable features of the stereocontrolled approach include an asymmetric reverse prenylation of an alcohol using a method of Krische, a thiazolium catalyzed transformation of an epoxyaldehyde as described by Bode, and a late-stage oxazole formation from advanced intermediates. This route demonstrates the applicability of these methodologies in complex natural product synthesis.
Co-reporter:Elke Persch, Teodora Basile, Svenja Bockelmann, Markus Huss, Helmut Wieczorek, Teresa Carlomagno, Dirk Menche
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 24) pp:7735-7738
Publication Date(Web):15 December 2012
DOI:10.1016/j.bmcl.2012.09.081
The water-solubility of the highly potent V-ATPase inhibitors archazolid A and the glucosylated derivative archazolid C was studied in the presence of a wide range of cosolvents, revealing very low solubilites. The first water-soluble analogue was then designed, synthesized, and evaluated for V-ATPase inhibitory activity in vitro.The water-solubility of the highly potent V-ATPase inhibitors archazolid A and the glucosylated derivative archazolid C was studied in the presence of a wide range of cosolvents, revealing very low solubilites. The first water-soluble analogue was then designed, synthesized, and evaluated for V-ATPase inhibitory activity in vitro.
Co-reporter:Mario Altendorfer, Herbert Irschik, Dirk Menche
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 17) pp:5731-5734
Publication Date(Web):1 September 2012
DOI:10.1016/j.bmcl.2012.06.070
Novel simplified side chains of the potent RNA polymerase inhibitor etnangien were designed, synthesized and evaluated for antibacterial activity against Gram-positive bacteria and one Gram-negative bacterium.
Co-reporter:Liang Wang and Dirk Menche
The Journal of Organic Chemistry 2012 Volume 77(Issue 23) pp:10811-10823
Publication Date(Web):November 9, 2012
DOI:10.1021/jo302102x
Biologically significant tetrahydropyrans (THP) were synthesized by a Tandem oxa-Michael/Tsuji–Trost reaction. Different Michael acceptors were investigated, and optimal results in terms of diastereoselectivities and yields were obtained with nitro olefins. The influence of the reaction parameters, substrate patterns, and type of metal counterions on the yield and stereochemical outcome of this process is discussed, and an explanation for the observed stereoselectivities is proposed.
Co-reporter:Michael Dieckmann, Sven Rudolph, Sandra Dreisigacker, and Dirk Menche
The Journal of Organic Chemistry 2012 Volume 77(Issue 23) pp:10782-10788
Publication Date(Web):November 16, 2012
DOI:10.1021/jo302134y
A highly convergent synthesis of the central dimeric core of the potent antibiotic macrolide rhizopodin is reported. Notable features of the highly concise route include an effective preparation of the key C8-C22 building block based on an iridium-catalyzed Krische allylation and a chemoselective cross-coupling approach toward the macrocycle involving a highly advantageous Heck reaction for macrocyclization.
Co-reporter:Dr. Liang Wang;Dr. Dirk Menche
Angewandte Chemie International Edition 2012 Volume 51( Issue 37) pp:9425-9427
Publication Date(Web):
DOI:10.1002/anie.201203911
Co-reporter:Dr. Liang Wang;Dr. Dirk Menche
Angewandte Chemie 2012 Volume 124( Issue 37) pp:9559-9562
Publication Date(Web):
DOI:10.1002/ange.201203911
Co-reporter:Dipl.-Chem. Michael Dieckmann;Dipl.-Chem. Manuel Kretschmer;Dr. Pengfei Li;Dr. Sven Rudolph;Dipl.-Chem. Daniel Herkommer;Dr. Dirk Menche
Angewandte Chemie International Edition 2012 Volume 51( Issue 23) pp:5667-5670
Publication Date(Web):
DOI:10.1002/anie.201201946
Co-reporter:Dipl.-Chem. Michael Dieckmann;Dipl.-Chem. Manuel Kretschmer;Dr. Pengfei Li;Dr. Sven Rudolph;Dipl.-Chem. Daniel Herkommer;Dr. Dirk Menche
Angewandte Chemie 2012 Volume 124( Issue 23) pp:5765-5768
Publication Date(Web):
DOI:10.1002/ange.201201946
Co-reporter:Tongtong Wang; Michael Schrempp; Andreas Berndhäuser; Olav Schiemann
Organic Letters () pp:
Publication Date(Web):August 7, 2015
DOI:10.1021/acs.orglett.5b01845
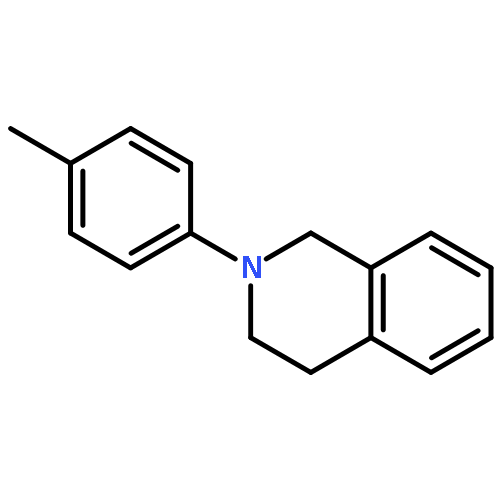
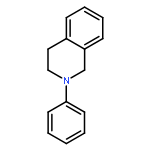
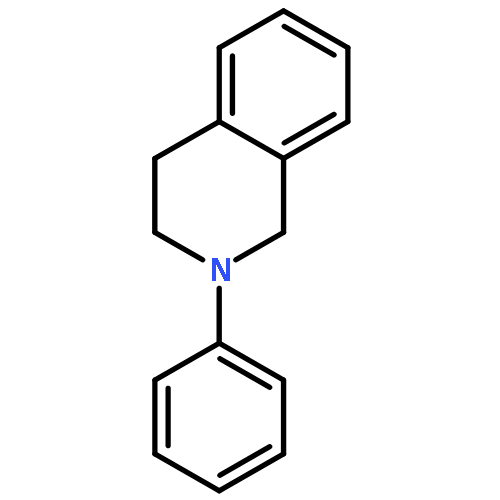
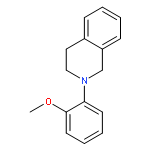
A general new method for the highly concise synthesis of C-1-alkylated tetrahydroisoquinolines (THIQ) is reported. The CuCl2-catalyzed procedure is based on a coupling of nonfunctionalized THIQs with organozinc reagents under aerobic conditions. It proceeds in high yields and is broadly applicable to a wide range of substrates. It relies on a regioselective sp3 C–H bond activation allowing for an sp3–sp3 bond union under mild reaction conditions in a rapid and effective manner. Mechanistically it involves an iminium ion intermediate that is formed via an organic radical involving a single-electron-transfer process. For the first time for this type of reaction a radical intermediate has been proven by EPR spectroscopy.
Co-reporter:Thomas Debnar, Sandra Dreisigacker and Dirk Menche
Chemical Communications 2013 - vol. 49(Issue 7) pp:NaN727-727
Publication Date(Web):2012/11/30
DOI:10.1039/C2CC37678D
An efficient protocol for the highly regioselective opening of aliphatic zirconacyclopentadienes is reported. The one-pot process involves a zirconocene-mediated cyclization of 1,6-diynes and highly selective cleavage of the metallacycles with NBS and enables a concise synthesis of the tetrahydrofuran-core of the leupyrrins.
Co-reporter:Mario Altendorfer, Aruna Raja, Florenz Sasse, Herbert Irschik and Dirk Menche
Organic & Biomolecular Chemistry 2013 - vol. 11(Issue 13) pp:NaN2139-2139
Publication Date(Web):2012/11/19
DOI:10.1039/C2OB26906F
An efficient procedure for the concise synthesis of hetero-bis-metallated alkenes as useful building blocks for the modular access to highly elaborate polyenes and stabilized analogues is reported. By applying these bifunctional olefins in convergent Stille/Suzuki–Miyaura couplings, novel, carefully selected side chain analogues of the potent RNA polymerase inhibitor etnangien were synthesized by a modular late stage coupling strategy and evaluated for antibacterial and antiproliferative activities.
Co-reporter:Mario Altendorfer and Dirk Menche
Chemical Communications 2012 - vol. 48(Issue 66) pp:NaN8269-8269
Publication Date(Web):2012/07/02
DOI:10.1039/C2CC34052F
An efficient synthesis of diverse hetero-bis-metallated alkenes with conjugated and isolated olefin subunits is reported. Relying on those useful tin/boron reagents a convergent, palladium-catalyzed fragment coupling strategy has been developed as an elegant methodology to construct highly conjugated polyenes and stabilized olefinic analogues thereof. The utility of these reagents was further demonstrated in a concise and modular construction of extended polyene side chains of the potent polyketide antibiotic etnangien.

![Ruthenium, [1,3-bis(2,4,6-triMethylphenyl)-2-imidazolidinylidene]dichloro[[2-(1-Methylethoxy-κO)-5-nitrophenyl]Methylene-κC]-, (SP-5-41)-](/data/chemimg/1247800/502964-52-5.png)
![Ruthenium, [1,3-bis(2,4,6-triMethylphenyl)-2-imidazolidinylidene]dichloro[[2-(1-Methylethoxy-κO)-5-nitrophenyl]Methylene-κC]-, (SP-5-41)-](/data/chemimg/1247800/502964-52-5_b.png)








![5-Hexen-3-ol, 1-[(4-methoxyphenyl)methoxy]-4,4-dimethyl-, (3S)-](http://img.cochemist.com/ccimg/693300/693221-73-7.png)
![5-Hexen-3-ol, 1-[(4-methoxyphenyl)methoxy]-4,4-dimethyl-, (3S)-](http://img.cochemist.com/ccimg/693300/693221-73-7_b.png)


![2-BUTANOL, 4-[(4-METHOXYPHENYL)METHOXY]-](http://img.cochemist.com/ccimg/671300/671232-60-3.png)
![2-BUTANOL, 4-[(4-METHOXYPHENYL)METHOXY]-](http://img.cochemist.com/ccimg/671300/671232-60-3_b.png)


![2-Oxazolidinethione, 4-(phenylmethyl)-3-[(2-propenyloxy)acetyl]-, (4S)-](http://img.cochemist.com/ccimg/501700/501644-55-9.png)
![2-Oxazolidinethione, 4-(phenylmethyl)-3-[(2-propenyloxy)acetyl]-, (4S)-](http://img.cochemist.com/ccimg/501700/501644-55-9_b.png)
![2-Butenoic acid, 4-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-3-methyl-, ethylester, (2E)-](http://img.cochemist.com/ccimg/497200/497144-17-9.png)
![2-Butenoic acid, 4-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-3-methyl-, ethylester, (2E)-](http://img.cochemist.com/ccimg/497200/497144-17-9_b.png)






![Zinc, bromo[(4-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/398500/398456-86-5.png)
![Zinc, bromo[(4-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/398500/398456-86-5_b.png)


![5-Hexen-1-ol, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (3S)-](http://img.cochemist.com/ccimg/381000/380909-95-5.png)
![5-Hexen-1-ol, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (3S)-](http://img.cochemist.com/ccimg/381000/380909-95-5_b.png)
![5-Hexen-3-ol, 1-[(4-methoxyphenyl)methoxy]-, (3S)-](http://img.cochemist.com/ccimg/381000/380909-94-4.png)
![5-Hexen-3-ol, 1-[(4-methoxyphenyl)methoxy]-, (3S)-](http://img.cochemist.com/ccimg/381000/380909-94-4_b.png)
![5-Hexen-3-ol, 1-[(4-methoxyphenyl)methoxy]-, (3R)-](http://img.cochemist.com/ccimg/325200/325172-29-0.png)
![5-Hexen-3-ol, 1-[(4-methoxyphenyl)methoxy]-, (3R)-](http://img.cochemist.com/ccimg/325200/325172-29-0_b.png)
![2-Butanone, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-3-methyl-, (3S)-](http://img.cochemist.com/ccimg/308300/308240-90-6.png)
![2-Butanone, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-3-methyl-, (3S)-](http://img.cochemist.com/ccimg/308300/308240-90-6_b.png)


![Benzenesulfonamide,2,4,6-trimethyl-N-[(1S,2R)-1-methyl-2-(1-oxopropoxy)-2-phenylethyl]-N-(phenylmethyl)-](http://img.cochemist.com/ccimg/187400/187324-66-9.png)
![Benzenesulfonamide,2,4,6-trimethyl-N-[(1S,2R)-1-methyl-2-(1-oxopropoxy)-2-phenylethyl]-N-(phenylmethyl)-](http://img.cochemist.com/ccimg/187400/187324-66-9_b.png)
![Phosphine,1,1'-[(1R)-5,5'-dichloro-6,6'-dimethoxy[1,1'-biphenyl]-2,2'-diyl]bis[1,1-diphenyl-](/data/chemimg/110700/185913-97-7.png)
![Phosphine,1,1'-[(1R)-5,5'-dichloro-6,6'-dimethoxy[1,1'-biphenyl]-2,2'-diyl]bis[1,1-diphenyl-](/data/chemimg/110700/185913-97-7_b.png)


![9,10-Anthracenedione,1,4-bis[[(8a,9R)-10,11-dihydro-6'-methoxycinchonan-9-yl]oxy]-](http://img.cochemist.com/ccimg/176100/176097-24-8.png)
![9,10-Anthracenedione,1,4-bis[[(8a,9R)-10,11-dihydro-6'-methoxycinchonan-9-yl]oxy]-](http://img.cochemist.com/ccimg/176100/176097-24-8_b.png)


![Propanal, 3-[(4-methoxyphenyl)methoxy]-2-methyl-, (2R)-](http://img.cochemist.com/ccimg/160300/160238-46-0.png)
![Propanal, 3-[(4-methoxyphenyl)methoxy]-2-methyl-, (2R)-](http://img.cochemist.com/ccimg/160300/160238-46-0_b.png)




![Serine,N,N,O-trimethyl-,(3E,5E,8R,9S,10R,11R,14S,15E,18R,20R,21E,24S)-24-[(1S,2S,3S,6R,7S,8R,9R,10E)-8-(acetyloxy)-6-[2-(dimethylamino)-1-oxopropoxy]-11-(formylmethylamino)-2-hydroxy-1,3,7,9-tetramethyl-10-undecen-1-yl]-10-hydroxy-14,20-dimethoxy-9,11,15,18-tetramethyl-2-oxooxacyclotetracosa-3,5,15,21-tetraen-8-ylester](http://img.cochemist.com/ccimg/152000/151923-84-1.png)
![Serine,N,N,O-trimethyl-,(3E,5E,8R,9S,10R,11R,14S,15E,18R,20R,21E,24S)-24-[(1S,2S,3S,6R,7S,8R,9R,10E)-8-(acetyloxy)-6-[2-(dimethylamino)-1-oxopropoxy]-11-(formylmethylamino)-2-hydroxy-1,3,7,9-tetramethyl-10-undecen-1-yl]-10-hydroxy-14,20-dimethoxy-9,11,15,18-tetramethyl-2-oxooxacyclotetracosa-3,5,15,21-tetraen-8-ylester](http://img.cochemist.com/ccimg/152000/151923-84-1_b.png)
![Formamide,N-[11-(14,16-dihydroxy-10-methoxy-11,21-dimethyl-12,18-dioxo-3,7,19,27-tetraoxa-29,30,31-triazatetracyclo[24.2.1.12,5.16,9]hentriaconta-2(31),4,6(30),8,24,26(29),28-heptaen-20-yl)-4,10-dimethoxy-5,9-dimethyl-6-oxo-1-undecen-1-yl]-N-methyl-](http://img.cochemist.com/ccimg/149500/149420-76-8.png)
![Formamide,N-[11-(14,16-dihydroxy-10-methoxy-11,21-dimethyl-12,18-dioxo-3,7,19,27-tetraoxa-29,30,31-triazatetracyclo[24.2.1.12,5.16,9]hentriaconta-2(31),4,6(30),8,24,26(29),28-heptaen-20-yl)-4,10-dimethoxy-5,9-dimethyl-6-oxo-1-undecen-1-yl]-N-methyl-](http://img.cochemist.com/ccimg/149500/149420-76-8_b.png)


![Propanal, 3-[(4-methoxyphenyl)methoxy]-2-methyl-, (2S)-](http://img.cochemist.com/ccimg/133000/132969-60-9.png)
![Propanal, 3-[(4-methoxyphenyl)methoxy]-2-methyl-, (2S)-](http://img.cochemist.com/ccimg/133000/132969-60-9_b.png)
![Urea,N-(1-benzo[b]thien-2-ylethyl)-N'-hydroxy-](http://img.cochemist.com/ccimg/132900/132880-11-6.png)
![Urea,N-(1-benzo[b]thien-2-ylethyl)-N'-hydroxy-](http://img.cochemist.com/ccimg/132900/132880-11-6_b.png)
![Propanal, 3-[(4-methoxyphenyl)methoxy]-](http://img.cochemist.com/ccimg/128500/128461-65-4.png)
![Propanal, 3-[(4-methoxyphenyl)methoxy]-](http://img.cochemist.com/ccimg/128500/128461-65-4_b.png)








![Butanoic acid, 2-methoxy-4-[[(phenylmethoxy)carbonyl]amino]-, (S)-](http://img.cochemist.com/ccimg/110900/110880-96-1.png)
![Butanoic acid, 2-methoxy-4-[[(phenylmethoxy)carbonyl]amino]-, (S)-](http://img.cochemist.com/ccimg/110900/110880-96-1_b.png)





![Formamide,N-[(1E,3R,4R,5R,9S,10S)-4-(acetyloxy)-11-[(10S,16S,20S,21R,22S,24E)-16-hydroxy-22-methoxy-10,21-dimethyl-12,18-dioxo-3,7,19,27-tetraoxa-29,30,31-triazatetracyclo[24.2.1.12,5.16,9]hentriaconta-2(31),4,6(30),8,24,26(29),28-heptaen-20-yl]-10-methoxy-3,5,9-trimethyl-6-oxo-1-undecen-1-yl]-N-methyl-](http://img.cochemist.com/ccimg/100100/100045-73-6.png)
![Formamide,N-[(1E,3R,4R,5R,9S,10S)-4-(acetyloxy)-11-[(10S,16S,20S,21R,22S,24E)-16-hydroxy-22-methoxy-10,21-dimethyl-12,18-dioxo-3,7,19,27-tetraoxa-29,30,31-triazatetracyclo[24.2.1.12,5.16,9]hentriaconta-2(31),4,6(30),8,24,26(29),28-heptaen-20-yl]-10-methoxy-3,5,9-trimethyl-6-oxo-1-undecen-1-yl]-N-methyl-](http://img.cochemist.com/ccimg/100100/100045-73-6_b.png)
![10,32,45,46-Tetraoxatricyclo[39.3.1.119,23]hexatetraconta-5,7,21,27,29,43-hexaene-9,31-dione,3,13,15,25,35,37-hexahydroxy-11,33-bis[(1S,2S,3S)-2-hydroxy-1,3-dimethyl-5-[(2S,4R,6S)-tetrahydro-4-methoxy-6-methyl-2H-pyran-2-yl]pentyl]-17,39-dimethoxy-6,12,16,28,34,38-hexamethyl-,(1R,3S,5E,7E,11S,12S,13R,15S,16S,17S,19S,23R,25S,27E,29E,33S,34S,35R,37S,38S,39S,41S)-](http://img.cochemist.com/ccimg/96000/95927-67-6.png)
![10,32,45,46-Tetraoxatricyclo[39.3.1.119,23]hexatetraconta-5,7,21,27,29,43-hexaene-9,31-dione,3,13,15,25,35,37-hexahydroxy-11,33-bis[(1S,2S,3S)-2-hydroxy-1,3-dimethyl-5-[(2S,4R,6S)-tetrahydro-4-methoxy-6-methyl-2H-pyran-2-yl]pentyl]-17,39-dimethoxy-6,12,16,28,34,38-hexamethyl-,(1R,3S,5E,7E,11S,12S,13R,15S,16S,17S,19S,23R,25S,27E,29E,33S,34S,35R,37S,38S,39S,41S)-](http://img.cochemist.com/ccimg/96000/95927-67-6_b.png)


![Propanal, 3-[(4-methoxyphenyl)methoxy]-2,2-dimethyl-](http://img.cochemist.com/ccimg/92200/92156-87-1.png)
![Propanal, 3-[(4-methoxyphenyl)methoxy]-2,2-dimethyl-](http://img.cochemist.com/ccimg/92200/92156-87-1_b.png)


![Butanal, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/87200/87184-81-4.png)
![Butanal, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/87200/87184-81-4_b.png)
![Butanoic acid, 3-hydroxy-4-[[(phenylmethoxy)carbonyl]amino]-, (R)-](http://img.cochemist.com/ccimg/80600/80503-94-2.png)
![Butanoic acid, 3-hydroxy-4-[[(phenylmethoxy)carbonyl]amino]-, (R)-](http://img.cochemist.com/ccimg/80600/80503-94-2_b.png)















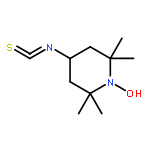
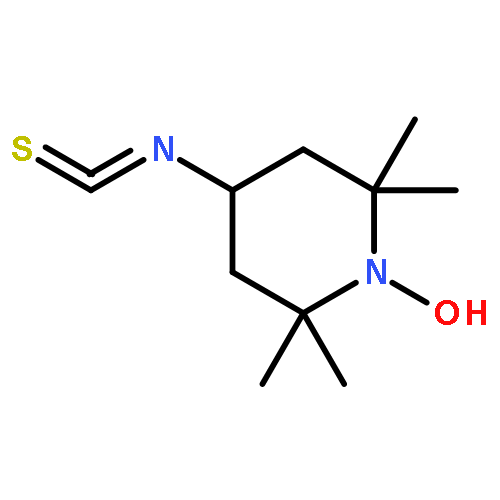




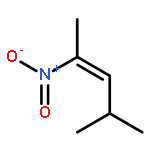
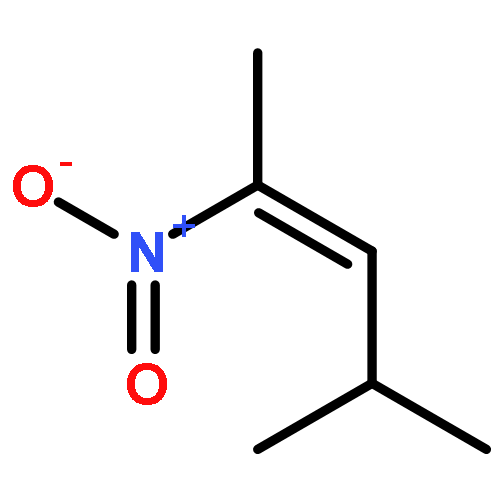










![N-ETHYLMALEIMIDE, [ETHYL-1-14C]](http://img.cochemist.com/ccimg/7100/7013-07-2.png)
![N-ETHYLMALEIMIDE, [ETHYL-1-14C]](http://img.cochemist.com/ccimg/7100/7013-07-2_b.png)
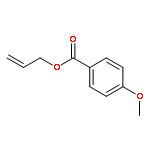
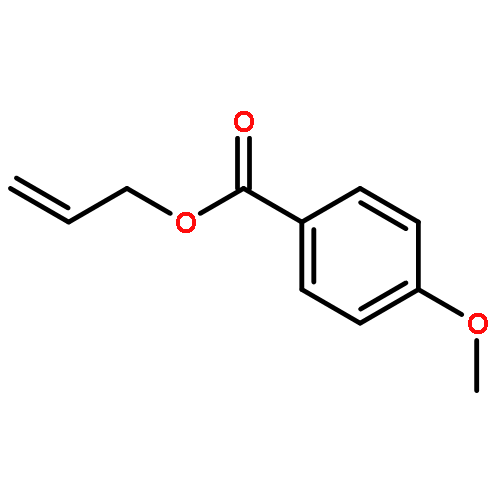
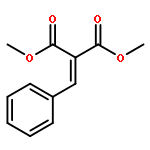
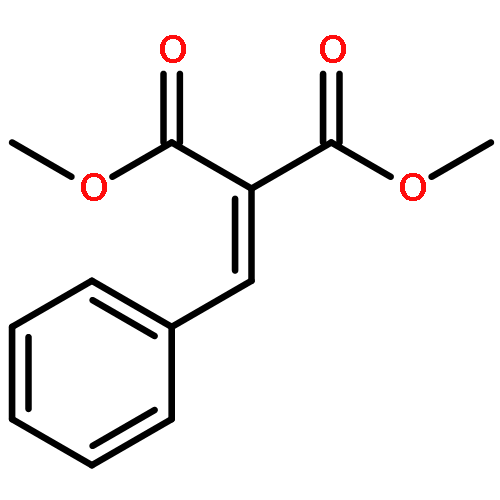





![5-HEXEN-3-OL, 1-[(4-METHOXYPHENYL)METHOXY]-2,2-DIMETHYL-, (3S)-](http://img.cochemist.com/ccimg/885700/885674-86-2.png)
![5-HEXEN-3-OL, 1-[(4-METHOXYPHENYL)METHOXY]-2,2-DIMETHYL-, (3S)-](http://img.cochemist.com/ccimg/885700/885674-86-2_b.png)
![2-BUTANONE, 4-[(4-METHOXYPHENYL)METHOXY]-3-METHYL-, (3S)-](http://img.cochemist.com/ccimg/410100/410077-41-7.png)
![2-BUTANONE, 4-[(4-METHOXYPHENYL)METHOXY]-3-METHYL-, (3S)-](http://img.cochemist.com/ccimg/410100/410077-41-7_b.png)


![5-Hexenal, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (3S)-](http://img.cochemist.com/ccimg/381000/380909-42-2.png)
![5-Hexenal, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (3S)-](http://img.cochemist.com/ccimg/381000/380909-42-2_b.png)


![Phosphine,[(1S)-5,5'-dichloro-6,6'-dimethoxy[1,1'-biphenyl]-2,2'-diyl]bis[diphenyl-](/data/chemimg/110700/185913-98-8.png)
![Phosphine,[(1S)-5,5'-dichloro-6,6'-dimethoxy[1,1'-biphenyl]-2,2'-diyl]bis[diphenyl-](/data/chemimg/110700/185913-98-8_b.png)
![1-Piperidinyloxy, 4-[[(cyclohexylamino)thioxomethyl]amino]-2,2,6,6-tetramethyl-](http://img.cochemist.com/ccimg/111200/111103-86-7.png)
![1-Piperidinyloxy, 4-[[(cyclohexylamino)thioxomethyl]amino]-2,2,6,6-tetramethyl-](http://img.cochemist.com/ccimg/111200/111103-86-7_b.png)
![2-Butanone, 4-[(4-methoxyphenyl)methoxy]-](http://img.cochemist.com/ccimg/85900/85846-38-4.png)
![2-Butanone, 4-[(4-methoxyphenyl)methoxy]-](http://img.cochemist.com/ccimg/85900/85846-38-4_b.png)


![1-Piperidinyloxy,4-[(cyclohexylcarbonimidoyl)amino]-2,2,6,6-tetramethyl-](http://img.cochemist.com/ccimg/42300/42249-40-1.png)
![1-Piperidinyloxy,4-[(cyclohexylcarbonimidoyl)amino]-2,2,6,6-tetramethyl-](http://img.cochemist.com/ccimg/42300/42249-40-1_b.png)




![2-HEPTYN-4-OL, 1-[[(1,1-DIMETHYLETHYL)DIMETHYLSILYL]OXY]-6-METHYL-, (4S)-](http://img.cochemist.com/ccimg/817300/817204-12-9.png)
![2-HEPTYN-4-OL, 1-[[(1,1-DIMETHYLETHYL)DIMETHYLSILYL]OXY]-6-METHYL-, (4S)-](http://img.cochemist.com/ccimg/817300/817204-12-9_b.png)
![11,14-Epoxy-15,18-iminofuro[2,3-n][1,16,5]dioxaazacycloeicosine-2,5,8(3H)-trione, 12-[(Z)-[(2S,4Z)-dihydro-4-(4-methoxy-1-methylbutylidene)-2-(2-methylpropyl)-3(2H)-furanylidene]methyl]-3a,10,11,12,19,21a-hexahydro-3-(hydroxymethyl)-3,21-dimethyl-7-(2-methylpropyl)-, (3R,3aR,6E,11S,12R,20E,21aS)- (9CI)](/data/chemimg/3721500/618911-32-3.png)
![11,14-Epoxy-15,18-iminofuro[2,3-n][1,16,5]dioxaazacycloeicosine-2,5,8(3H)-trione, 12-[(Z)-[(2S,4Z)-dihydro-4-(4-methoxy-1-methylbutylidene)-2-(2-methylpropyl)-3(2H)-furanylidene]methyl]-3a,10,11,12,19,21a-hexahydro-3-(hydroxymethyl)-3,21-dimethyl-7-(2-methylpropyl)-, (3R,3aR,6E,11S,12R,20E,21aS)- (9CI)](/data/chemimg/3721500/618911-32-3_b.png)
![Formamide,N,N'-[(5S,9S,11R,12E,14E,17S,25S,29S,31R,32E,34E,37S)-3,9,23,29-tetrahydroxy-11,17,31,37-tetramethoxy-4,4,24,24-tetramethyl-7,27-dioxo-6,20,26,40-tetraoxa-41,42-diazatricyclo[36.2.1.118,21]dotetraconta-1(41),12,14,18,21(42),32,34,38-octaene-5,25-diyl]bis[(1E,4R,5R,9S,10S)-4,10-dimethoxy-5,9-dimethyl-6-oxo-1-undecene-11,1-diyl]bis[N-methyl-](http://img.cochemist.com/ccimg/150400/150346-23-9.png)
![Formamide,N,N'-[(5S,9S,11R,12E,14E,17S,25S,29S,31R,32E,34E,37S)-3,9,23,29-tetrahydroxy-11,17,31,37-tetramethoxy-4,4,24,24-tetramethyl-7,27-dioxo-6,20,26,40-tetraoxa-41,42-diazatricyclo[36.2.1.118,21]dotetraconta-1(41),12,14,18,21(42),32,34,38-octaene-5,25-diyl]bis[(1E,4R,5R,9S,10S)-4,10-dimethoxy-5,9-dimethyl-6-oxo-1-undecene-11,1-diyl]bis[N-methyl-](http://img.cochemist.com/ccimg/150400/150346-23-9_b.png)
![11,14-Epoxy-15,18-iminofuro[2,3-n][1,16,5]dioxaazacycloeicosine-2,5,8(3H)-trione, 12-[(Z)-[(2S,4Z)-dihydro-4-(4-methoxy-1-methylbutylidene)-2-(2-methylpropyl)-3(2H)-furanylidene]methyl]-3a,6,7,10,11,12,19,21a-octahydro-3-(hydroxymethyl)-3,21-dimethyl-7-(2-methylpropyl)-, (3R,3aR,7S,11S,12R,20E,21aS)- (9CI)](/data/chemimg/3721500/618911-30-1.png)
![11,14-Epoxy-15,18-iminofuro[2,3-n][1,16,5]dioxaazacycloeicosine-2,5,8(3H)-trione, 12-[(Z)-[(2S,4Z)-dihydro-4-(4-methoxy-1-methylbutylidene)-2-(2-methylpropyl)-3(2H)-furanylidene]methyl]-3a,6,7,10,11,12,19,21a-octahydro-3-(hydroxymethyl)-3,21-dimethyl-7-(2-methylpropyl)-, (3R,3aR,7S,11S,12R,20E,21aS)- (9CI)](/data/chemimg/3721500/618911-30-1_b.png)